

Epidemiological and Genetic Characteristics of Respiratory Viral Coinfections with Different Variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Specimen Sampling
2.2. Methodology for the Detection of Respiratory Co-Pathogens
- Extraction
- Multiplex real-time polymerase chain reaction (PCR)
2.3. Sequencing Methodology and Data Analysis
- Multiplex Next-Generation Sequencing
- Genome, phylogenetic, and amino acid analyses
2.4. Statistical Analysis
3. Results
3.1. Patient Characteristics
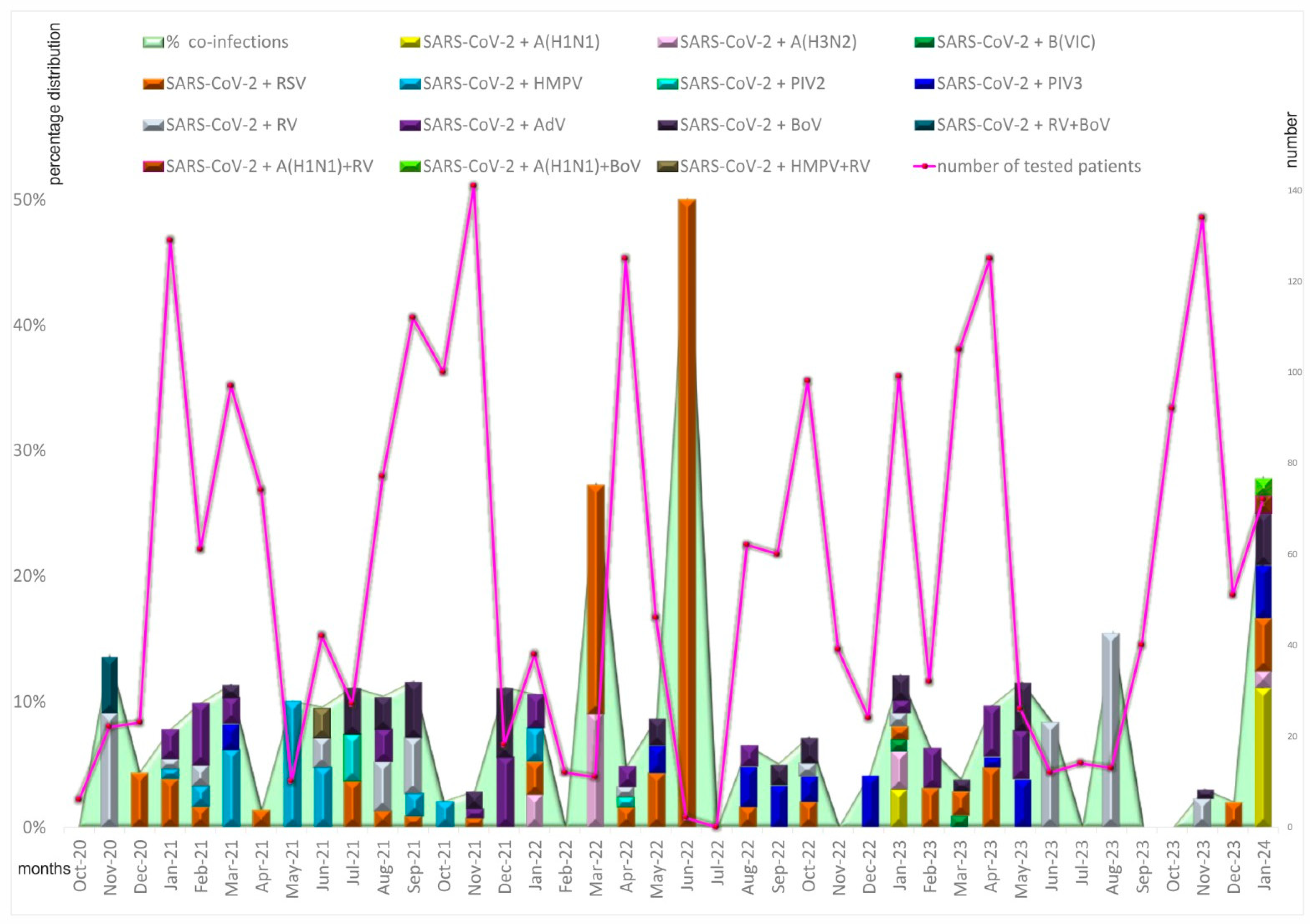
3.2. Seasonal and Age Distribution of Cases with Proven Coinfections
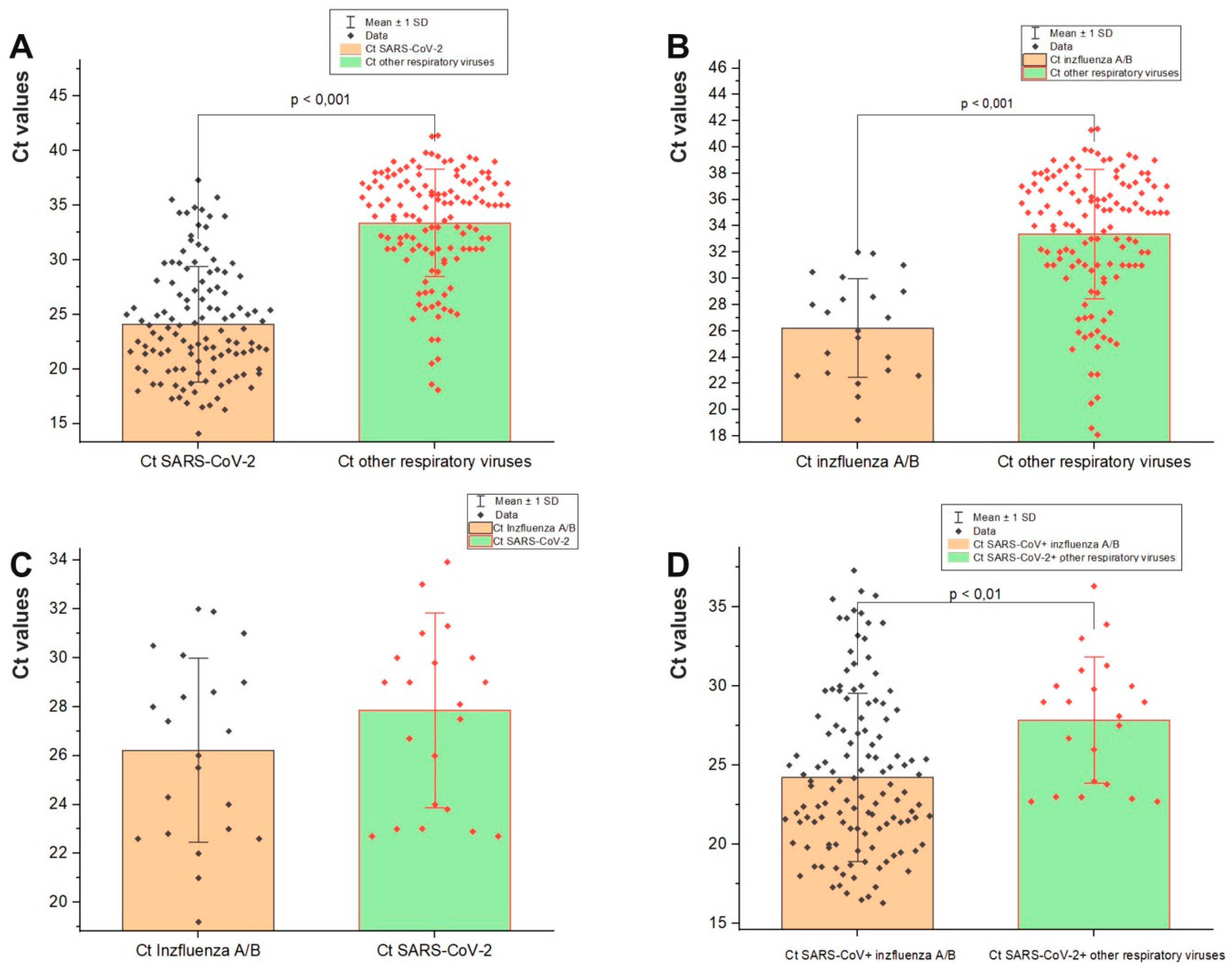
3.3. Viral Load in Mixed Infections
3.4. Sequencing of Respiratory Viral Co-Pathogens
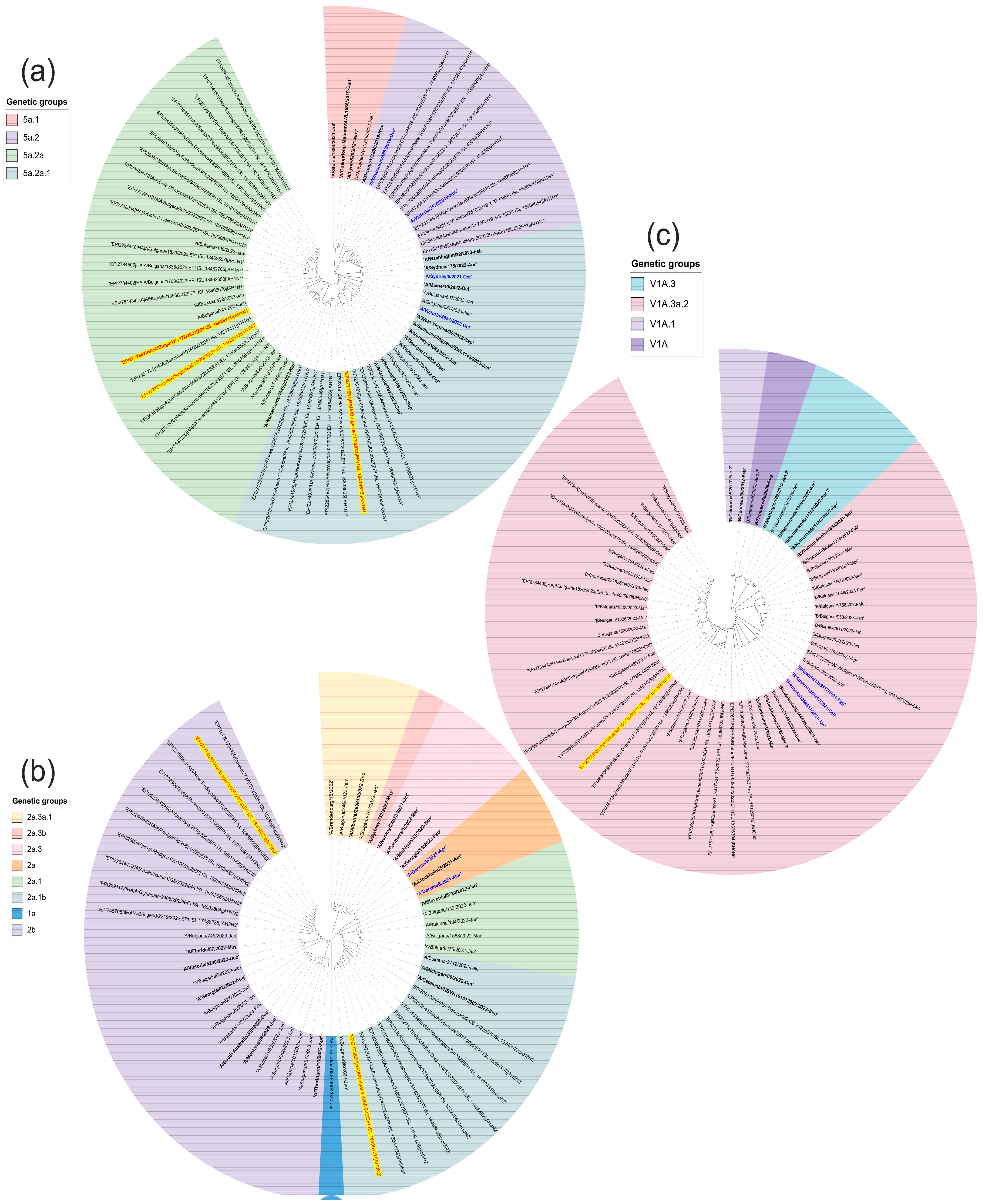
3.5. Phylogenetic Analysis and Amino Acid Analyses of Influenza Viruses Involved in Mixed Infections with SARS-CoV-2
3.6. Genetic Analysis of RSV-B Involved in SARS-CoV-2 Mixed Infections
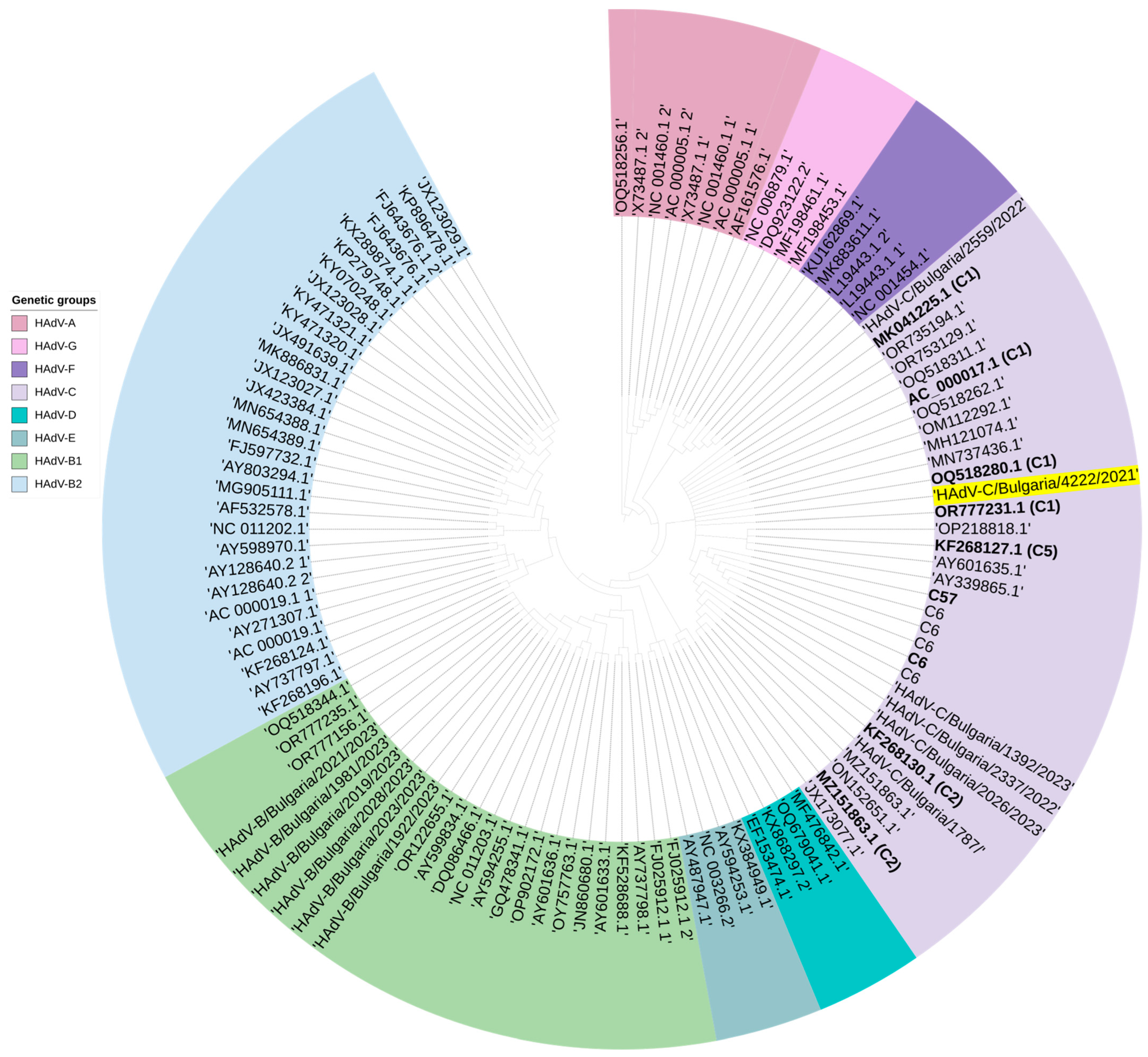
3.7. Genetic Analysis of AdV Involved in Mixed Infection with SARS-CoV-2
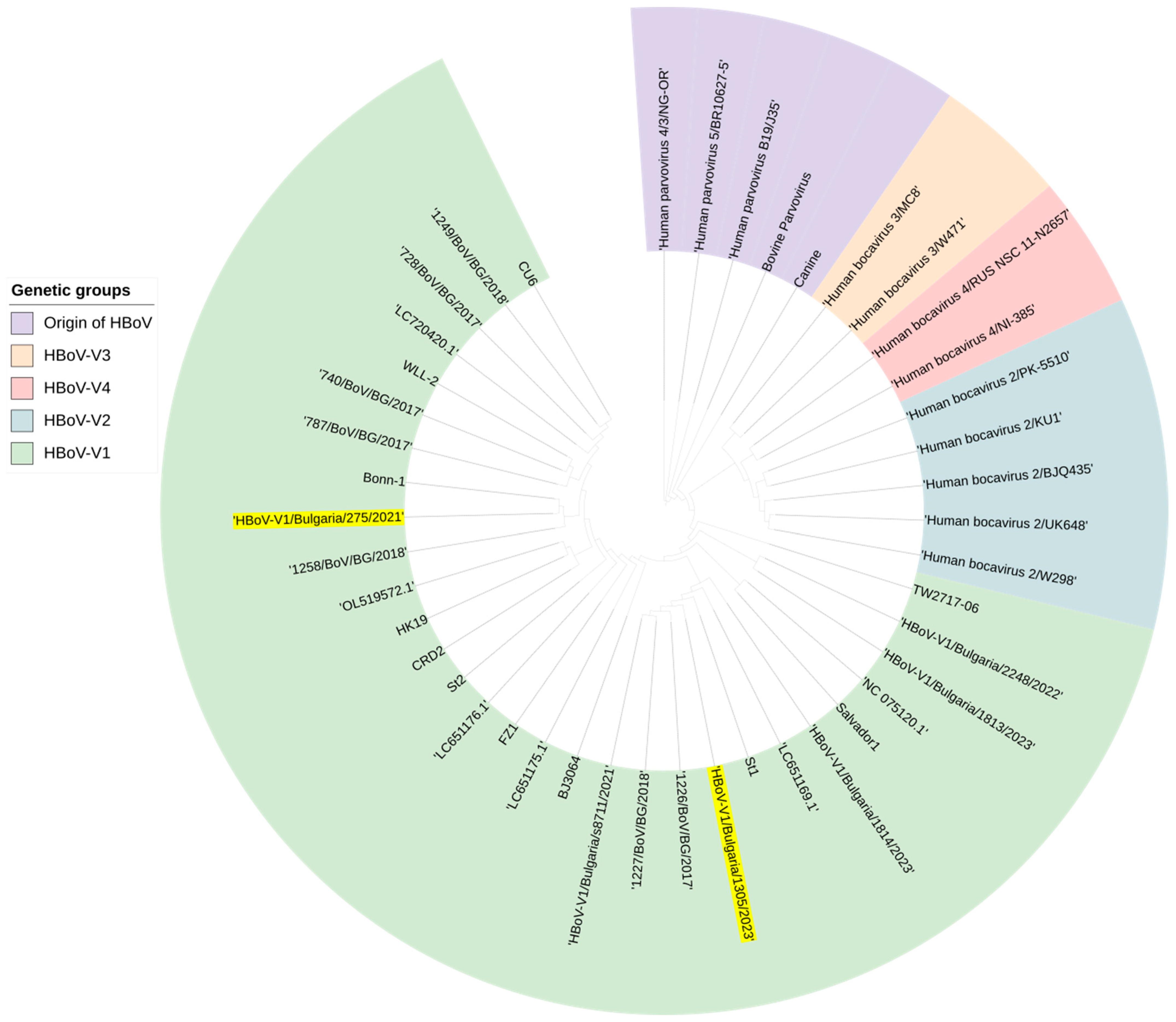
3.8. Genetic Analysis of HBoV Involved in Mixed Infection with SARS-CoV-2
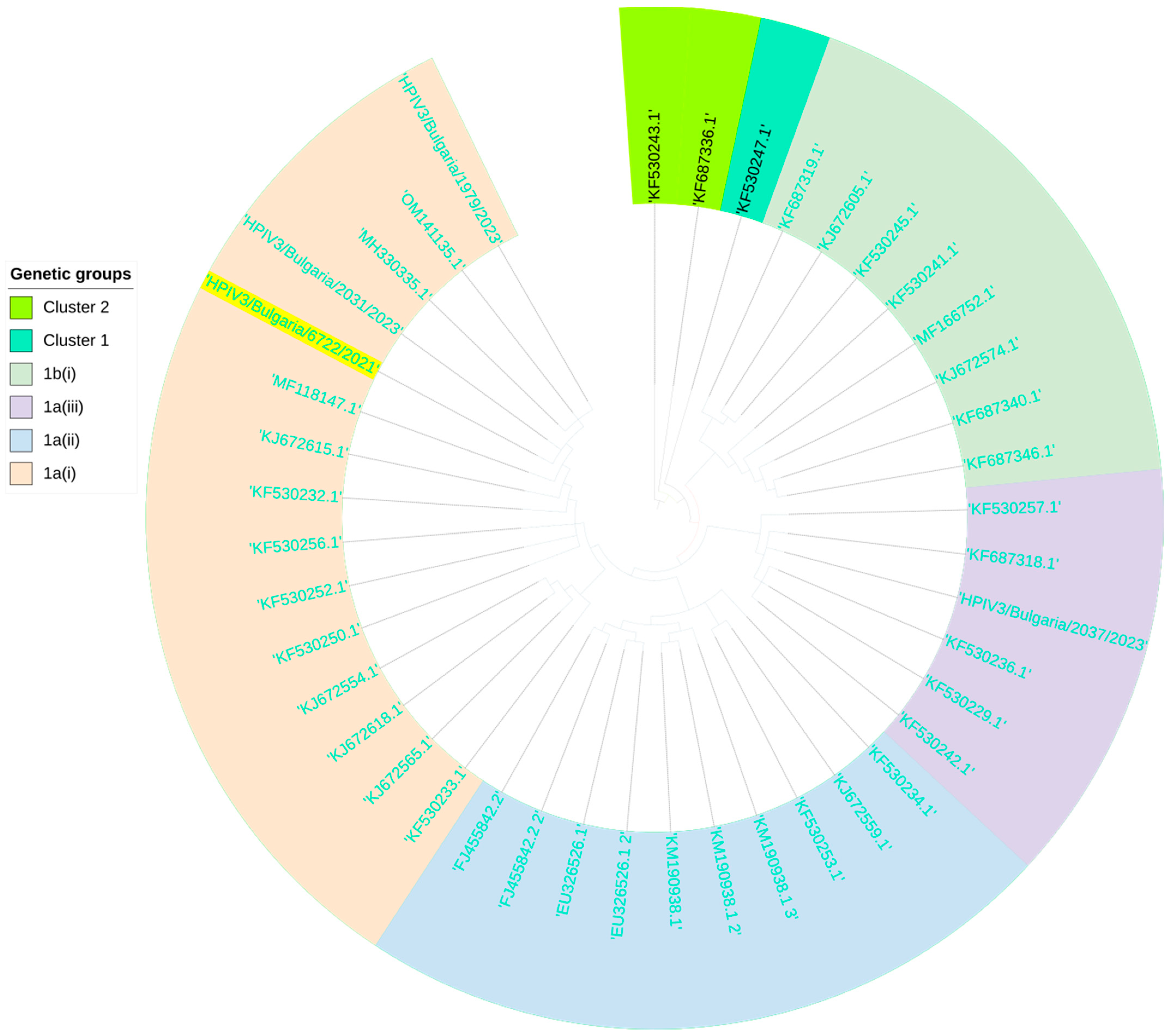
3.9. Genetic Analysis of PIV3 Involved in Mixed Infection with SARS-CoV-2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, P.; Zhang, Y.; Shen, W.; Shi, S.; Zhao, Z. dbGSRV: A manually curated database of genetic susceptibility to respiratory virus. PLoS ONE 2022, 17, e0262373. [Google Scholar] [CrossRef]
- Wu, X.; Cai, Y.; Huang, X.; Yu, X.; Zhao, L.; Wang, F.; Li, Q.; Gu, S.; Xu, T.; Li, Y.; et al. Co-infection with SARS-CoV-2 and Influenza A Virus in Patient with Pneumonia, China. Emerg. Infect. Dis. 2020, 26, 1324–1326. [Google Scholar] [CrossRef]
- Cuadrado-Payán, E.; Montagud-Marrahi, E.; Torres-Elorza, M.; Bodro, M.; Blasco, M.; Poch, E.; Soriano, A.; Piñeiro, G.J. SARS-CoV-2 and influenza virus co-infection. Lancet 2020, 395, e84. [Google Scholar] [CrossRef] [PubMed]
- Maltezou, H.C.; Papanikolopoulou, A.; Vassiliu, S.; Theodoridou, K.; Nikolopoulou, G.; Sipsas, N.V. COVID-19 and Respiratory Virus Co-Infections: A Systematic Review of the Literature. Viruses 2023, 15, 865. [Google Scholar] [CrossRef] [PubMed]
- Rubin, R. Influenza’s Unprecedented Low Profile During COVID-19 Pandemic Leaves Experts Wondering What This Flu Season Has in Store. JAMA 2021, 326, 899–900. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.S.; Wood, T.; Jelley, L.; Jennings, T.; Jefferies, S.; Daniells, K.; Nesdale, A.; Dowell, T.; Turner, N.; Campbell-Stokes, P.; et al. Impact of the COVID-19 nonpharmaceutical interventions on influenza and other respiratory viral infections in New Zealand. Nat. Commun. 2021, 12, 1001. [Google Scholar] [CrossRef]
- Fujita, D.M.; Dos Santos Soares, G.; Sartori, G.P.; Henrique da Silva Nali, L. COVID-19 and Influenza coinfection: The rise of Ômicron and H3N2 in Brazil—2022. Travel. Med. Infect. Dis. 2022, 46, 102262. [Google Scholar] [CrossRef]
- Principi, N.; Autore, G.; Ramundo, G.; Esposito, S. Epidemiology of Respiratory Infections during the COVID-19 Pandemic. Viruses 2023, 15, 1160. [Google Scholar] [CrossRef]
- Maltezou, H.C.; Theodoridou, K.; Poland, G. Influenza immunization and COVID-19. Vaccine 2020, 38, 6078–6079. [Google Scholar] [CrossRef]
- Khasawneh, A.I.; Himsawi, N.M.; Abu-Raideh, J.A.; Sammour, A.; Abu Safieh, H.; Obeidat, A.; Azab, M.; Tarifi, A.A.; Al Khawaldeh, A.; Al-Momani, H.; et al. Prevalence of SARS-CoV-2 and other respiratory pathogens among a Jordanian subpopulation during Delta-to-Omicron transition: Winter 2021/2022. PLoS ONE 2023, 18, e0283804. [Google Scholar] [CrossRef]
- Kim, E.H.; Nguyen, T.Q.; Casel, M.A.B.; Rollon, R.; Kim, S.M.; Kim, Y.I.; Yu, K.M.; Jang, S.G.; Yang, J.; Poo, H.; et al. Coinfection with SARS-CoV-2 and Influenza A Virus Increases Disease Severity and Impairs Neutralizing Antibody and CD4+ T Cell Responses. J. Virol. 2022, 96, e0187321. [Google Scholar] [CrossRef] [PubMed]
- Bordi, L.; Vulcano, A.; Sberna, G.; Nonis, M.; Giacomini, P.; Maggi, F.; Fontana, C.; Lalle, E. Co-Circulation of SARS-CoV-2 and Other Respiratory Pathogens in Upper and Lower Respiratory Tracts during Influenza Season 2022–2023 in Lazio Region. Microorganisms 2023, 11, 2239. [Google Scholar] [CrossRef] [PubMed]
- Lansbury, L.; Lim, B.; Baskaran, V.; Lim, W.S. Co-infections in people with COVID-19: A systematic review and meta-analysis. J. Infect. 2020, 81, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Le Hingrat, Q.; Bouzid, D.; Choquet, C.; Laurent, O.; Lescure, F.X.; Timsit, J.F.; Houhou-Fidouh, N.; Casalino, E.; Lucet, J.C.; Descamps, D.; et al. Viral epidemiology and SARS-CoV-2 co-infections with other respiratory viruses during the first COVID-19 wave in Paris. France. Influenza Other Respir. Viruses 2021, 15, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Negi, A.; Negi, S.; Diksha, D.; Kandwal, D.; Singh, A.; Singh, A.; Gupta, P.; Kalita, D. Incidence of SARS-CoV-2 Co-infections During the Second Wave in Sub-Himalayan Region. India. Cureus 2023, 15, e36215. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.D.; Sordillo, E.M.; Gitman, M.R.; Paniz Mondolfi, A.E. Coinfection in SARS-CoV-2 infected patients: Where are influenza virus and rhinovirus/enterovirus? J. Med. Virol. 2020, 92, 1699–1700. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. The role of co-infections and secondary infections in patients with COVID-19. Pneumonia 2021, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Varela, F.H.; Sartor, I.T.S.; Polese-Bonatto, M.; Azevedo, T.R.; Kern, L.B.; Fazolo, T.; de David, C.N.; Zavaglia, G.O.; Fernandes, I.R.; Krauser, J.R.M.; et al. Rhinovirus as the main co-circulating virus during the COVID-19 pandemic in children. J. Pediatr. 2022, 98, 579–586. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Campogiani, L.; Iannetta, M.; Iannazzo, R.; Imeneo, A.; Alessio, G.; D’Aquila, V.; Massa, B.; Fato, I.; Rindi, L.V.; et al. The Impact of Viral and Bacterial Co-Infections and Home Antibiotic Treatment in SARS-CoV-2 Hospitalized Patients at the Policlinico Tor Vergata Hospital. Rome. Italy. Antibiotics 2023, 12, 1348. [Google Scholar] [CrossRef]
- Trifonova, I.; Christova, I.; Madzharova, I.; Angelova, S.; Voleva, S.; Yordanova, R.; Tcherveniakova, T.; Krumova, S.; Korsun, N. Clinical significance and role of coinfections with respiratory pathogens among individuals with confirmed severe acute respiratory syndrome coronavirus-2 infection. Front. Public Health 2022, 10, 959319. [Google Scholar] [CrossRef]
- Sheehan, N.P.; Leonelli, S.; Botta, F. From Collection to Analysis: A Comparison of GISAID and the COVID-19 Data Portal. bioRxiv 2023. [Google Scholar] [CrossRef]
- Motayo, B.O.; Oluwasemowo, O.O.; Olusola, B.A.; Akinduti, P.A.; Arege, O.T.; Obafemi, Y.D.; Faneye, A.O.; Isibor, P.O.; Aworunse, O.S.; Oranusi, S.U. Evolution and genetic diversity of SARS-CoV-2 in Africa using whole genome sequences. Int. J. Infect. Dis. 2021, 103, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Wong, S.Y.; Liew, N.W.C.; Joseph, N.; Zakaria, Z.; Nurulfiza, I.; Soe, H.J.; Kairon, R.; Amin-Nordin, S.; Chee, H.Y. Whole genome sequencing analysis of SARS-CoV-2 from Malaysia: From alpha to Omicron. Front. Med. 2022, 9, 1001022. [Google Scholar] [CrossRef] [PubMed]
- Public Health England. SARS-CoV-2 Variants of Concern and Variants under Investigation in England. 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/984274/Variants_of_Concern_VOC_Technical_Briefing_10_England.pdf (accessed on 10 June 2024).
- Ward, T.; Glaser, A.; Johnsen, A.; Xu, F.; Hall, I.; Pellis, L. Growth, reproduction numbers and factors affecting the spread of SARS-CoV-2 novel variants of concern in the UK from October 2020 to July 2021: A modelling analysis. BMJ Open 2021, 11, e056636. [Google Scholar] [CrossRef] [PubMed]
- Tallei, T.E.; Alhumaid, S.; AlMusa, Z.; Fatimawali Kusumawaty, D.; Alynbiawi, A.; Alshukairi, A.N.; Rabaan, A.A. Update on the omicron sub-variants BA.4 and BA.5. Rev. Med. Virol. 2023, 33, e2391. [Google Scholar] [CrossRef] [PubMed]
- Vogel, L. What to know about Omicron XBB.1.5. CMAJ 2023, 195, E127–E128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef] [PubMed]
- Looi, M.K. What do we know about the Arcturus XBB.1.16 subvariant? BMJ 2023, 381, 1074. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: The XBB.1.5 (‘Kraken’) Subvariant of Omicron SARS-CoV-2 and its Rapid Global Spread. Med. Sci. Monit. 2023, 29, e939580. [Google Scholar] [CrossRef]
- Ramos, N.; Panzera, Y.; Frabasile, S.; Tomás, G.; Calleros, L.; Marandino, A.; Goñi, N.; Techera, C.; Grecco, S.; Fuques, E.; et al. A multiplex-NGS approach to identifying respiratory RNA viruses during the COVID-19 pandemic. Arch. Virol. 2023, 168, 87. [Google Scholar] [CrossRef]
- Tsibane, T.; Ekiert, D.C.; Krause, J.C.; Martinez, O.; Crowe, J.E., Jr.; Wilson, I.A.; Basler, C.F. Influenza human monoclonal antibody 1F1 interacts with three major antigenic sites and residues mediating human receptor specificity in H1N1 viruses. PLoS Pathog. 2012, 8, e1003067. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Wang, G.; Li, A.; Zhang, Q.; Wu, C.; Zhang, R.; Cai, Q.; Song, W.; Yuen, K.Y. The hemagglutinin structure of an avian H1N1 influenza A virus. Virology 2009, 392, 73–81. [Google Scholar] [CrossRef]
- Burrel, S.; Hausfater, P.; Dres, M.; Pourcher, V.; Luyt, C.E.; Teyssou, E.; Soulié, C.; Calvez, V.; Marcelin, A.G.; Boutolleau, D. Co-infection of SARS-CoV-2 with other respiratory viruses and performance of lower respiratory tract samples for the diagnosis of COVID-19. Int. J. Infect. Dis. 2021, 102, 10–13. [Google Scholar] [CrossRef]
- Morales-Jadán, D.; Muslin, C.; Viteri-Dávila, C.; Coronel, B.; Castro-Rodríguez, B.; Vallejo-Janeta, A.P.; Henríquez-Trujillo, A.R.; Garcia-Bereguiain, M.A.; Rivera-Olivero, I.A. Coinfection of SARS-CoV-2 with other respiratory pathogens in outpatients from Ecuador. Front. Public Health 2023, 11, 1264632. [Google Scholar] [CrossRef]
- Moumbeket Yifomnjou, M.H.; Monamele, G.C.; Njankouo-Ripa, M.; Fatawou Modiyinji, A.; Ngoupo, P.A.; Boyomo, O.; Njouom, R. Viral co-infection with human respiratory syncytial virus in suspected acute and severe respiratory tract infections during COVID-19 pandemic in Yaoundé, Cameroon, 2020–2021. Influenza Other Respir. Viruses 2023, 17, e13131. [Google Scholar] [CrossRef] [PubMed]
- Pucarelli-Lebreiro, G.; Venceslau, M.T.; Cordeiro, C.C.; Maciel, F.Q.; Anachoreta, T.D.; de Abreu, T.F.; Frota, A.C.C.; Castiñeiras, T.M.P.P.; da Costa, A.M.; Lopes, A.C.D.L.; et al. Clinical Manifestations and Complications of Children with COVID-19 Compared to Other Respiratory Viral Infections: A Cohort Inpatient Study from Rio de Janeiro, Brazil. Front. Pediatr. 2022, 10, 934648. [Google Scholar] [CrossRef]
- Nunziata, F.; Salomone, S.; Catzola, A.; Poeta, M.; Pagano, F.; Punzi, L.; Lo Vecchio, A.; Guarino, A.; Bruzzese, E. Clinical Presentation and Severity of SARS-CoV-2 Infection Compared to Respiratory Syncytial Virus and Other Viral Respiratory Infections in Children Less than Two Years of Age. Viruses 2023, 15, 717. [Google Scholar] [CrossRef]
- Samal, J.; Agarwal, R.; Soni, A.; Pandey, A.; Thapar, S.; Gupta, E. Co-infection of SARS-CoV-2 with other respiratory pathogens in patients with liver disease. Access Microbiol. 2022, 4, acmi000456. [Google Scholar] [CrossRef]
- Zhu, X.; Tian, F.; Li, Y.; Lu, Q.; Long, Q.; Long, X.; Cao, D. High Prevalence of Respiratory Co-Infections and Risk Factors in COVID-19 Patients at Hospital Admission During an Epidemic Peak in China. Infect. Drug Resist. 2023, 16, 6781–6793. [Google Scholar] [CrossRef] [PubMed]
- Kırca, F.; Aydoğan, S.; Gozalan, A.; Güler, E.; Uyan Erten, A.Z.; Özşen Uygur, A.S.; Doğan, A.; Dinc, B. Impact of non-pharmaceutical interventions on circulating respiratory viruses during the COVID-19 pandemic in Turkey. Ann. Saudi Med. 2023, 43, 143–153. [Google Scholar] [CrossRef]
- Uhteg, K.; Amadi, A.; Forman, M.; Mostafa, H.H. Circulation of Non-SARS-CoV-2 Respiratory Pathogens and Coinfection with SARS-CoV-2 Amid the COVID-19 Pandemic. Open Forum Infect. Dis. 2021, 9, ofab618. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Guan, W.; Liu, Y.; Lin, Z.; Liang, W.; Liang, J.; Chen, B.; Wu, T.; Wang, Y.; Yang, C.; et al. Different Circulation Pattern of Multiple Respiratory Viruses in Southern China During the COVID-19 Pandemic. Front. Microbiol. 2022, 12, 801946. [Google Scholar] [CrossRef] [PubMed]
- Korsun, N.; Trifonova, I.; Dobrinov, V.; Madzharova, I.; Grigorova, I.; Christova, I. Low prevalence of influenza viruses and predominance of A(H3N2) virus with respect to SARS-CoV-2 during the 2021–2022 season in Bulgaria. J. Med. Virol. 2023, 95, e28489. [Google Scholar] [CrossRef] [PubMed]
- Inaida, S.; Paul, R.E.; Matsuno, S. Viral transmissibility of SARS-CoV-2 accelerates in the winter, similarly to influenza epidemics. Am. J. Infect. Control 2022, 50, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Nichols, G.L.; Gillingham, E.L.; Macintyre, H.L.; Vardoulakis, S.; Hajat, S.; Sarran, C.E.; Amankwaah, D.; Phalkey, R. Coronavirus seasonality, respiratory infections and weather. BMC Infect. Dis. 2021, 21, 1101. [Google Scholar] [CrossRef] [PubMed]
- Korsun, N.; Trifonova, I.; Madzharova, I.; Alexiev, I.; Uzunova, I.; Ivanov, I.; Velikov, P.; Tcherveniakova, T.; Christova, I. Resurgence of respiratory syncytial virus with dominance of RSV-B during the 2022–2023 season. Front. Microbiol. 2024, 15, 1376389. [Google Scholar] [CrossRef] [PubMed]
- Mandelia, Y.; Procop, G.W.; Richter, S.S.; Worley, S.; Liu, W.; Esper, F. Dynamics and predisposition of respiratory viral co-infections in children and adults. Clin. Microbiol. Infect. 2021, 27, 631.e1–631.e6. [Google Scholar] [CrossRef]
- Pigny, F.; Wagner, N.; Rohr, M.; Mamin, A.; Cherpillod, P.; Posfay-Barbe, K.M.; Kaiser, L.; Eckerle, I.; L’Huillier, A.G.; Geneva Pediatric COVID Group. Viral co-infections among SARS-CoV-2-infected children and infected adult household contacts. Eur. J. Pediatr. 2021, 180, 1991–1995. [Google Scholar] [CrossRef]
- Kuchar, E.; Załęski, A.; Wronowski, M.; Krankowska, D.; Podsiadły, E.; Brodaczewska, K.; Lewicka, A.; Lewicki, S.; Kieda, C.; Horban, A.; et al. Children were less frequently infected with SARS-CoV-2 than adults during 2020 COVID-19 pandemic in Warsaw, Poland. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 541–547. [Google Scholar] [CrossRef]
- Davies, N.G.; Klepac, P.; Liu, Y.; Prem, K.; Jit, M.; CMMID COVID-19 working group. Eggo RM. Age-dependent effects in the transmission and control of COVID-19 epidemics. Nat. Med. 2020, 26, 1205–1211. [Google Scholar] [CrossRef]
- Bayhan, G.İ.; Özkubat Korkmaz, I.; Şahiner, E.S.; Tekeli, N.; Akdağ, A.G.; Uyan Erten, A.Z.; Taşbaşı, A.B.; Göre, B.; Doğru, M.; Kırca, F.; et al. Are SARS-CoV-2 viral loads in children lower than in adults? J. Infect. 2023, 86, e13–e14. [Google Scholar] [CrossRef] [PubMed]
- Dhochak, N.; Singhal, T.; Kabra, S.K.; Lodha, R. Pathophysiology of COVID-19: Why Children Fare Better than Adults? Indian J. Pediatr. 2020, 87, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Manti, S.; Licari, A.; Montagna, L.; Votto, M.; Leonardi, S.; Brambilla, I.; Castagnoli, R.; Foiadelli, T.; Marseglia, G.L.; Cardinale, F.; et al. SARS-CoV-2 infection in pediatric population. Acta Biomed. 2020, 91, e2020003. [Google Scholar] [CrossRef]
- Han, S.B.; Shin, J.A.; Kim, S.K.; Lee, J.W.; Lee, D.G.; Chung, N.G.; Cho, B.; Jeong, D.C.; Kang, J.H. Respiratory Viral Infections in Children and Adolescents with Hematological Malignancies. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019006. [Google Scholar] [CrossRef] [PubMed]
- Trapani, S.; Caporizzi, A.; Ricci, S.; Indolfi, G. Human Bocavirus in Childhood: A True Respiratory Pathogen or a “Passenger” Virus? A Comprehensive Review. Microorganisms 2023, 11, 1243. [Google Scholar] [CrossRef] [PubMed]
- Cong, B.; Deng, S.; Wang, X.; Li, Y. The role of respiratory co-infection with influenza or respiratory syncytial virus in the clinical severity of COVID-19 patients: A systematic review and meta-analysis. J. Glob. Health 2022, 12, 05040. [Google Scholar] [CrossRef]
- Cheng, Y.; Ma, J.; Wang, H.; Wang, X.; Hu, Z.; Li, H.; Zhang, H.; Liu, X. Co-infection of influenza A virus and SARS-CoV-2: A retrospective cohort study. J. Med. Virol. 2021, 93, 2947–2954. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Malhotra, B.; Sharma, P.; Bhomia, N.; Deeba, F.; Sharma, H.; Parsoya, D. Co-infection of respiratory pathogens in SARS-CoV-2 positive patients at North West India, Rajasthan. Indian J. Med. Microbiol. 2023, 44, 100371. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Li, C.; Wang, Y.; Wu, F.; Liang, C.; Deng, W.; Wang, Y. Targeted next-generation sequencing for pulmonary infection diagnosis in patients unsuitable for bronchoalveolar lavage. Front. Med. 2023, 10, 1321515. [Google Scholar] [CrossRef]
- Guo, W.; Cui, X.; Wang, Q.; Wei, Y.; Guo, Y.; Zhang, T.; Zhan, J. Clinical evaluation of metagenomic next-generation sequencing for detecting pathogens in bronchoalveolar lavage fluid collected from children with community-acquired pneumonia. Front. Med. 2022, 9, 952636. [Google Scholar] [CrossRef]
- Kanji, J.N.; Zelyas, N.; Pabbaraju, K.; Granger, D.; Wong, A.; Murphy, S.A.; Buss, E.; MacDonald, C.; Berenger, B.M.; Diggle, M.A.; et al. Respiratory virus coinfections with severe acute respiratory coronavirus virus 2 (SARS-CoV-2) continue to be rare one year into the coronavirus disease 2019 (COVID-19) pandemic in Alberta, Canada (June 2020–May 2021). Infect. Control Hosp. Epidemiol. 2023, 44, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Deveson, I.W.; Pang, C.N.I.; Yeang, M.; Naing, Z.; Adikari, T.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Verich, A.; et al. Respiratory viral co-infections among SARS-CoV-2 cases confirmed by virome capture sequencing. Sci. Rep. 2021, 11, 3934. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Njau, A.; Petty, Z.; Chen, J.; Ananth, S.; Ahluwalia, P.; Williams, C.; Ross, T.M.; Chaubey, A.; et al. High-Throughput Next-Generation Sequencing Respiratory Viral Panel: A Diagnostic and Epidemiologic Tool for SARS-CoV-2 and Other Viruses. Viruses 2021, 13, 2063. [Google Scholar] [CrossRef] [PubMed]
- Pipoli da Fonseca, J.; Kornobis, E.; Turc, E.; Enouf, V.; Lemée, L.; Cokelaer, T.; Monot, M. Capturing SARS-CoV-2 from patient samples with low viral abundance: A comparative analysis. Sci. Rep. 2022, 12, 19274. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bhattacharya, M.; Nag, S.; Dhama, K.; Chakraborty, C. A Detailed Overview of SARS-CoV-2 Omicron: Its Sub-Variants, Mutations and Pathophysiology, Clinical Characteristics, Immunological Landscape, Immune Escape, and Therapies. Viruses 2023, 15, 167. [Google Scholar] [CrossRef] [PubMed]
- Madzharova, I.; Tifonova, I.; Dobrinov, V.; Grigorova, I.; Alexiev, I.; Grigorova, L.; Dimitrova, R.; Ivan, I.; Ivan, S.; Dean, D.; et al. Dynamic of SARS-CoV-2 spread in Bulgaria, 2020–2022. Probl. Infect. Parasit. Dis. 2023, 51, 5–10. [Google Scholar] [CrossRef]
- Liang, M.; Fan, Y.; Zhang, D.; Yang, L.; Wang, X.; Wang, S.; Xu, J.; Zhang, J. Metagenomic next-generation sequencing for accurate diagnosis and management of lower respiratory tract infections. Int. J. Infect. Dis. 2022, 122, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qiu, X.; Wang, T.; Zhang, J. The Diagnostic Value of Metagenomic Next-Generation Sequencing in Lower Respiratory Tract Infection. Front. Cell Infect. Microbiol. 2021, 11, 694756. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, C.M.; Kistner, O.; Montomoli, E.; Viviani, S.; Marchi, S. Influenza Viruses and Vaccines: The Role of Vaccine Effectiveness Studies for Evaluation of the Benefits of Influenza Vaccines. Vaccines 2022, 10, 714. [Google Scholar] [CrossRef]
- Fratty, I.S.; Jurkowicz, M.; Zuckerman, N.; Nemet, I.; Atari, N.; Kliker, L.; Gur-Arie, L.; Rosenberg, A.; Glatman-Freedman, A.; Lustig, Y.; et al. Influenza vaccine compatibility among hospitalized patients during and after the COVID-19 pandemic. Front. Microbiol. 2024, 14, 1296179. [Google Scholar] [CrossRef]
- Igarashi, M.; Ito, K.; Yoshida, R.; Tomabechi, D.; Kida, H.; Takada, A. Predicting the antigenic structure of the pandemic (H1N1) 2009 influenza virus hemagglutinin. PLoS ONE 2010, 5, e8553. [Google Scholar] [CrossRef] [PubMed]
- Zolotarova, O.; Budzanivska, I.; Leibenko, L.; Radchenko, L.; Mironenko, A. Antigenic Site Variation in the Hemagglutinin of Pandemic Influenza A(H1N1)pdm09 Viruses between 2009–2017 in Ukraine. Pathogens 2019, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, R.; Tamburrino, G.; Schewe, K.E.; Crowe, J.; Nuccitelli, A.; Dibben, O. Haemagglutinin substitutions N125D, D127E, D222G and R223Q improve replicative fitness and vaccine effectiveness of an A/H1N1pdm09 live attenuated influenza vaccine virus by enhancing α-2,6 receptor binding. PLoS Pathog. 2022, 18, e1010585. [Google Scholar] [CrossRef] [PubMed]
- Byrd-Leotis, L.; Galloway, S.E.; Agbogu, E.; Steinhauer, D.A. Influenza hemagglutinin (HA) stem region mutations that stabilize or destabilize the structure of multiple HA subtypes. J. Virol. 2015, 89, 4504–4516. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Influenza Virus Characterization Summary Report, Europe, December 2023 (No. WHO/EURO: 2023 6189 45954 68789); World Health Organization, Regional Office for Europe: Copenhagen, Denmark, 2023. [Google Scholar]
- Fall, A.; Han, L.; Yunker, M.; Gong, Y.N.; Li, T.J.; Norton, J.M.; Abdullah, O.; Rothman, R.E.; Fenstermacher, K.Z.J.; Morris, C.P.; et al. Evolution of Influenza A(H3N2) Viruses in 2 Consecutive Seasons of Genomic Surveillance, 2021–2023. Open Forum Infect. Dis. 2023, 10, ofad577. [Google Scholar] [CrossRef] [PubMed]
- Bolton, M.J.; Ort, J.T.; McBride, R.; Swanson, N.J.; Wilson, J.; Awofolaju, M.; Furey, C.; Greenplate, A.R.; Drapeau, E.M.; Pekosz, A.; et al. Antigenic and virological properties of an H3N2 variant that continues to dominate the 2021–2022 Northern Hemisphere influenza season. Cell Rep. 2022, 39, 110897. [Google Scholar] [CrossRef] [PubMed]
- Gulati, U.; Hwang, C.C.; Venkatramani, L.; Gulati, S.; Stray, S.J.; Lee, J.T.; Laver, W.G.; Bochkarev, A.; Zlotnick, A.; Air, G.M. Antibody epitopes on the neuraminidase of a recent H3N2 influenza virus (A/Memphis/31/98). J. Virol. 2002, 76, 12274–12280. [Google Scholar] [CrossRef] [PubMed]
- Koul, P.A. Influenza vaccination: A case for removal of B/Yamagata from the quadrivalent vaccine. Lung India 2024, 41, 149–150. [Google Scholar] [CrossRef]
- Vajo, Z.; Torzsa, P. Extinction of the Influenza B Yamagata Line during the COVID Pandemic-Implications for Vaccine Composition. Viruses 2022, 14, 1745. [Google Scholar] [CrossRef]
- Dreyfus, C.; Laursen, N.S.; Kwaks, T.; Zuijdgeest, D.; Khayat, R.; Ekiert, D.C.; Lee, J.H.; Metlagel, Z.; Bujny, M.V.; Jongeneelen, M.; et al. Highly conserved protective epitopes on influenza B viruses. Science 2012, 337, 1343–1348. [Google Scholar] [CrossRef]
- Oakley, A.J.; Barrett, S.; Peat, T.S.; Newman, J.; Streltsov, V.A.; Waddington, L.; Saito, T.; Tashiro, M.; McKimm-Breschkin, J.L. Structural and functional basis of resistance to neuraminidase inhibitors of influenza B viruses. J. Med. Chem. 2010, 53, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Kang, D.S.; Zheng, A.; Liu, S.T.H.; Broecker, F.; Simon, V.; Krammer, F.; Palese, P. Antibody Responses toward the Major Antigenic Sites of Influenza B Virus Hemagglutinin in Mice, Ferrets, and Humans. J. Virol. 2019, 93, e01673-18. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Ray, W.C.; Peeples, M.E. Structure and function of respiratory syncytial virus surface glycoproteins. Curr. Top. Microbiol. Immunol. 2013, 372, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Quan, F.S.; Basak, S.; Chu, K.B.; Kim, S.S.; Kang, S.M. Progress in the development of virus-like particle vaccines against respiratory viruses. Expert Rev. Vaccines 2020, 19, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.A.; Holland, S.C.; Smith, M.F.; Leonard, V.R.; Murugan, V.; Nordstrom, L.; Mulrow, M.; Salgado, R.; White, M.; Lim, E.S. Genomic Sequencing Surveillance to Identify Respiratory Syncytial Virus Mutations, Arizona, USA. Emerg. Infect. Dis. 2023, 29, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.J.; Mousa, J.J. Structural basis for respiratory syncytial virus and human metapneumovirus neutralization. Curr. Opin. Virol. 2023, 61, 101337. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Abu-Raya, B.; Bonanni, P.; Cahn-Sellem, F.; Flanagan, K.L.; Martinon Torres, F.; Mejias, A.; Nadel, S.; Safadi, M.A.P.; Simon, A. Coadministration of Anti-Viral Monoclonal Antibodies with Routine Pediatric Vaccines and Implications for Nirsevimab Use: A White Paper. Front. Immunol. 2021, 12, 708939. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sun, X.; Ye, X.; Feng, Y.; Wang, J.; Zheng, X.; Liu, X.; Yi, C.; Hao, M.; Wang, Q.; et al. Hexon and fiber of adenovirus type 14 and 55 are major targets of neutralizing antibody but only fiber-specific antibody contributes to cross-neutralizing activity. Virology 2018, 518, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, A.C.; Stehle, T. Human adenovirus binding to host cell receptors: A structural view. Med. Microbiol. Immunol. 2020, 209, 325–333. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, J.; Lan, W.; Quan, L.; Ou, J.; Zhao, W.; Wu, J.; Woo, P.C.Y.; Seto, D.; Zhang, Q. Molecular Typing and Rapid Identification of Human Adenoviruses Associated with Respiratory Diseases Using Universal PCR and Sequencing Primers for the Three Major Capsid Genes: Penton Base, Hexon, and Fiber. Front. Microbiol. 2022, 13, 911694. [Google Scholar] [CrossRef]
- Dudding, B.A.; Wagner, S.C.; Zeller, J.A.; Gmelich, J.T.; French, G.R.; Top, F.H., Jr. Fatal pneumonia associated with adenovirus type 7 in three military trainees. N. Engl. J. Med. 1972, 286, 1289–1292. [Google Scholar] [CrossRef]
- Yamamoto, D.; Okamoto, M.; Lupisan, S.; Suzuki, A.; Saito, M.; Tamaki, R.; Tandoc, A., 3rd; Mercado, E.; Sombrero, L.; Olveda, R.; et al. Impact of human adenovirus serotype 7 in hospitalized children with severe fatal pneumonia in the Philippines. Jpn. J. Infect. Dis. 2014, 67, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Wen, L.; Wu, Z.; Liu, N.; Yang, C.; Liu, W.; Ba, Z.; Wang, J.; Yi, S.; Li, H.; et al. Human adenovirus type 7 infection associated with severe and fatal acute lower respiratory illness and nosocomial transmission. J. Clin. Microbiol. 2015, 53, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Ogimi, C.; Martin, E.T.; Xie, H.; Campbell, A.P.; Waghmare, A.; Jerome, K.R.; Leisenring, W.M.; Milano, F.; Englund, J.A.; Boeckh, M. Role of Human Bocavirus Respiratory Tract Infection in Hematopoietic Cell Transplant Recipients. Clin. Infect. Dis. 2021, 73, e4392–e4399. [Google Scholar] [CrossRef]
- Nantachit, N.; Kochjan, P.; Khamrin, P.; Kumthip, K.; Maneekarn, N. Human bocavirus genotypes 1, 2, and 3 circulating in pediatric patients with acute gastroenteritis in Chiang Mai, Thailand, 2012–2018. J. Infect. Public Health 2021, 14, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Korsun, N.; Angelova, S.; Trifonova, I.; Voleva, S.; Grigorova, I.; Tzotcheva, I.; Mileva, S.; Perenovska, P. Prevalence and Genetic Characteristics of Human Bocaviruses Detected in Patients with Acute Respiratory Infections in Bulgaria. Int. J. Microbiol. 2021, 2021, 7035081. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Liu, J.; Zhao, Y.; Xie, Z.; Shen, J.; Tan, W. Genetic characterization of human bocavirus among children with severe acute respiratory infection in China. J. Infect. 2016, 73, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ljubin-Sternak, S.; Slović, A.; Mijač, M.; Jurković, M.; Forčić, D.; Ivković-Jureković, I.; Tot, T.; Vraneš, J. Prevalence and Molecular Characterization of Human Bocavirus Detected in Croatian Children with Respiratory Infection. Viruses 2021, 13, 1728. [Google Scholar] [CrossRef]
- Abozahra, R.; Abdelhamid, S.M.; Khairy, K.; Baraka, K. Detection and phylogenetic analysis of Human bocavirus in children diagnosed with acute respiratory tract infection. J. Med. Microbiol. 2020, 69, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Piralla, A.; Zampiero, A.; Bianchini, S.; Umbrello, G.; Scala, A.; Bosis, S.; Fossali, E.; Baldanti, F.; Esposito, S. Bocavirus Infection in Otherwise Healthy Children with Respiratory Disease. PLoS ONE 2015, 10, e0135640. [Google Scholar] [CrossRef]
- Arnott, A.; Vong, S.; Rith, S.; Naughtin, M.; Ly, S.; Guillard, B.; Deubel, V.; Buchy, P. Human bocavirus amongst an all-ages population hospitalised with acute lower respiratory infections in Cambodia. Influenza Other Respir. Viruses 2013, 7, 201–210. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. Rates of molecular evolution in RNA viruses: A quantitative phylogenetic analysis. J. Mol. Evol. 2002, 54, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Smielewska, A.; Emmott, E.; Ranellou, K.; Popay, A.; Goodfellow, I.; Jalal, H. UK circulating strains of human parainfluenza 3: An amplicon based next generation sequencing method and phylogenetic analysis. Wellcome Open Res. 2018, 3, 118. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Liu, B.; Xiao, Y.; Wang, X.; Ren, L.; Dong, J.; Sun, L.; Zhu, Y.; Zhang, T.; Yang, F. Genetic Characteristics of Human Parainfluenza Virus Types 1–4 from Patients with Clinical Respiratory Tract Infection in China. Front. Microbiol. 2021, 12, 679246. [Google Scholar] [CrossRef] [PubMed]
- Coelingh, K.V.; Winter, C.C. Naturally occurring human parainfluenza type 3 viruses exhibit divergence in amino acid sequence of their fusion protein neutralization epitopes and cleavage sites. J. Virol. 1990, 64, 1329–1334. [Google Scholar] [CrossRef]
- He, Y.; Liu, W.J.; Jia, N.; Richardson, S.; Huang, C. Viral respiratory infections in a rapidly changing climate: The need to prepare for the next pandemic. EBioMedicine 2023, 93, 104593. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Kash, J.C. Insights on influenza pathogenesis from the grave. Virus Res. 2011, 162, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Potter, R.N.; Cantrell, J.A.; Mallak, C.T.; Gaydos, J.C. Adenovirus-associated deaths in US military during postvaccination period, 1999–2010. Emerg. Infect. Dis. 2012, 18, 507–509. [Google Scholar] [CrossRef]
- Carballal, G.; Videla, C.; Misirlian, A.; Requeijo, P.V.; Aguilar Mdel, C. Adenovirus type 7 associated with severe and fatal acute lower respiratory infections in Argentine children. BMC Pediatr. 2002, 2, 6. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coinfections with SARS-CoV-2 and Other Respiratory Viruses | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AGE GROUPS | A(H1N1) | A(H3N2) | B-Victoria | RSV | RV | HMPV | PIV2 | PIV3 | BoV | AdV | Total |
| 0–16 years of age, n = 527, n (%) | 7 (1.3) | 6 (1.1) | 0 (0) | 16 (3) | 16 (3) | 5 (0.9) | 1 (0.2) | 3 (0.5) | 20 (3.8) | 7 (1.3.) | 81 (15.4) |
| 17–64 years of age, n = 473, n (%) | 6 (1.3) | 1 (0.5) | 2 (0.4) | 3 (0.6) | 5 (1) | 2 (0.4) | 0 (0) | 6 (1.3) | 3 (0.6) | 5 (1) | 33 (6.9) |
| >65 years of age, n = 1158, n (%) | 0 (0) | 0 (0) | 0 (0) | 19 (1.6) | 4 (0.3) | 10 (0.9) | 1 (0.1) | 4 (0.3) | 4 (0.3) | 12 (1) | 54 (4.6) |
| Total coinfection cases, n = 2277, n (%) | 13 (0.6) | 7 (0.3) | 2 (0.08) | 38 (1.7) | 25 (1.1) | 17 (0.7) | 2 (0.08) | 13 (0.6) | 27 (1.2) | 24 (1) | 168 (7.4) |
| Two Co-Pathogens | Number of Coinfections | Three Co-Pathogens | Number of Coinfections | More Than Three Co-Pathogens | Number of Coinfections | |
|---|---|---|---|---|---|---|
| Coinfections with the participation of SARS-CoV-2 | A(H1N1) | 3 | A(H1N1) + RSV-B | 3 | RSV-B + AdV-C2 + BoV-V1 | 1 |
| A(H3N2) | 2 | A(H1N1) + HKU-1 | 1 | RSV-B + HMPV + BoV-V1 | 1 | |
| B-Victoria | 2 | A(H3N2) + RSV-B | 2 | A(H3N2) + RSV-B + PIV3 + AdV-C2 + BoV-V1 | 1 | |
| RSV-A | 1 | RSV-B + AdV-E4 | 1 | |||
| RSV-B | 6 | RSV-B + PIV3 | 1 | |||
| PIV3-2 | 2 | RSV-B + BoV-V1 | 1 | |||
| AdV-C2 | 3 | HMPV + AdV-C2 | 1 | |||
| HPeV | 1 | HMPV + BoV-V1 | 1 | |||
| BoV-V1 | 2 | |||||
| Total number of coinfections | 22 | 11 | 3 |
| Sampling Date | Variant | Pango Lineage | Co-Pathogens |
|---|---|---|---|
| 2021-03-01 | Alpha (B.1.1.7-like) | B.1.1.7 | PIV3 |
| 2021-03-02 | Alpha (B.1.1.7-like) | B.1.1.7 | AdV-C2 |
| 2021-03-20 | Alpha (B.1.1.7-like) | B.1.1.7 | PIV3 |
| 2021-04-01 | Alpha (B.1.1.7-like) | B.1.1.7 | RSV-A |
| 2021-07-12 | Delta (B.1.617.2-like) | B.1.617.2 | BoV-V1 |
| 2021-09-23 | Delta (B.1.617.2-like) | AY.9.2 | BoV-V1 |
| 2022-04-26 | Omicron (BA.2-like) | A.2 | AdV-C2 |
| 2022-11-10 | Delta (B.1.617.2-like) | B.1.617.2 | RSV-B |
| 2022-11-10 | Delta (B.1.617.2-like) | AY.92 | RSV-B |
| 2022-12-05 | Delta (B.1.617.2-like) | B.1.617.2 | A(H3N2) + RSV-B |
| 2022-12-07 | Delta (B.1.617.2-like) | AY.92 | RSV-B |
| 2022-12-15 | Delta (B.1.617.2-like) | B.1.617.2 | A(H3N2) + RSV-B + AdV-C2 + BoV-V2 |
| 2022-12-19 | Delta (B.1.617.2-like) | B.1.617.2 | RSV-B |
| 2023-01-01 | Omicron (BA.2-like) | CH.1.1 | RSV-B |
| 2023-01-06 | Omicron (BA.5-like) | CK.1 | B-Victoria |
| 2023-01-11 | Omicron (BA.5-like) | BA.5.1 | A(H3N2) |
| 2023-01-16 | Omicron (BA.5-like) | EH.1 | A(H1N1) |
| 2023-01-16 | Omicron (BA.5-like) | EH.1 | A(H1N1) |
| 2023-01-16 | Omicron (Unassigned) | BQ.1.1.23 | A(H3N2) |
| 2023-02-22 | Omicron (XBB.1.5-like) | XBB.1.5.20 | A(H1N1) |
| 2023-04-13 | Omicron (XBB.1-like) | EG.1 | A(H1N1) |
| 2023-04-18 | Omicron (XBB.1.5-like) | XBB.1.5 | RSV-B |
| 2023-05-02 | Omicron (XBB.1.5-like) | EU.1.1 | RSV-B |
| Alignment Against a Reference Genome in HA A/Victoria/4897/2022(H1N1) | Bulgarian A(H1N1) Sequences | ||
|---|---|---|---|
| HA_A/Bulgaria/s215/2023_18428912 | HA_A/Bulgaria/s372/2023_1842891 | HA_A/Bulgaria/773/2023_18414675 | |
| Clade | 6B.1A.5a.2a (5a.2a) | 6B.1A.5a.2a (5a.2a) | 6B.1A.5a.2a.1 (5a.2a.1) |
| List of mutations/antigenic sites | S137P, R142K/Sa, A216T, R240Q, E260D, V272I, A277T, N356Y, L407Y, N408Y, H451N | S137P, R142K/Sa, A216T, R240Q, E260D, D269N V272I, A277T, N356Y, N431I, H451N | S157T/Ca2, A216T, R223Q, H438Q, D439G, S440I, N441stop, V442C, K443E, N444E, Y446V, E447stop, V449S, R450K, H451T, Q452S, L453V |
| % AA identity | 98.1 | 98.2 | 97.3 |
| Alignment against a reference genome in NA A/Victoria/4897/2022 (H1N1) | NA_A/Bulgaria/s215/2023_18428912 | NA_A/Bulgaria/s372/2023_18428911 | NA_A/Bulgaria/773/2023_18414675 |
| List of mutations | I7K, I8T, N21S, D50N, S200N, E382G | D50N, S200N, E382G | I13V, D50N, S339L, E382G |
| % AA identity | 99.1 | 99.3 | 99.1 |
| Alignment Against the HA of Vaccine virus A/Darwin/9/2021 | Bulgarian A(H3N2) Sequences | ||
|---|---|---|---|
| HA_A/Bulgaria/325/2023_18406107 | HA_A/Bulgaria/605/2023_18406223 | ||
| Clade | 2a.1b | 2b | |
| Amino acid substitutions/antigenic sites | D53G/C β, D104G/α, N186D/D G225D/220loop, K276R | E50I/C β F79V/α I140K/β S156H/B | N186D D G225D/220loop I242M L202V |
| % AA identity | 99.1 | 98.6 | |
| Alignment against NA of vaccine virus A/Darwin/9/2021 | NA_A/Bulgaria/325/2023_18406107 | NA_A/Bulgaria/605/2023_18406223 | |
| Amino acid substitutions | T238A, D463N, N465S | V215I, D221N | G346S, S370T |
| V263I | P386H | ||
| R315S | E435K | ||
| % AA identity | 99.4 | 99.3 | |
| Amino Acid Changes in the Bulgarian Strain | Frequency in the Bulgarian Strain, n (%) | Countries Where the Mutation Occurred (n) | Frequency of All Samples from Other Countries (%) |
|---|---|---|---|
| A74V | 10 (100) | 13 | 8.51 |
| T131A | 10 (100) | 11 | 4.55 |
| P214S | 10 (100) | 15 | 5.68 |
| P221L | 10 (100) | 16 | 5.94 |
| I252T | 10 (100) | 18 | 10.92 |
| K256N | 10 (100) | 11 | 4.27 |
| I268T | 10 (100) | 18 | 7.48 |
| S275P | 10 (100) | 14 | 6.27 |
| Y285H | 10 (100) | 20 | 8.68 |
| I137T | 9 (90) | 11 | 4.25 |
| S100G | 8 (80) | 9 | 2.87 |
| P120L | 2 (20) | NF * | NF |
| D251N | 2 (20) | 8 | 0.59 |
| H277R | 2 (20) | 2 | 0.09 |
| L71P | 2 (20) | 3 | 0.10 |
| T225N | 1 (10) | 5 | 0.10 |
| S309Y | 1 (10) | NF | NF |
| T117A | 1 (10) | 1 | 0.02 |
| S295T | 1 (10) | 1 | 0.02 |
| T296H | 1 (10) | 1 | 0.02 |
| Q297P | 1 (10) | 1 | 0.02 |
| P299del | 1 (10) | NF | NF |
| T292I | 1 (10) | 2 | 0.03 |
| I59T | 1 (10) | 1 | 0.02 |
| T139A | 1 (10) | NF | NF |
| N143S | 1 (10) | NF | NF |
| K145R | 1 (10) | NF | NF |
| S147G | 1 (10) | NF | NF |
| K153R | 1 (10) | NF | NF |
| T209K | 1 (10) | 1 | 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trifonova, I.; Korsun, N.; Madzharova, I.; Alexiev, I.; Ivanov, I.; Levterova, V.; Grigorova, L.; Stoikov, I.; Donchev, D.; Christova, I. Epidemiological and Genetic Characteristics of Respiratory Viral Coinfections with Different Variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Viruses 2024, 16, 958. https://doi.org/10.3390/v16060958
Trifonova I, Korsun N, Madzharova I, Alexiev I, Ivanov I, Levterova V, Grigorova L, Stoikov I, Donchev D, Christova I. Epidemiological and Genetic Characteristics of Respiratory Viral Coinfections with Different Variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Viruses. 2024; 16(6):958. https://doi.org/10.3390/v16060958
Chicago/Turabian StyleTrifonova, Ivelina, Neli Korsun, Iveta Madzharova, Ivailo Alexiev, Ivan Ivanov, Viktoria Levterova, Lyubomira Grigorova, Ivan Stoikov, Dean Donchev, and Iva Christova. 2024. "Epidemiological and Genetic Characteristics of Respiratory Viral Coinfections with Different Variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)" Viruses 16, no. 6: 958. https://doi.org/10.3390/v16060958