1. Introduction
Bovine coronavirus (BCoV) belongs to the
Embecovirus subgenus of the
Betacoronavirus genus within the
Orthocoronavirinae subfamily of the
Coronaviridae family. Betacoronaviruses, along with alphacoronaviruses, infect exclusively mammals, whereas gammacoronaviruses and deltacoronaviruses primarily infect avian species [
1]. BCoV was initially identified by Mebus et al., who discovered coronavirus-like particles in a diarrheic calf’s feces by electron microscopy in Nebraska in 1972 [
2,
3]. Subsequent experiments confirmed that the BCoV found in the fecal material caused diarrhea in infected calves and was closely related to bovine-like CoVs in other mammals such as dogs, sheep, and deer [
3,
4,
5,
6,
7]. BCoV viral particles are enveloped and pleomorphic with a diameter of around 65–210 nm and spike (S) and hemagglutinin-esterase (HE) protein projections [
4,
8,
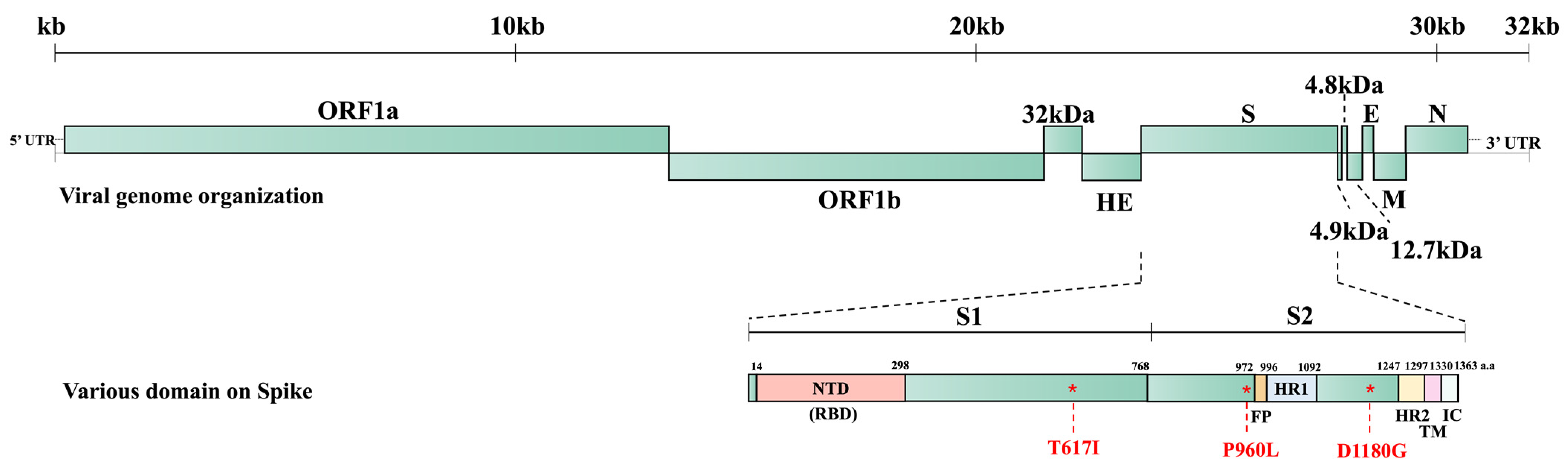
9]. The BCoV genome possesses open reading frame (ORF) 1a, ORF1b, and the ORFs for the structural and accessory proteins, including the 32 kDa, HE, S, 4.9 kDa, 4.8 kDa, envelope (E), membrane (M), and nucleocapsid (N) proteins [
10,
11,
12]. BCoV possesses the unique HE gene, which is absent from other subgenera of betacoronaviruses, alphacoronaviruses, gammacoronaviruses, and deltacoronaviruses.
BCoV infects both the upper and lower respiratory and gastrointestinal tracts of cattle, being shed in feces and nasal secretions [
4,
8]. Coronaviruses undergo continuous evolution through the accumulation of point mutations, insertions and deletions, and recombination events, resulting in the emergence of new variants capable of evading host immune responses [
13]. Despite being half a century since the discovery of the first BCoV, significant knowledge gaps exist regarding BCoV pathogenesis and evolution.
The S protein of coronaviruses serves as the receptor-binding protein and is a major target for inducing protective immunity. It shows significant genetic variations among strains due to immune pressure. Changes in tissue tropism among coronaviruses often correlate with mutations in the S protein. For instance, the deletion of the N-terminal domain of the S protein, and sometimes together with deletions in ORF3a and ORF3b of transmissible gastroenteritis virus (TGEV), an alphacoronavirus, shifted the viral major tropism from enteric to the respiratory tract, resulting in porcine respiratory coronavirus (PRCV) [
14,
15,
16,
17]. Therefore, a comprehensive investigation of BCoV S protein sequence variation and domain function is essential for the understanding of the virus adaptation and transmission mechanisms.
Furthermore, BCoV serves as a valuable model for molecular studies of coronavirus. Critical questions, such as the mechanisms underlying the manifestation of enteric or respiratory diseases, and the role of the HE protein, remain unanswered. Thus, gaining insights into the current BCoV genomes and the isolation of contemporary strains are imperative steps toward establishing a reverse genetics platform in our laboratory.
In this study, we detected and isolated contemporary BCoVs from cattle samples. Then, we performed sequence analyses of BCoVs at the genomic, S protein, and HE protein levels. Additionally, by comparing four pairs of fecal and nasal samples, we found potential associations between the virus strain tissue origin and viral genetic markers. These results provide insights into the BCoV strain genetic diversity and molecular characteristics.
2. Materials and Methods
2.1. Sample Collection, Screening, and Preparation
Sample collection information is listed in
Table 1. Ten pairs of fecal and nasal swab samples were collected in March 2021 from 10 veal calves, which were separated from their mother and supplemented with milk and had no apparent clinical signs on a veal farm in Ohio (OH). An additional 51 samples were obtained in April–May 2023 from various age groups of cattle, including pre-weaned calves (n = 27), nursing beef (n = 1), weaned feeder calves (n = 20), and unknown-age cattle (n = 3) of Holstein, Jersey, Nigerian Red, and Angus breeds from three farms in Georgia (GA1, GA2, and GA3). Samples were diluted tenfold with phosphate-buffered saline without Mg2+ and Ca2+ [PBS(−)] (Gibco, Carlsbad, CA, USA), vortexed, and then centrifuged at 2000×
g at 4 °C for 10 min. The supernatant was subsequently filtered through 0.22 μm pore size filters (Millipore, Chicago, IL, USA) to remove bacteria. The filtered samples were then stored at −80 °C until further analysis. Diluted samples underwent TaqMan real-time reverse transcription-PCR (RT-qPCR) assay targeting the conserved M gene of BCoV for the detection of viral RNA titers [
18].
2.2. Virus Isolation and Propagation
The cloned human ileocecal colorectal human rectal tumor-18 (HRT-18) cell line (provided by Dr. Linda Saif, The Ohio State University, Wooster, OH, USA) was selected for the isolation of BCoVs from the RT-qPCR positive samples from Ohio and Georgia [
19]. The growth medium (GM) for HRT-18 cells consisted of advanced minimum essential medium (AMEM) (Gibco, Carlsbad, CA, USA), 1% antibiotic–antimycotic (Gibco, Carlsbad, CA, USA), 1% l-glutamine (Gibco, Carlsbad, CA, USA), and 10% heat-inactivated fetal bovine serum (FBS, Hyclone, Logan, UT, USA).
Two-day-old HRT-18 cells reaching two-thirds confluency in 24-well plates were used for virus inoculation. First, the GM was replaced with AMEM and incubated for 1 h at 37 °C. Then, the cell monolayers were inoculated with 200 μL per well of diluted sample, with one sample being added to four wells. After incubating the plate for 60 min at 37 °C with 5% CO2, the supernatant was removed from two wells followed by gentle washing using PBS(−) twice [Inoc (−) condition], while the supernatant was retained in the other two wells [Inoc (+) condition]. AMEM supplemented with 5 μg/mL of trypsin (Gibco, Carlsbad, CA, USA) was added to all wells. At this stage, 50 μL of supernatant was collected from each well for RNA extraction to establish a baseline at 1 h post-inoculation (hpi) for RNA measurement.
Cytopathogenic effects (CPEs) were monitored in the following days. When either 80% CPE was observed, or at four days post-inoculation (dpi) without CPE, plates were frozen at −80 °C and thawed once. All harvested supernatants were subjected to RNA extraction and tested for BCoV using RT-qPCR to calculate the difference in cycle threshold (ΔCt) values between the samples at 1 hpi and harvested time.
Samples with ΔCt > 1 (n = 15) were subjected to serial passages, with the first three passages conducted in the same manner to confirm the successful isolation of the virus in HRT-18 cells. In serial passaging, the procedure was modified to utilize two wells per sample, with 200 μL of harvested supernatant inoculated per well in 12-well plates. After a 60-minute incubation in a 5% CO2 incubator at 37 °C, the inoculum was removed, and AMEM with trypsin was added directly.
2.3. Viral RNA Extraction
Viral RNA was extracted from 50 μL of the field sample suspension and the culture supernatants using the 5× MagMAX-96 Viral Kit (Invitrogen, Carlsbad, CA, USA) and the MagMax™ Express machine (Applied Biosystems, Bedford, MA, USA), following the manufacturer’s instructions. Finally, 50 μL of RNA was obtained in an elution buffer.
2.4. TaqMan RT-qPCR and Conventional PCR
For the RT-qPCR targeting the conserved M gene, the previously reported forward primer (5′-CTGGAAGTTGGTGGAGTT-3′), reverse primer (5′-ATTATCGGCCTAACATACATC-3′), and probe (6-carboxyfluorescein-CCTTCATATCTATACACATCAAGTTGTT-black hole quencher 1) [
18] were synthesized by Integrated DNA Technologies (
https://www.idtdna.com/pages, accessed on 25 April 2024), and the reaction system was implemented using 2 μL of RNA and the Qiagen OneStep RT-PCR kit (Qiagen, Valencia, CA, USA) on RealPlex real-time thermocyclers (Eppendorf, Barkhausenweg, Hamburg, Germany). Both viral genomic and several subgenomic RNAs that contain the M gene are detected by this assay. The whole genome sequence (WGS) of the BC18 strain was obtained using conventional PCR with multiple primers (shown in
Supplementary Table S1) followed by Sanger sequencing at the Comprehensive Cancer Center of The Ohio State University or Oxford nanopore sequencing at Plasmidsaurus (
https://www.plasmidsaurus.com/, accessed on 25 April 2024). Initially, viral RNA was reverse transcribed to cDNA using the SuperScript™ IV cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA) employing a strategy combining oligo(dT) and random hexamers’ priming. Subsequently, the cDNA was amplified with the BCoV-specific primers using the high-fidelity PrimeSTAR GXL DNA polymerase (TaKaRa, Toshima-ku, Tokyo, Japan). The PCR reaction mixture (50 μL) consisted of 2 μL of cDNA, 10 μL of 5× PCR buffer, 4 μL of deoxynucleotide triphosphates (dNTPs) (2.5 mM each), 2 μL of GXL PCR enzyme, and 1 μL each of forward and reverse primers (10 µM each). The PCR reaction followed thermal cycling conditions: 98 °C for 30 s, 30 cycles of 98 °C for 10 s, 55 °C/60 °C for 15 s, and 68 °C for 1–3 min, with a final extension step at 68 °C for 5 min. PCR products were analyzed using 1% agarose gel electrophoresis to confirm the proper sizes of amplicons. Subsequently, PCR products of the correct size were purified using the QIAquick Gel extraction kit (Qiagen, Valencia, CA, USA) for subsequent sequencing.
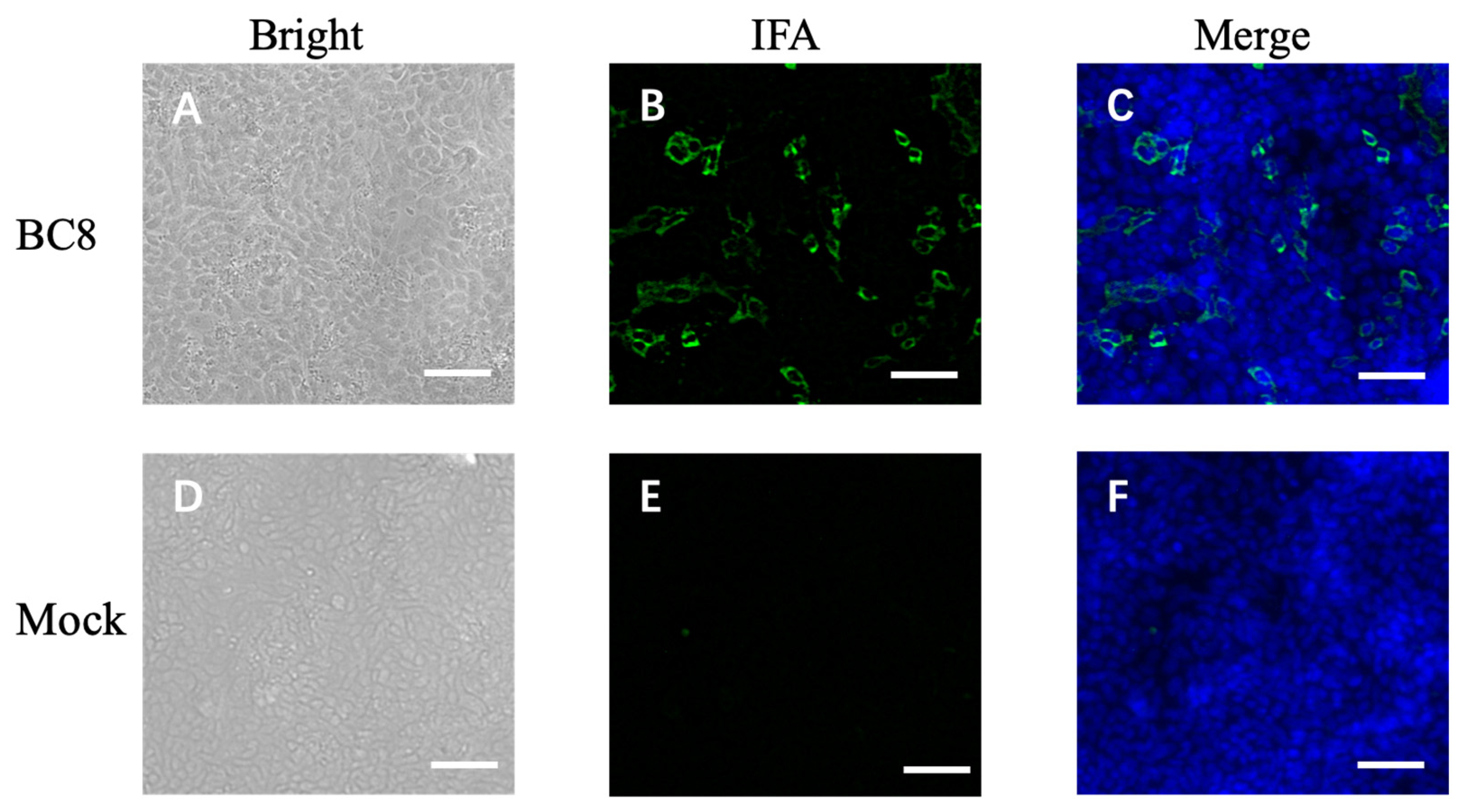
2.5. Immunofluorescence Assay (IFA)
BCoV-infected HRT-18 cells in 6-well plates were fixed and permeabilized with 100% methanol at −20 °C for 15 min. Subsequently, the cells were washed five times with PBS and then incubated with 5% bovine serum albumin (BSA) at room temperature for one hour to block nonspecific binding sites. The primary antibody, guinea pig hyperimmune antiserum to the Mebus strain of BCoV, NR455 (diluted 1:2000) (provided by Dr. Linda Saif, The Ohio State University), was added to the fixed cells and incubated at room temperature for one hour. After washing, the secondary antibody, fluorescein AF488-conjugated goat anti-guinea pig IgG (diluted 1:1000) (Invitrogen, Carlsbad, CA, USA), was applied and incubated at room temperature for one hour in the dark. After incubation, the cells were washed five times with PBS. Subsequently, staining of the nuclei was performed using 4′,6-diamidino-2-phenylindole (DAPI). The plates were then observed using an IX-70 fluorescence microscope (Olympus, Center Valley, PA, USA).
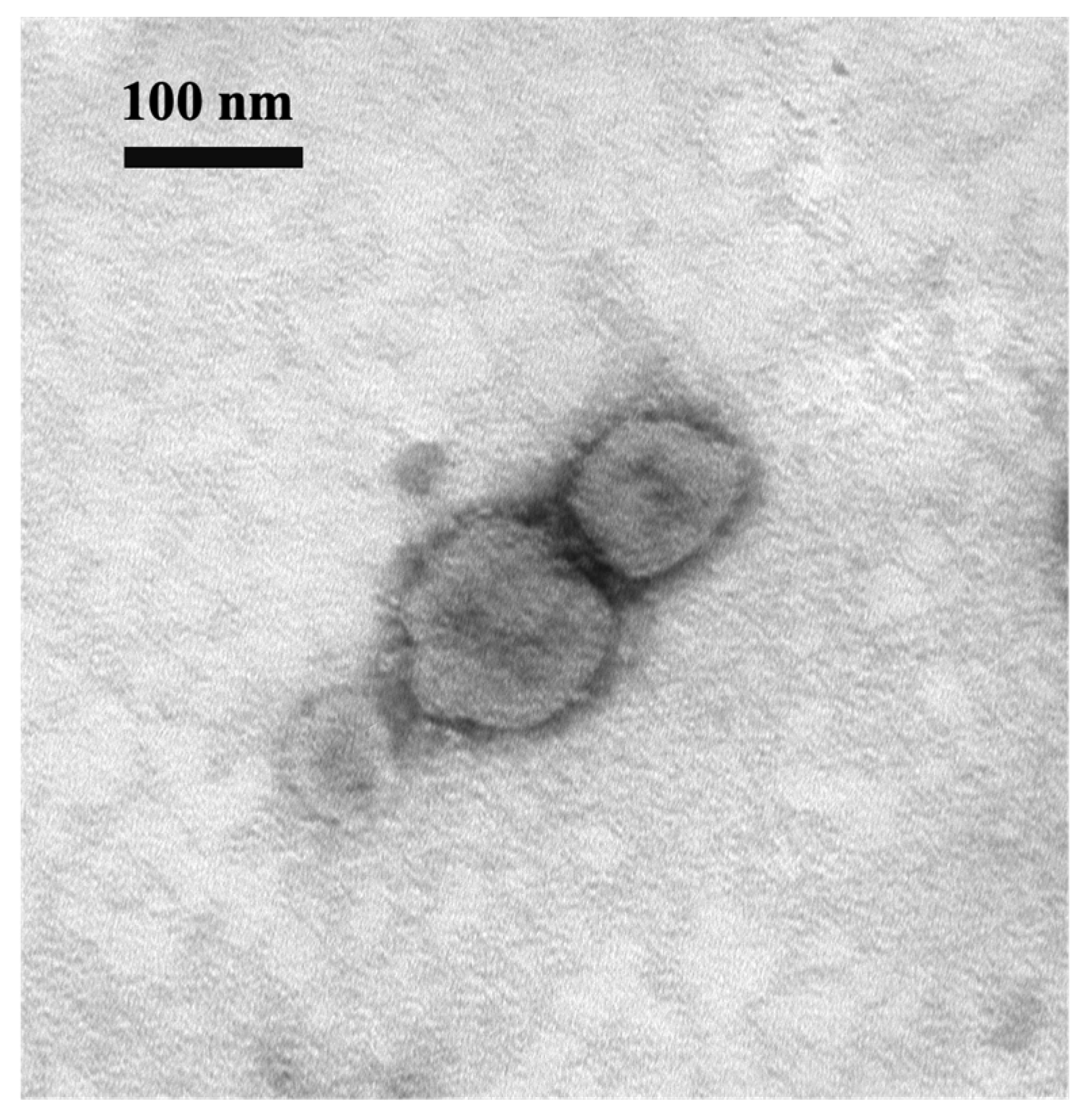
2.6. Immunoelectron Microscopy (IEM)
For sample preparation, BCoV BC8-infected HRT-18 cell cultures were collected at 48 hpi and centrifuged at 2095× g for 30 min at 4 °C, and supernatants were collected. The guinea pig hyperimmune antiserum NR455 was diluted 1:1000 in PBS, ultracentrifuged at 17,000× g for 30 min at 4 °C using SW41 Ti rotor (Beckman Coulter, Brea, CA, USA), and filtered through a 0.22 µm filter before incubation with the samples. Cell culture supernatants were filtered through a 0.45 µm filter and then mixed with the diluted antiserum with gentle shaking at 4 °C for 12–18 h. The samples were subsequently ultracentrifuged at 14,000× g at 4 °C for 1.5 h using the SW32 Ti rotor, washed once with AMEM, and then ultracentrifuged again. To detect virion particles, samples were stained with an equal volume of uranyl acetate (2% in ddH2O), incubated for 1 min, and applied to a 300 mesh formvar-coated copper grid. After incubating on the grid for 3–5 min, the excess sample was absorbed using filter paper, and the viral particles were visualized using an H7500 electron microscope (Hitachi High Technologies, Minato-ku, Tokyo, Japan).
2.7. Plaque Assay for BCoV Titration and Purification of the BC8 Strain
HRT-18 cells were seeded in 6-well plates, reaching semi-confluency on the first day. Once the cells reached 100% confluency, the growth medium (GM) was replaced with AMEM and incubated for one hour at 37 °C. Subsequently, the AMEM was removed, and the cell monolayers were inoculated with a 10-fold serially diluted and well-vortexed sample (500 μL/well), with duplicates per dilution. Plates were gently shaken every 15 min. After one hour of incubation, the inoculum was removed, and the cell monolayer was washed twice with PBS(−). A mixture of equal volumes of 1.5% SeaPlaque agarose (Lonza, Walkersville, MD, USA) and 2× MEM containing 2% antibiotic–antimycotic, 2% glutamine, and 10 µg/mL trypsin was prepared. The 0.75% agarose mixture was poured onto the cell monolayers, and the plates were kept in a hood for approximately 20 min until the agarose solidified. Subsequently, the plates were inverted and incubated at 37 °C for a maximum of five days, with staining occurring on the last day.
For the titration of a sample, on the fourth day, the cells were fixed using 10% neutral buffered formalin for 15 min, followed by staining with 1% crystal violet (Invitrogen, Carlsbad, CA, USA) for one minute to visualize obvious plaques. Virus titers in plaque forming units (PFUs)/mL were calculated based on the last two dilutions that showed plaques. For plaque purification, the culture was directly stained with 0.33% neutral red (Invitrogen, Carlsbad, CA, USA), with incubation for a maximum of three hours. After incubation, the dye was removed, and the plaques were visualized. Individual plaques were confirmed using an LED light box and an optical microscope (Olympus, Hachioji-shi, Tokyo, Japan). Individual plaques were picked using sterile pipette tips and transferred to 500 μL of AMEM. The plaque purification process was repeated at least twice for each isolated virus strain to ensure virus purity.
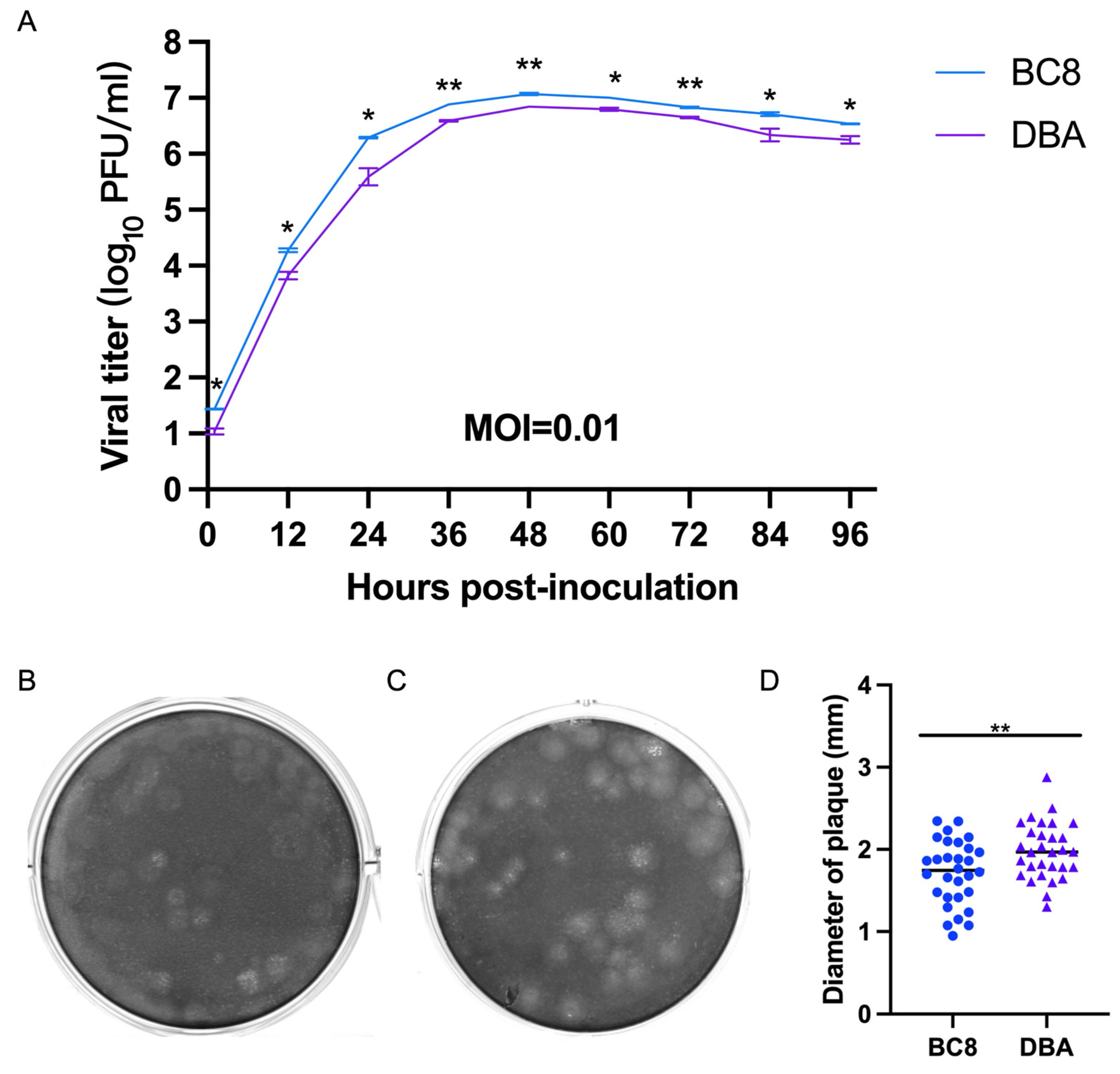
2.8. Growth Kinetics and Plaque Size Calculation
HRT-18 cells were inoculated with BCoV at a multiplicity of infection (MOI) of 0.01 and incubated for one hour at 37 °C. The culture supernatants were then collected at various time points post-infection: 1 hpi, 12 hpi, 24 hpi, 36 hpi, 48 hpi, 60 hpi, 72 hpi, 84 hpi, and 96 hpi, respectively. Infectious viral titers were determined by plaque assay. The growth kinetics of one of the newly isolated BCoV strains, BC8, were compared with those of the historical DBA BCoV strain [
20]. The diameter of 30 plaques was measured for each strain to calculate plaque size.
2.9. Sequencing, Protein Structure Prediction, and Phylogenetic Analysis
We isolated five strains: BC7, BC8, BC9, BC39, and BC47. Because BC7 was from the same farm (Ohio) as BC8 and BC9, we just sent BC8 and BC9 for whole genome sequencing using next-generation sequencing (NGS) at the University of Illinois at Urbana-Champaign. We chose the strain BC8 at passage 2 (BC8-P2), BC9-P3, BC39-P5, and BC47-P3 because the original fecal samples had low viral RNA titers (with a Ct > 26) and reached high titers after passaging in HRT-18 cells (with a Ct < 25). Nucleic acids of these isolates were extracted on KingFisher Flex using the MagMAX™ Pathogen RNA/DNA Kit (ThermoFisher, Waltham, MA, USA), following the kit manual. Nucleic acid samples were subject to sequence-independent, single-primer amplification (SISPA). In detail, the nucleic acids were reverse transcribed into cDNA using Superscript III (ThermoFisher, Waltham, MA, USA) and a random octamer primer (GACCATCTAGCGACCTCCACNNNNNNNN), converted into dsDNA by Klenow polymerase (NEB, Ipswich, MA, USA), and further amplified using a single primer (GACCATCTAGCGACCTCCAC) and the Advantage 2 PCR kit (Takara Bio, Ann Arbor, MI, USA). The PCR products were purified using the QIAquick PCR Purification Kit (QIAGEN, Germantown, MD, USA) and quantified using Qubit broad-range and high-sensitivity kits (ThermoFisher, Waltham, MA, USA). The NGS library of each sample was prepared using the Nextera XT kit (Illumina, San Diego, CA, USA) and sequenced on Illumina MiSeq. FASTQ files of each isolate were assembled using SPAdes version v3.14.0 and the assembled sequences were blasted against the NCBI NT local blast database. For strain BC18, WGS was acquired using conventional PCR with the primers shown in
Table S1. Purified PCR products were sent to PlasmidSaurus (
https://www.plasmidsaurus.com/, accessed on 25 April 2024) to obtain lineage DNA sequences, and overlapping DNA sequences were then analyzed to assemble the complete WGS.
Sequence alignments of the S proteins and HE proteins of three historical pairs (EF424615 and EF424617, AF391541 and AF391542, and EF424619, and EF424620) and one pair (OR502440 and OR502442) from this study of respiratory and enteric strains were performed using Clustal Omega (
https://www.ebi.ac.uk/Tools/msa/clustalo/, accessed on 25 April 2024) [
14,
21]. The 3D structures of the S protein trimers of the BC8, BC18, LUN, and ENT strains were predicted using SWISS-MODEL (
https://swissmodel.expasy.org/) and visualized using PyMOL2.6 software (
https://pymol.org/2/, accessed on 25 April 2024). Phylogenetic analysis was performed using MEGA11 software (
http://www.megasoftware.net/, accessed on 25 April 2024) with a bootstrap test of 1000 replicates.
2.10. Statical Analysis
Student’s t-test was conducted to compare the two BCoV strains’ infectious titers in PFU/mL at different time points and diameters of plaque sizes utilizing GraphPad Prism, version 9.0 (
https://www.graphpad.com/, accessed on 25 April 2024). Significance levels were set at a
p < 0.05.
2.11. Nucleotide Sequence Accession Numbers
The genomic information for strains BC8, BC9, BC18, BC39, and BC47 was submitted to GenBank (accession no. OR502440-OR502444).
4. Discussion
It has been several decades since the discovery of the first BCoV Mebus strain in 1972 [
23]. However, there remains a pressing need to investigate the evolution of American strains of BCoVs and isolate contemporary strains. BCoV causes both respiratory and enteric diseases, yet the underlying mechanisms remain poorly understood despite extensive research efforts on strain and disease characterization [
14,
21].
From our study, we obtained genome information for five current BCoV strains, shedding light on the current evolutionary status of BCoVs. We isolated five BCoV strains using HRT-18 cells. Interestingly, BCoV-infected cells exhibited minimal CPE until subjected to serial passages. For studies reliant on observing CPE, alternative cell types such as Vero or Madin–Darby bovine kidney (MDBK) cells might yield more pronounced effects [
24,
25]. In terms of RT-qPCR detection, although some samples showed low Ct values, corresponding to high viral RNA levels, the virus isolation and infectivity titers might not correspond well with mRNA levels. For instance, samples BC8 and BC18 exhibited Ct values of 35 and 22, respectively. However, only BC8, a fecal sample, could be successfully isolated in HRT-18 cells; BC18, a nasal swab sample, could not. This suggests that sample storage conditions or other factors may influence isolation success. Exploring alternative cell lines could also be beneficial. BCoV respiratory samples might replicate better in bovine/human respiratory epithelial cells, which mimic the viral natural growth environment. In this study, isolation experiments were conducted under two conditions: Inoc (+) and Inoc (−). The Inoc (+) condition retained the inoculum, probably containing bacterial metabolites, bile acids, and other factors from the original intestinal environment that could promote virus replication. In contrast, the Inoc (−) condition was performed to avoid potential toxic materials in the original sample that could be toxic to the cells and inhibit virus isolation. Unlike the porcine epidemic diarrhea virus [
26], the five isolated BCoV strains exhibited similar replication under both conditions, although the inoculated cells in the Inoc (+) condition detached more quickly, which might have been due to the presence of large intestinal contents.
The detection of BCoV yielded a positive rate of 21% (15/71), indicating that BCoV remains circulating and represents a risk to herds’ health. This underscores the continued importance of addressing BCoV as a major infectious agent in farming operations. Investigating BCoV co-infections with other pathogens, such as
Cryptosporidium parvum, rotavirus,
Salmonella enterica, and
E. coli K99, is essential for understanding the impact of BCoV co-infections on bovine health and farm biosafety. The S protein and HE protein play crucial roles in virus binding to host cell surface receptors [
27,
28]. BCoV utilizes N-acetyl-9-O-acetylneuraminic acid as a receptor, not only for the agglutination of erythrocytes but also for infecting cultured cells [
29,
30]. The S protein exhibited pairwise identities ranging from 90.15% to 99.85%, while the HE protein showed pairwise identities ranging from 93.40% to 100%. This indicates a higher degree of conservation in the HE protein compared with the S protein. These findings suggest a complex evolutionary history of BCoVs, with strains from different geographical regions and phenotypes showing mixed clustering patterns rather than distinct separation based on disease types.
In our virological experiments, we observed that the BC8 strain exhibited similar growth kinetics to DBA but had higher titers than DBA. However, despite its higher titers, BC8 had smaller plaque sizes than DBA. This suggested that viral titer may not necessarily correlate with the spread of a virus to neighboring cells, as factors such as cell–cell junctions, host cytoskeleton, and other unknown virus–host interaction factors also play a role in determining infectivity [
31]. One interesting phenomenon observed in the plaque assays was that BCoVs did not exhibit high virulence sufficient to induce complete cell detachment and primarily caused cell damage, which may be the pathogenic characteristic of BCoVs.
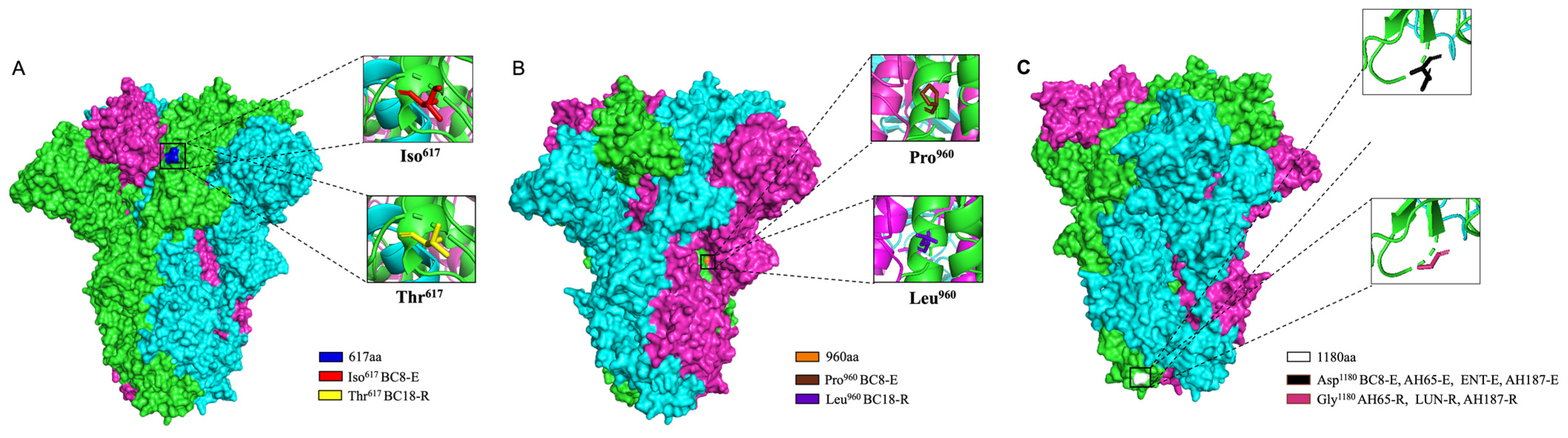
Given that samples from different animals exhibited more variation, we focused on analyzing BC8 and BC18. They were both collected from the same calf, with BC8 coming from a fecal sample and BC18 from a nasal sample. The S protein acts as an important receptor-binding protein and contains a furin cleave site, facilitating membrane fusion and the release of the viral genome to the cytoplasm [
32,
33]. We noted that BCoV S proteins had the furin cleavage site 763KRRSRR768 (position based on BC8 S protein), which is related to viral infection and cell–cell fusion [
34,
35]. Amino acid residue 617, located in the S1 C-terminal domain, and residue 960, located in the S2 subunit, were different between BC8-P2 and wildtype BC18. These two residues may be related to BCoV respiratory and enteric tropisms or cell culture adaptation.
D1180 was consistently observed in the four enteric strains, while G1180 was predominant in respiratory strains (three of four) (
Table 2 and
Table S2). The D1180G mutation lies between the HR1 and HR2 regions of the S2 subunit (
Figure 5). Because the S1 but not the S2 subunit contains a receptor-binding domain, this D1180 mutation is less likely to lead to changes in tissue tropism. The D1180G mutation may simply provide an advantage for viral replication in the respiratory tract. Due to the limited number of paired spike sequences in this study, we cannot draw definitive conclusions. Further investigation using reverse genetics systems and animal studies is warranted to explore whether certain residues are related to BCoV replication efficiency in different tissues.
In summary, we successfully isolated five BCoV strains, BC7, BC8, BC9, BC39, and BC47, from feces and sequenced BC8, BC9, BC39, and BC47 and the BC18 respiratory strain directly from a nasal sample. Our findings contribute to the understanding of the current virological and sequence diversity of BCoVs. These findings offer valuable insights for future research endeavors aimed at elucidating BCoV pathogenesis, antigenicity, and tissue tropism. Furthermore, the data we have amassed lay the groundwork for the development of a BCoV reverse genetics platform, which promises to enhance our comprehension and management of this significant bovine pathogen. Besides this, we are currently constructing a BCoV reverse genetics platform that will enable us to edit viral genes and change targeted sites at the DNA level, a capability that has not been available for BCoV so far. The role of these predicted amino acid sites in BCoV tissue tropisms will be explored. This study was the foundation of our lab’s long-term BCoV studies.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}