
Diversity of Picorna-Like Viruses in the Teltow Canal, Berlin, Germany



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Workup
2.2. Illumina Sequencing, Sequence Data Processing, and Virus Sequence Analyses
3. Results
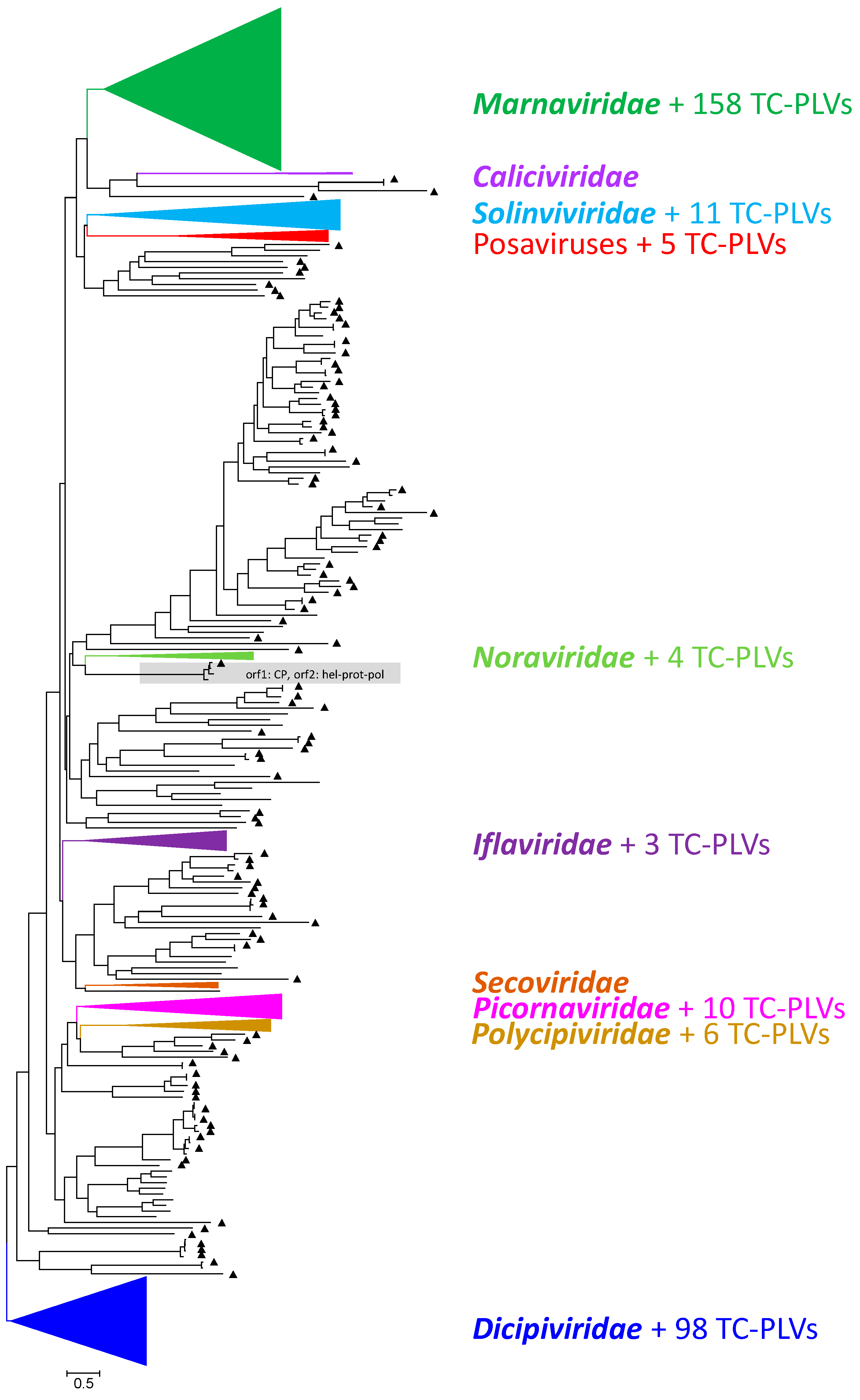
3.1. Sequencing, Assembly, and Identification of Viral Sequences
3.2. Phylogenetic Analyses
3.2.1. Marnaviridae and Related Viruses
3.2.2. Dicistroviridae and Related Viruses
3.2.3. Solinviviridae and Related Viruses
3.2.4. Iflaviridae and Related Viruses
3.2.5. Polycipiviridae and Related Viruses
3.2.6. Picornaviridae and Related Viruses
3.2.7. Noraviridae and Related Viruses
3.2.8. Posaviruses and Related Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Current ICTV Taxonomy Release. Available online: https://ictv.global/taxonomy (accessed on 12 May 2024).
- Le Gall, O.; Christian, P.; Fauquet, C.M.; King, A.M.Q.; Knowles, N.J.; Nakashima, N.; Stanway, G.; Gorbalenya, A.E. Picornavirales, a proposed order of positive-sense single-stranded RNA viruses with a pseudo-T = 3 virion architecture. Arch. Virol. 2008, 153, 715–727. [Google Scholar] [CrossRef]
- Sanfacon, H.; Gorbalenya, A.E.; Knowles, N.J.; Chen, Y.P. Order Picornavirales. In Virus Taxonomy. Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012; pp. 835–839. [Google Scholar]
- Wolf, Y.I.; Kazlauskas, D.; Inranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and evolution of the global RNA virome. mBio 2018, 9, e02320-18. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Silas, S.; Wang, Y.; Wu, S.; Bocek, M.; Kazlauskas, D.; Krupovic, M.; Fire, A.; Dolja, V.V.; Koonin, E.V. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat. Microbiol. 2020, 5, 1262–1270. [Google Scholar] [CrossRef]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Picorna-like viruses of the Havel River, Germany. Front. Microbiol. 2022, 13, 865287. [Google Scholar] [CrossRef]
- Chen, Y.M.; Sadiq, S.; Tian, J.H.; Chen, X.; Lin, X.D.; Shen, J.J.; Chen, H.; Hao, Z.Y.; Wille, M.; Zhou, Z.C.; et al. RNA viromes from terrestrail sites across China expand environmental viral diversity. Nat. Microbiol. 2022, 7, 1312–1323. [Google Scholar] [CrossRef]
- Shan, T.; Yang, S.; Wang, H.; Wang, H.; Zhang, J.; Gong, G.; Xiao, Y.; Yang, J.; Wang, X.; Lu, J.; et al. Virome in the cloaca of wild and breeding birds revealed a diversity of significant viruses. Microbiome 2022, 10, 60. [Google Scholar] [CrossRef]
- Richard, J.C.; Blevins, E.; Dunn, C.D.; Leis, E.M.; Goldberg, T.L. Viruses of freshwater mussels during mass mortality events in Oregon and Washington, USA. Viruses 2023, 15, 1719. [Google Scholar] [CrossRef]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. High diversity of unknown picorna-like viruses in the sea. Nature 2003, 424, 1054–1105. [Google Scholar] [CrossRef]
- Culley, A.I.; Mueller, J.A.; Belcaid, M.; Wood-Charison, E.M.; Poisson, G.; Steward, G.F. The characterization of RNA virus in tropical seawater using targeted PCR and metagenomics. mBio 2014, 3, e01210-14. [Google Scholar] [CrossRef]
- Lang, A.S.; Rise, M.L.; Culley, A.I.; Steward, G.F. RNA viruses in the sea. FEMS Microbiol. Rev. 2018, 33, 295–323. [Google Scholar] [CrossRef] [PubMed]
- Peduzzi, P. Virus ecology of fluvial systems: A blank spot on the map? Biol. Rev. 2015, 91, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Hepeliviruses in two waterbodies in Berlin, Germany. Arch. Virol. 2023, 169, 9. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Exploring the diversity of plant-associated viruses and related viruses in riverine freshwater samples collected in Berlin, Germany. Pathogens 2023, 12, 1458. [Google Scholar] [CrossRef] [PubMed]
- Wyn-Jones, A.P.; Carducci, A.; Cook, N.; D’Agostino, M.; Divizia, M.; Fleischer, J.; Gantzer, C.; Gawler, A.; Girones, R.; Höller, C.; et al. Surveillance of adenoviruses and noroviruses in European recreational waters. Water Res. 2011, 45, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 2011, 17, 200. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Lang, A.S.; Vlok, M.; Culley, A.I.; Suttle, C.A.; Takao, Y.; Tomaru, Y.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Marnaviridae 2021. J. Gen. Virol. 2021, 102, 001633. [Google Scholar] [CrossRef]
- Takao, Y.; Mise, K.; Nagasaki, K.; Okuno, T.; Honda, D. Complete nucleotide sequence and genome organization of a single-stranded RNA virus infecting the marine fungoid protist Schizochytrium sp. J. Gen. Virol. 2006, 87, 723–733. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Dicistroviridae. J. Gen. Virol. 2017, 98, 355–356. [Google Scholar] [CrossRef]
- Brown, K.; Olendraite, I.; Valles, S.M.; Firth, A.E.; Chen, Y.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; et al. ICTV Virus Taxonomy Profile: Solinviviridae. J. Gen. Virol. 2019, 100, 736–737. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; ICTV Report consortium. ICTV Virus Taxonomy Profile: Iflaviridae. J. Gen. Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef]
- Olendraite, I.; Brown, K.; Valles, S.M.; Firth, A.E.; Chen, Y.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; et al. ICTV Virus Taxonomy Profile: Polycipiviridae. J. Gen. Virol. 2019, 100, 554–555. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Zell, R.; Knowles, N.J.; Simmonds, P. A proposed division of the family Picornaviridae into subfamilies based on phylogenetic relationships and functional genomic organization. Arch. Virol. 2021, 166, 2927–2935. [Google Scholar] [CrossRef]
- Habayeb, M.S.; Ekengren, S.K.; Hultmark, D. Nora virus, a persistent virus in Drosophila, defines a new picorna-like virus family. J. Gen. Virol. 2006, 87, 3045–3051. [Google Scholar] [CrossRef]
- Ekström, J.O.; Habayeb, M.S.; Srivastava, V.; Kieselbach, T.; Wingsle, G.; Hultmark, D. Drosophila Nora virus capsid proteins differ from those of other picorna-like viruses. Virus Res. 2011, 160, 51–58. [Google Scholar] [CrossRef]
- Hause, B.M.; Hesse, R.A.; Anderson, G.A. Identification of a novel Picornavirales virus distantly related ot posavirus in swine feces. Virus Genes 2015, 51, 144–147. [Google Scholar] [CrossRef]
- Hause, B.M.; Palinski, R.; Hesse, R.; Anderson, G. Highly diverse posaviruses in swine feces are aquatic in origin. J. Gen. Virol. 2016, 97, 1362–1367. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Cotten, M.; Deijs, M.; Jebbink, M.F.; Bakker, M.; Farsani, S.M.J.; Canuti, M.; Kellam, P.; van der Hoek, L. A novel genus in the order Picornavirales detected in human stool. J. Gen. Virol. 2015, 96, 3440–3443. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Phan, M.V.T.; the VIZIONS Consortium; Simmonds, P.; Koopmans, M.P.G.; Kellam, P.; van den Hoek, L.; Cotten, M. Characterization of posa and posa-like virus genomes in fecal samples from humans, pigs, rats, and bats collected from a single location in Vietnam. Virus Evol. 2017, 3, vex022. [Google Scholar] [CrossRef]
- Reuter, G.; Pankovics, P.; Delwart, E.; Boros, A. A novel posavirus-related single-stranded RNA virus from fish (Cyprinus carpio). Arch. Virol. 2015, 160, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Zell, R.; Deboutte, W.; Zeller, M.; Conceicao-Neto, N.; Heylen, E.; Maes, P.; Knowles, N.J.; Ghogomu, S.M.; Van Ranst, M.; et al. Highly diverse population of Picornaviridae and other members of the Picornavirales, in Cameroonian fruit bats. BMC Genom. 2017, 18, 249. [Google Scholar] [CrossRef]
- Negrey, J.D.; Thompson, M.E.; Langergraber, K.E.; Machanda, Z.P.; Mitani, J.C.; Muller, M.N.; Otali, E.; Owens, L.A.; Wrangham, R.W.; Goldberg, T.L. Demography, life-history trade-offs, and the gastrointestinal virome of wild chimpanzees. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190613. [Google Scholar] [CrossRef]
- Webster, C.L.; Longdon, B.; Lewis, S.H.; Obbard, D.J. Twenty-five new viruses associated with the Drosophilidae (Diptera). Evol. Bioinform. Online 2016, 12, 13–25. [Google Scholar] [CrossRef]
- van der Loos, L.M.; De Coninck, L.; Zell, R.; Lequime, S.; Willems, A.; De Clerck, O.; Matthijnssens, J. Highly divergent CRESS DNA and picorna-like viruses associated with bleached thalli of the green seaweed Ulva. Microbiol. Spectrum 2023, 11, e0025523. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Czech, B.; Crunk, A.; Wallace, A.; Mitreva, M.; Hannon, G.J.; Davis, R.E. Deep small RNA sequencing from the nematode Ascaris reveals conservation, functional diversification, and novel developmental profiles. Genome Res. 2011, 21, 1462–1477. [Google Scholar] [CrossRef]
- Yang, S.; Mao, Q.; Wang, Y.; He, J.; Yang, J.; Chen, X.; Xiao, Y.; He, Y.; Lu, J.; Yang, Z.; et al. Expanding known viral diversity in plants: Virome of 161 species alongside an ancient canal. Environ. Microbiome 2022, 17, 58. [Google Scholar] [CrossRef]
- Koonin, E.V.; Wolf, Y.I.; Nagasaki, K.; Dolja, V.V. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat. Rev. Microbiol. 2008, 6, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Arhab, Y.; Bulakhov, A.G.; Pestova, T.V.; Hellen, C.U.T. Dissemination of internal ribosomal entry sites (IRES) between viruses by horizontal gene transfer. Viruses 2020, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Bunke, J.; Receveur, K.; Oeser, A.C.; Fickenscher, H.; Zell, R.; Krumbholz, A. High genetic diversity of porcine enterovirus G in Schleswig-Holstein, Germany. Arch. Virol. 2017, 163, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Knutson, T.P.; Velayudhan, B.T.; Marthaler, D.G. A porcine enterovirus G associatred with enteric disease contains a novel papain-like cysteine protease. J. Gen. Virol. 2017, 98, 1305–1310. [Google Scholar] [CrossRef]
- Shang, P.; Misra, S.; Hause, B.; Fang, Y. A naturally occurring recombinant enterovirus expresses a torovirus deubiquitinase. J. Virol. 2017, 91, e00450-17. [Google Scholar] [CrossRef]
- Rosario, K.; Symonds, E.M.; Sinigalliano, C.; Stewart, C.; Breitbart, M. Pepper mild mottle virus as an indicator of fecal pollution. Appl. Environ. Microbiol. 2009, 75, 7261–7267. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Genus | No. of Sequences | Σ |
|---|---|---|---|
| Caliciviridae | 11 genera | 0 | 0 |
| Dicipiviridae * | Aparavirus | 0 | 98 |
| Cripavirus | 3 | ||
| Triatovirus | 0 | ||
| Untypeable | 95 | ||
| Iflaviridae * | Iflavirus | 3 | 3 |
| Marnaviridae * | Bacillarnavirus | 0 | 158 |
| Kusarnavirus | 0 | ||
| Labyrnavirus | 10 | ||
| Locarnavirus | 106 | ||
| Marnavirus | 3 | ||
| Salisharnavirus | 8 | ||
| Sogarnavirus | 11 | ||
| Untypeable | 20 | ||
| Noraviridae * | Orthonoravirus | 0 | 4 |
| Untypeable | 4 | ||
| Picornaviridae * | Ampivirus | 1 | 10 |
| Kobuvirus | 1 | ||
| 66 other genera | 0 | ||
| Untypeable | 8 | ||
| Polycipiviridae * | Chipolycivirus | 4 | 6 |
| Hupolycivirus | 0 | ||
| Sopolycivirus | 0 | ||
| Untypeable | 2 | ||
| Secoviridae | 8 genera | 0 | 0 |
| Solinviviridae * | Invictavirus | 0 | 11 |
| Nyfulvavirus | 0 | ||
| Untypeable | 11 | ||
| Unclassified family | Unclassified genus | 106 | 106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zell, R.; Groth, M.; Selinka, L.; Selinka, H.-C. Diversity of Picorna-Like Viruses in the Teltow Canal, Berlin, Germany. Viruses 2024, 16, 1020. https://doi.org/10.3390/v16071020
Zell R, Groth M, Selinka L, Selinka H-C. Diversity of Picorna-Like Viruses in the Teltow Canal, Berlin, Germany. Viruses. 2024; 16(7):1020. https://doi.org/10.3390/v16071020
Chicago/Turabian StyleZell, Roland, Marco Groth, Lukas Selinka, and Hans-Christoph Selinka. 2024. "Diversity of Picorna-Like Viruses in the Teltow Canal, Berlin, Germany" Viruses 16, no. 7: 1020. https://doi.org/10.3390/v16071020