
Multiomics Analysis Reveals Gut Virome–Bacteria–Metabolite Interactions and Their Associations with Symptoms in Patients with IBS-D
Abstract
:1. Introduction
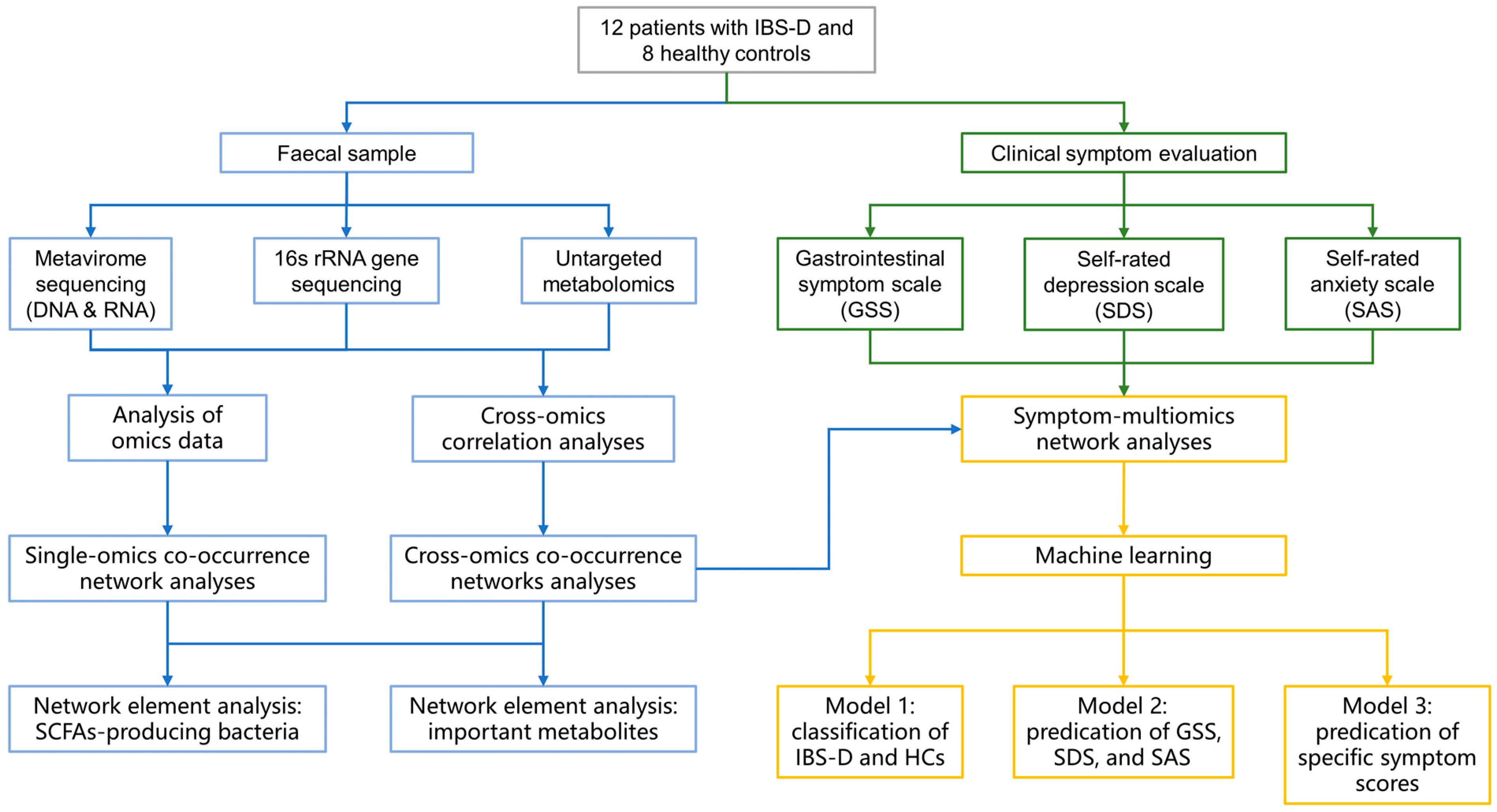
2. Materials and Methods
2.1. Study Cohorts
2.2. Metavirome Sequencing and Analysis
2.2.1. Virus-like Particle (VLP) Enrichment and Metavirome Sequencing
2.2.2. Data Filtering and Assembly
2.2.3. Identification and Classification Annotation of Viral Sequences
2.2.4. Viral Abundance Statistics and Diversity Analysis
2.2.5. Functional Analysis and Virus Host Prediction
2.3. 16S rRNA Gene Sequencing
2.4. Untargeted Metabolomics Analysis
2.5. Co-Occurrence Network Construction and Topological Analyses
2.6. Machine Learning Model Training
2.7. Statistical Analysis
3. Results
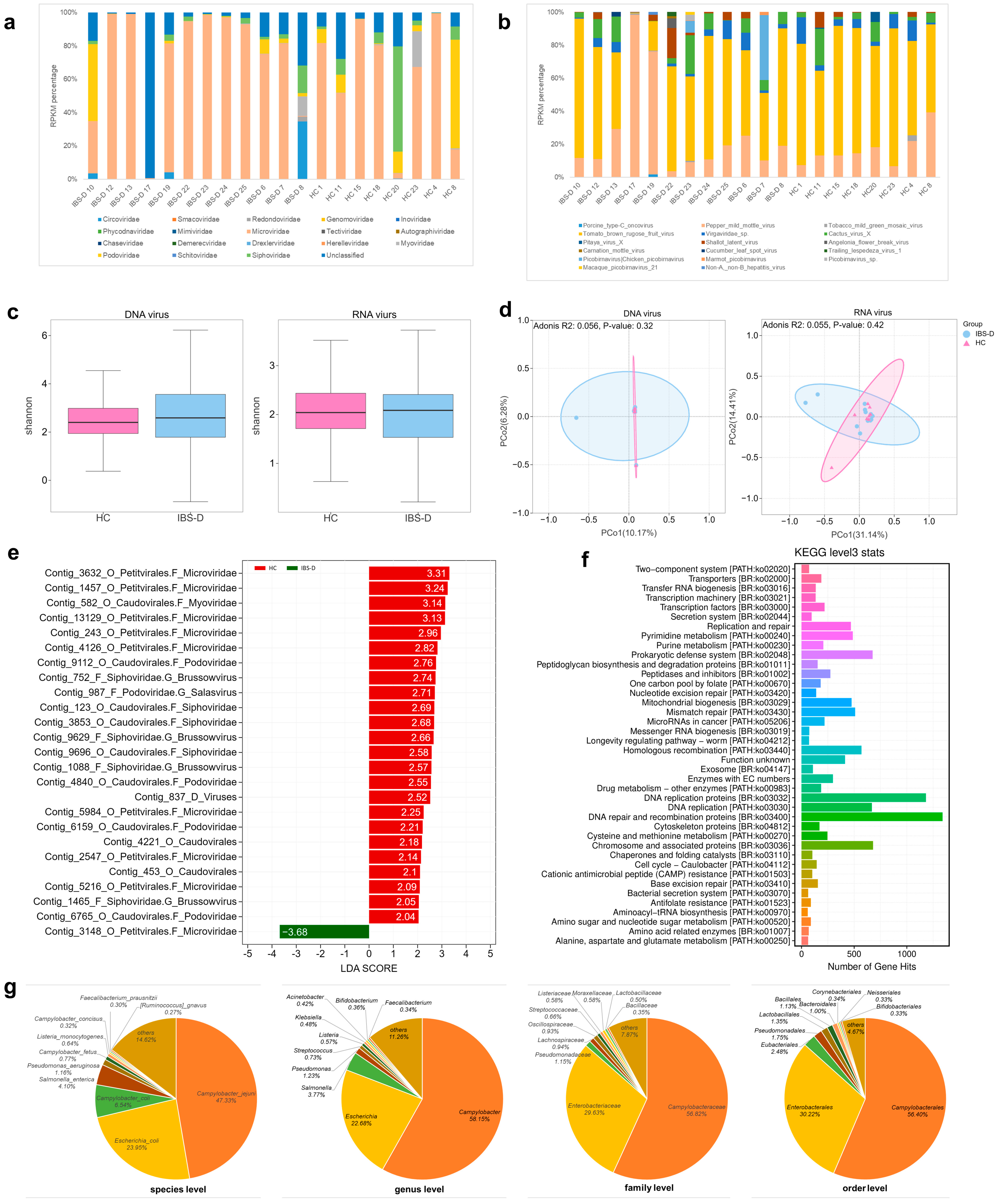
3.1. The Characteristics of the Gut Virome in IBS-D Patients
3.1.1. Sequencing and Identification of the Gut Virome
3.1.2. Alterations in the Composition of the Gut Virome in IBS-D Patients
3.1.3. Diversity of the Gut Virome in IBS-D Patients
3.1.4. Differences in the Abundances of Gut Viruses between the IBS-D and HCs Groups
3.1.5. Functional Annotation and Phage Host Prediction of the Gut Virome in IBS-D
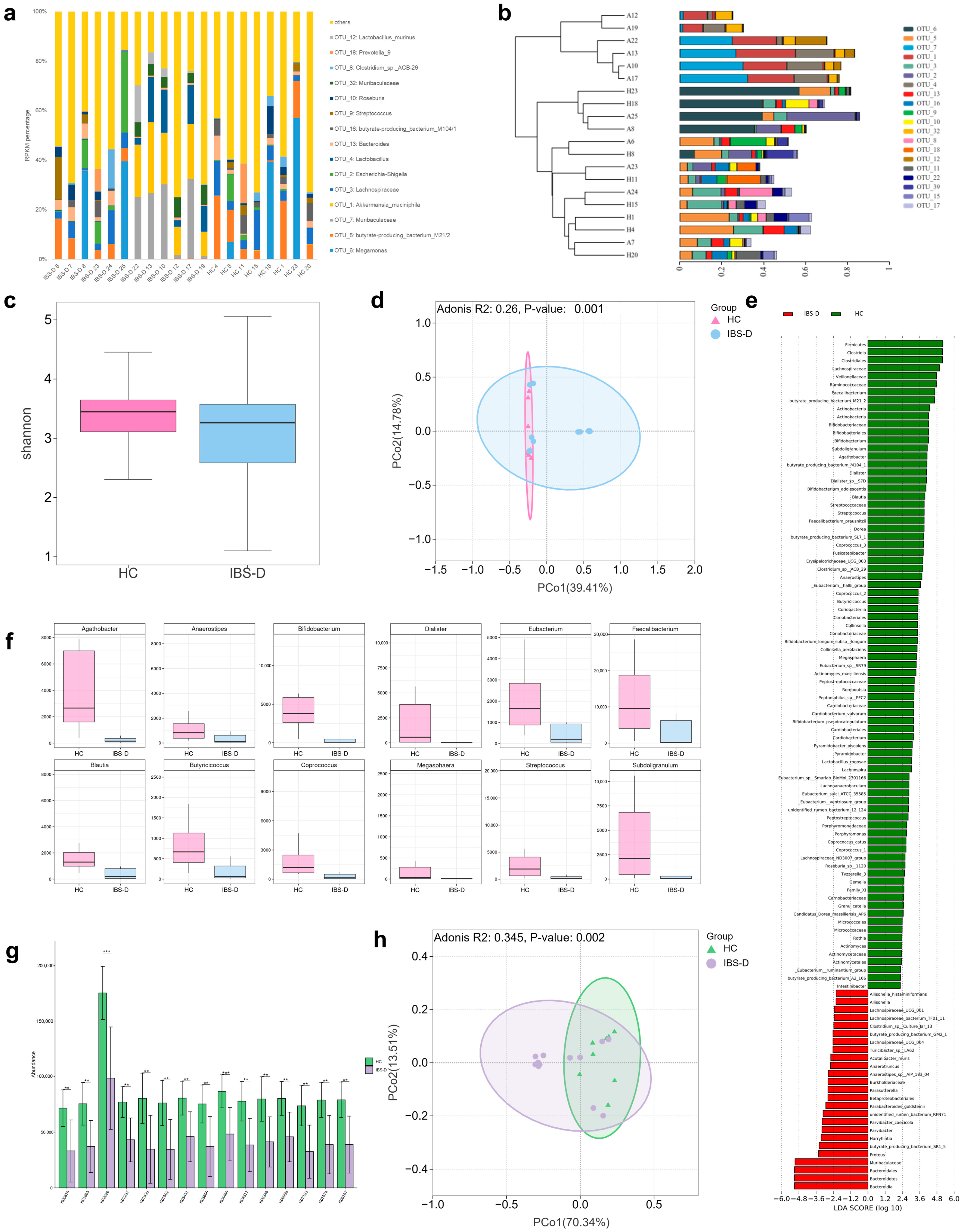
3.2. Differences in Gut Bacteria between IBS-D Patients and HCs Were Associated with SCFA-Producing Bacteria and Amino Acid Functional Pathway Alterations
3.3. Alterations in Gut Metabolites in IBS-D Patients Were Correlated with Disordered Amino Acid Metabolism
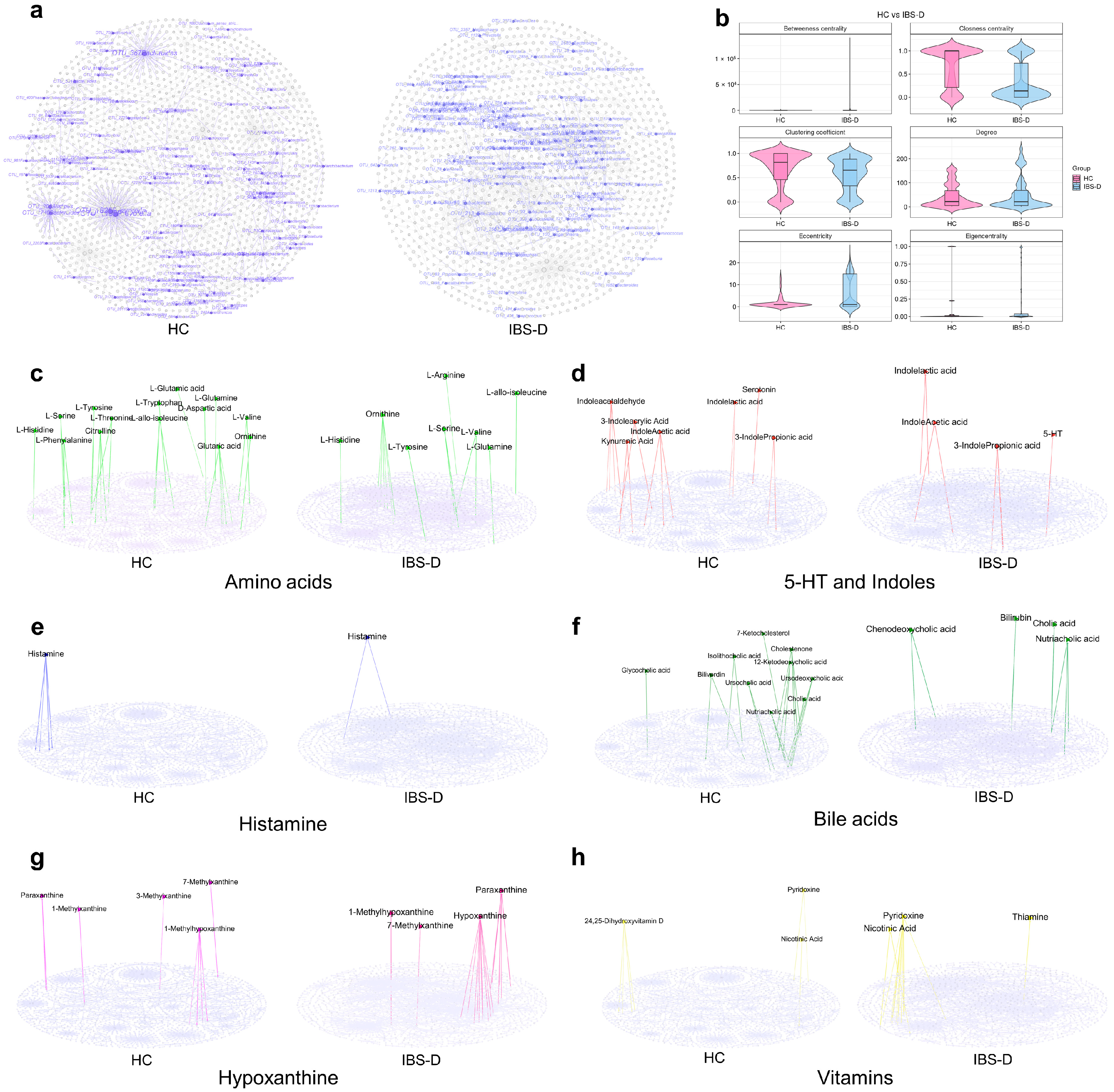
3.4. Differential Gut Viruses Were Significantly Associated with Clinical Symptoms, Gut Bacteria, and Metabolites in IBS-D Patients
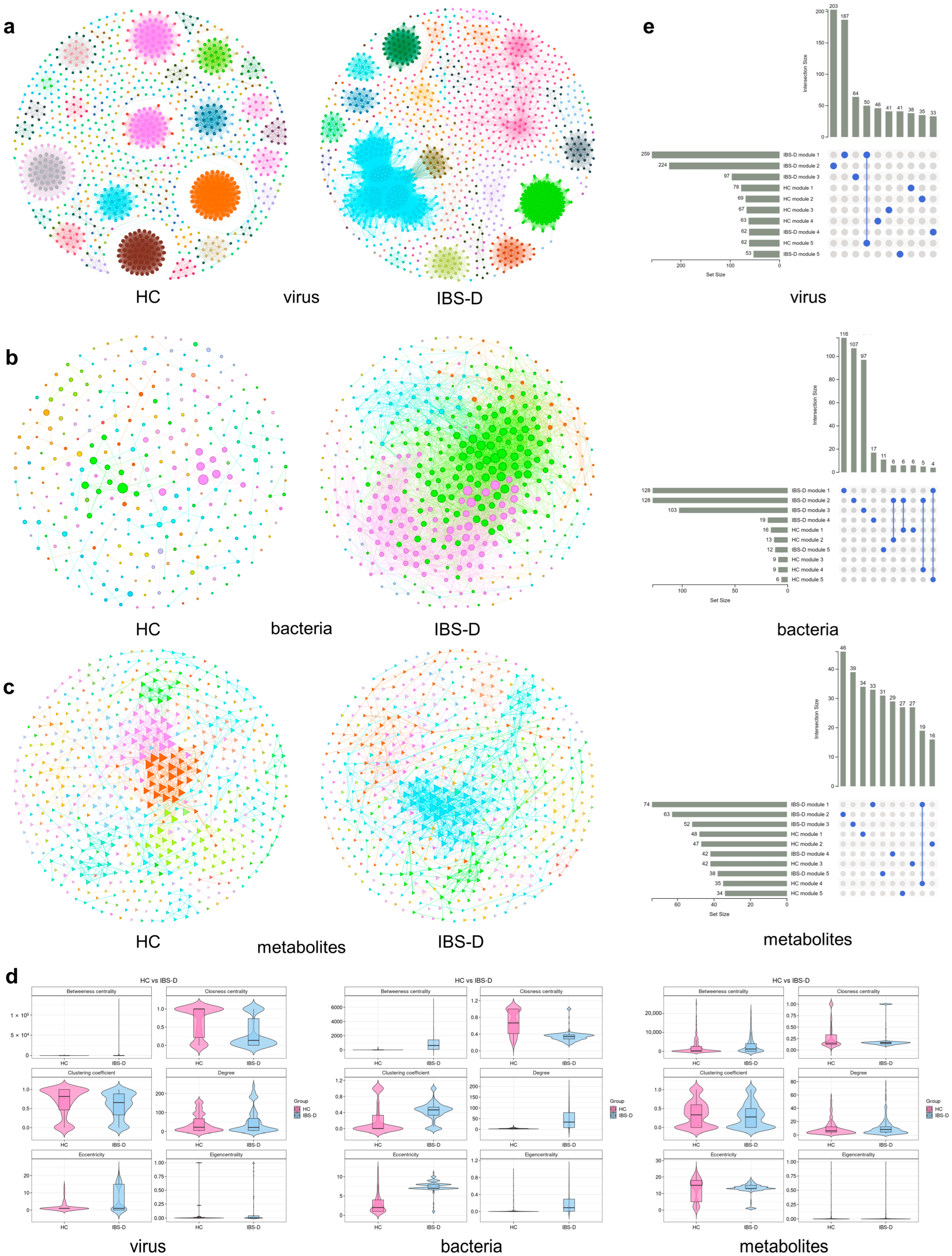
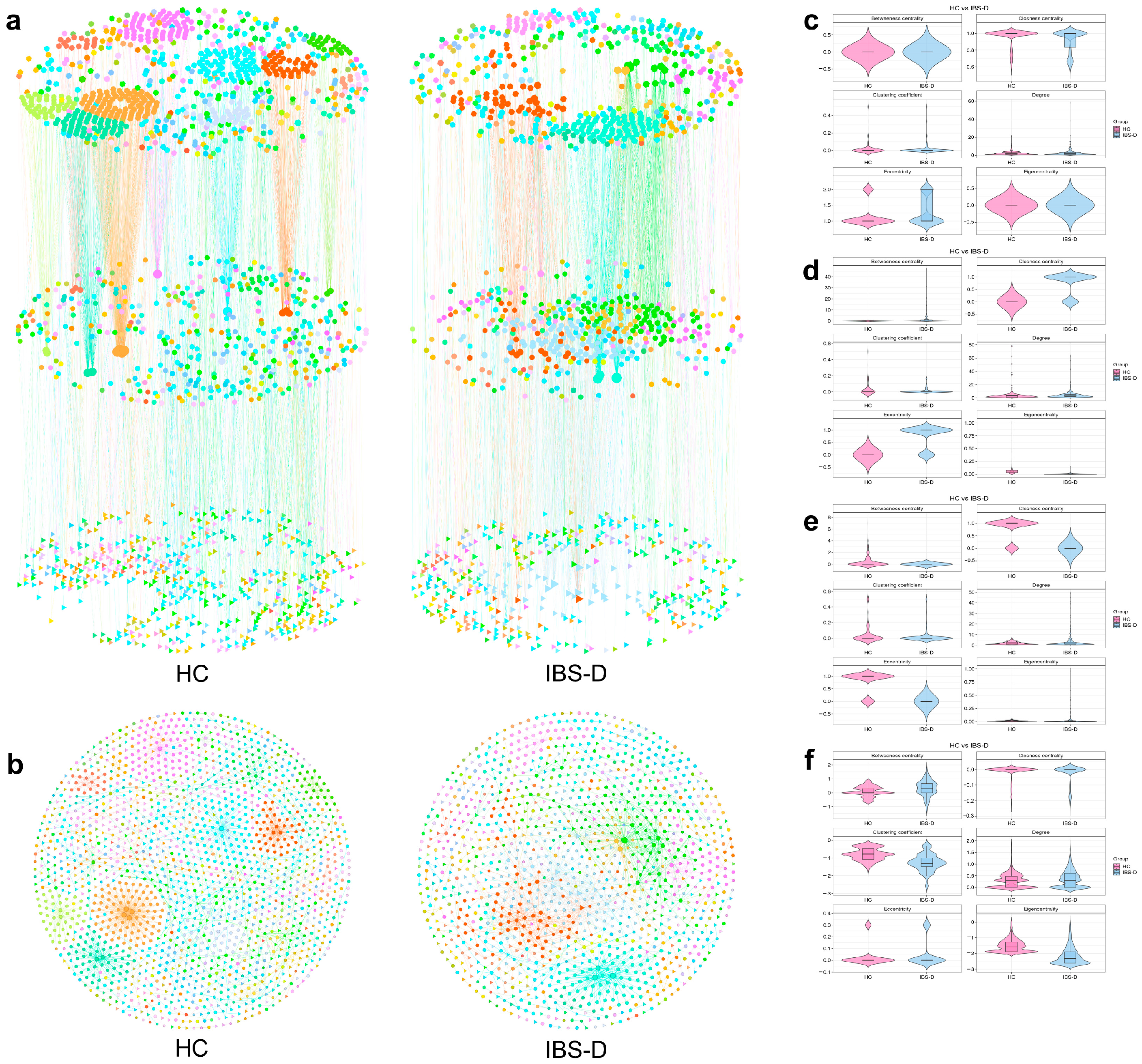
3.5. Single-Omics Co-Occurrence Network Analyses of Gut Viruses, Bacteria, and Metabolites
3.6. Multiomics Co-Occurrence Networks Integrating Gut Viruses, Bacteria, and Metabolites
3.7. SCFA-Producing Bacteria Occupied More Core Positions in Multiomics Networks but Had Fewer Links to Viruses
3.8. Amino Acids and Their Derivatives Showed Unique Connectivity Patterns and Centrality Features in the Multiomics Network of IBS-D
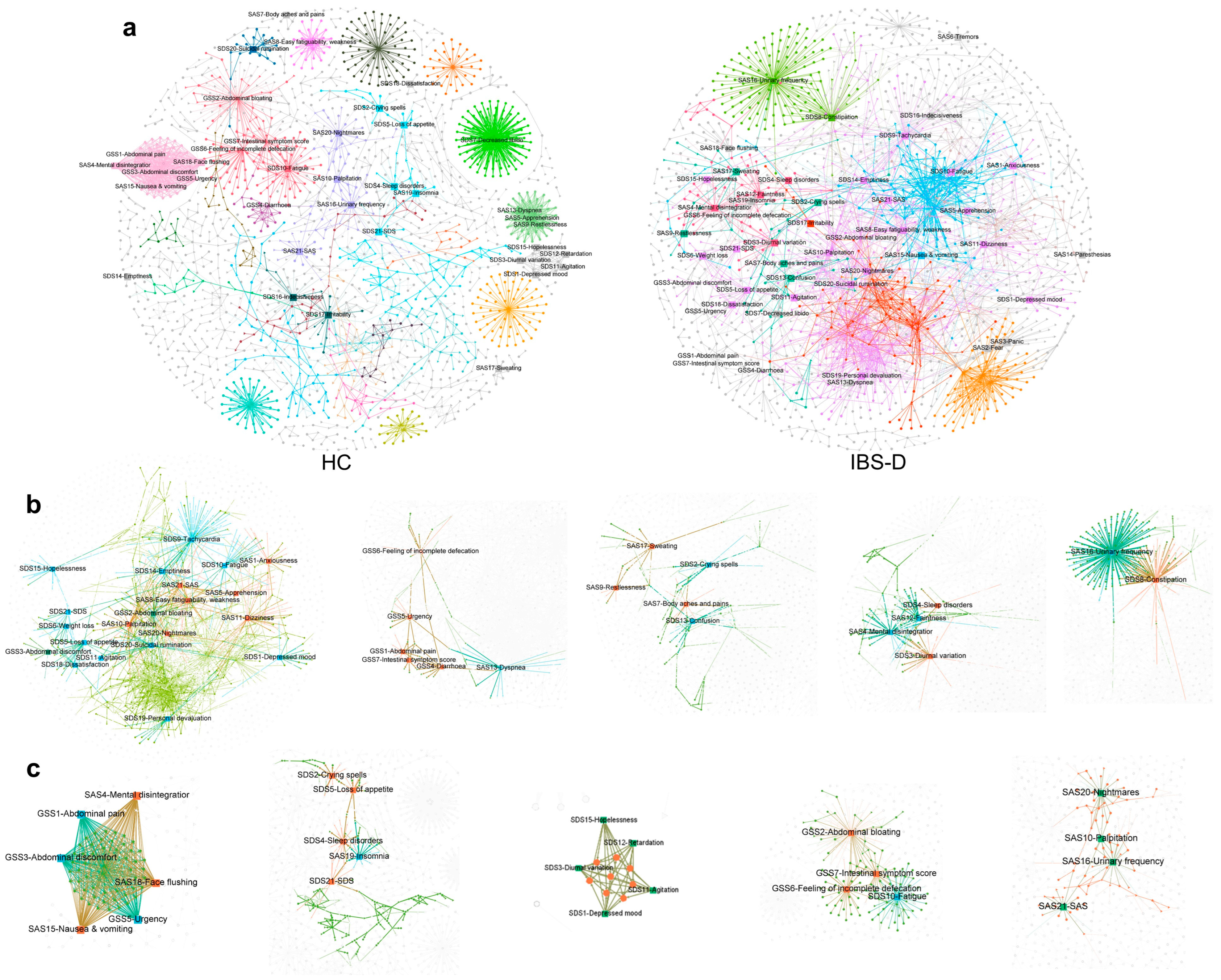
3.9. The Gastrointestinal and Psychological Symptom Factors of IBS-D Patients Were Highly Clustered in the Symptom-Multiomics Network
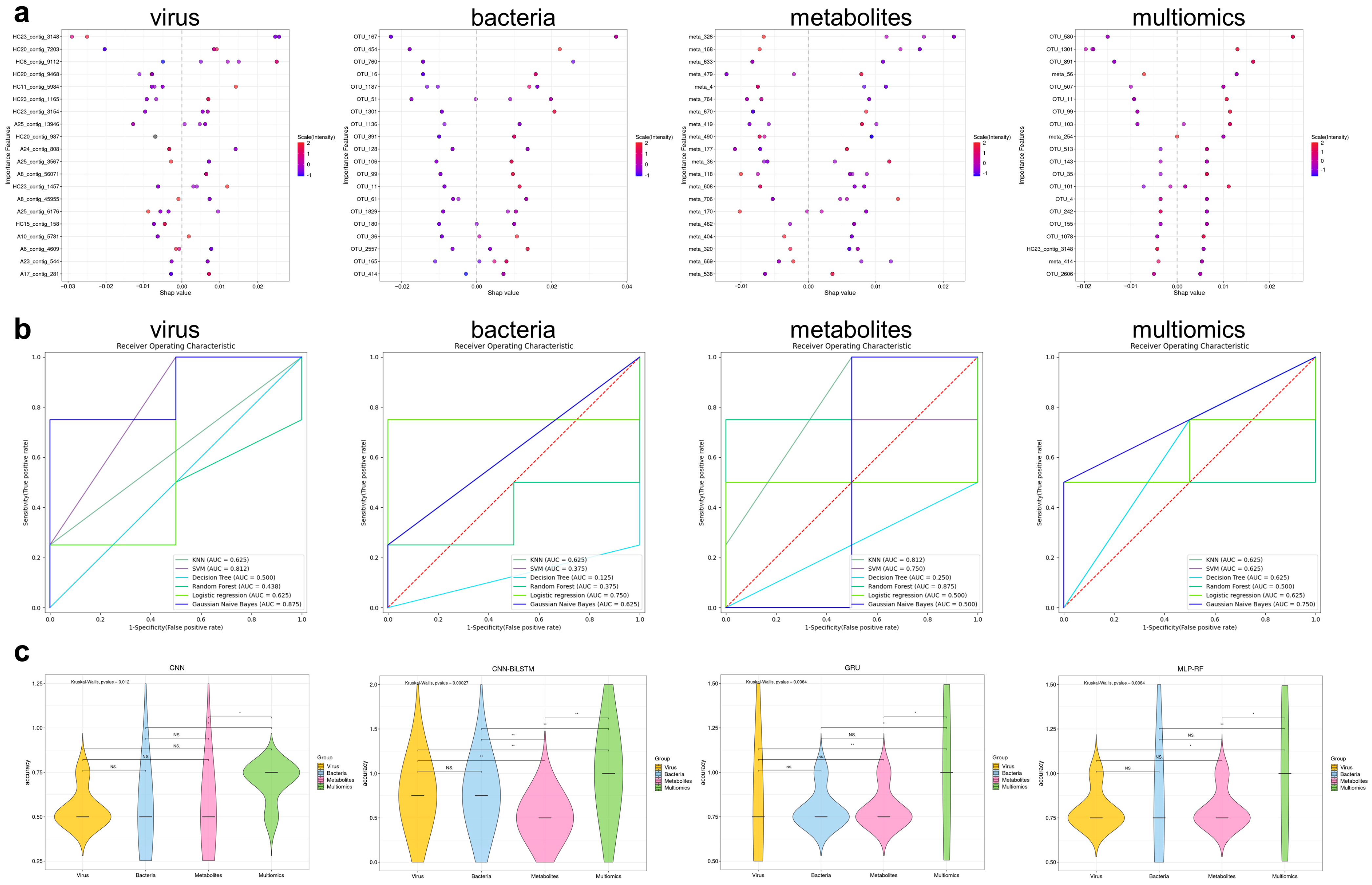
3.10. Machine Learning Models Based on Multiomics Data Could Distinguish IBS-D Patients from HCs and Predict Gastrointestinal and Psychological Symptom Scores
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oka, P.; Parr, H.; Barberio, B.; Black, C.J.; Savarino, E.V.; Ford, A.C. Global prevalence of irritable bowel syndrome according to Rome III or IV criteria: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.S.; Chen, M.H.; Chen, H.X.; Xu, A.G.; Wang, W.A.; Hu, P.J. A population-based epidemiologic study of irritable bowel syndrome in South China: Stratified randomized study by cluster sampling. Aliment. Pharmacol. Ther. 2004, 19, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, H.M.; Black, C.J.; Teasdale, S.B.; Mikocka-Walus, A.; Keefer, L. Irritable bowel syndrome and mental health comorbidity—Approach to multidisciplinary management. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xiang, W.; Li, C.-Y.; Li, S.-C. Economic burden of irritable bowel syndrome in China. World J. Gastroenterol. 2016, 22, 10450–10460. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.C.; Sperber, A.D.; Corsetti, M.; Camilleri, M. Functional Gastrointestinal Disorders 2 Irritable bowel syndrome. Lancet 2020, 396, 1675–1688. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.P.; Chin, V.K.; Looi, C.Y.; Wong, W.F.; Madhavan, P.; Yong, V.C. The Microbiome and Irritable Bowel Syndrome—A Review on the Pathophysiology, Current Research and Future Therapy. Front. Microbiol. 2019, 10, 424646. [Google Scholar] [CrossRef]
- Camilleri, M.; Boeckxstaens, G. Irritable bowel syndrome: Treatment based on pathophysiology and biomarkers. Gut 2023, 72, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Pittayanon, R.; Lau, J.T.; Yuan, Y.; Leontiadis, G.I.; Tse, F.; Surette, M.; Moayyedi, P. Gut Microbiota in Patients With Irritable Bowel Syndrome—A Systematic Review. Gastroenterology 2019, 157, 97–108. [Google Scholar] [CrossRef]
- Ford, A.C.; Harris, L.A.; Lacy, B.E.; Quigley, E.M.M.; Moayyedi, P. Systematic review with meta-analysis: The efficacy of prebiotics, probiotics, synbiotics and antibiotics in irritable bowel syndrome. Aliment. Pharmacol. Ther. 2018, 48, 1044–1060. [Google Scholar] [CrossRef]
- Wang, M.C.; Xie, X.F.; Zhao, S.B.; Ma, X.J.; Wang, Z.Y.; Zhang, Y.C. Fecal microbiota transplantation for irritable bowel syndrome: A systematic review and meta-analysis of randomized controlled trials. Front. Immunol. 2023, 14, 1136343. [Google Scholar] [CrossRef]
- Holtmann, G.J.; Ford, A.C.; Talley, N.J. Pathophysiology of irritable bowel syndrome. Lancet Gastroenterol. Hepatol. 2016, 1, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Sugimura, N.; Burgermeister, E.; Ebert, M.P.; Zuo, T.; Lan, P. The gut virome: A new microbiome component in health and disease. Ebiomedicine 2022, 81, 104113. [Google Scholar] [CrossRef] [PubMed]
- Veses-Garcia, M.; Liu, X.; Rigden, D.J.; Kenny, J.G.; McCarthy, A.J.; Allison, H.E. Transcriptomic analysis of Shiga-toxigenic bacteriophage carriage reveals a profound regulatory effect on acid resistance in Escherichia coli. Appl. Environ. Microbiol. 2015, 81, 8118–8125. [Google Scholar] [CrossRef] [PubMed]
- Popescu, M.; Van Belleghem, J.D.; Khosravi, A.; Bollyky, P.L. Bacteriophages and the Immune System. Annu. Rev. Virol. 2021, 8, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J. Missing a Phage: Unraveling Tripartite Symbioses within the Human Gut. Msystems 2019, 4. [Google Scholar] [CrossRef]
- Ansari, M.H.; Ebrahimi, M.; Fattahi, M.R.; Gardner, M.G.; Safarpour, A.R.; Faghihi, M.A.; Lankarani, K.B. Viral metagenomic analysis of fecal samples reveals an enteric virome signature in irritable bowel syndrome. BMC Microbiol. 2020, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, S.; Das, A.; O’Herlihy, E.; Shanahan, F.; O’Toole, P.W.; Jeffery, I.B. The gut virome in Irritable Bowel Syndrome differs from that of controls. Gut Microbes 2021, 13, 1887719. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Shiryan, I.; Borenstein, E. Multi-omic integration of microbiome data for identifying disease-associated modules. Nat. Commun. 2024, 15, 2621. [Google Scholar] [CrossRef]
- Zung, W.W. A self-rating depression scale. Arch. Gen. Psychiatry 1965, 12, 63–70. [Google Scholar] [CrossRef]
- Zung, W.W. A rating instrument for anxiety disorders. Psychosomatics 1971, 12, 371–379. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Hulo, C.; de Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Mirdita, M.; Levy Karin, E.; Norroy, C.; Galiez, C.; Söding, J. SpacePHARER: Sensitive identification of phages from CRISPR spacers in prokaryotic hosts. Bioinformatics 2021, 37, 3364–3366. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using Nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- The MathWorks Inc. MATLAB Version: 9.14.0 (R2023a); The MathWorks Inc.: Natick, MA, USA, 2023. [Google Scholar]
- The MathWorks Inc. Deep Learning Toolbox Version: 14.6 (R2023a); The MathWorks Inc.: Natick, MA, USA, 2022. [Google Scholar]
- The MathWorks Inc. Statistics and Machine Learning Toolbox Version: 12.5 (R2023a); The MathWorks Inc.: Natick, MA, USA, 2022. [Google Scholar]
- Lavelle, K.; Sadovskaya, I.; Vinogradov, E.; Kelleher, P.; Lugli, G.A.; Ventura, M.; van Sinderen, D.; Mahony, J. Brussowvirus SW13 Requires a Cell Surface-Associated Polysaccharide To Recognize Its Streptococcus thermophilus Host. Appl. Environ. Microbiol. 2022, 88, e01723-21. [Google Scholar] [CrossRef] [PubMed]
- Hanemaaijer, L.; Kelleher, P.; Neve, H.; Franz, C.; de Waal, P.; van Peij, N.; van Sinderen, D.; Mahony, J. Biodiversity of Phages Infecting the Dairy Bacterium Streptococcus thermophilus. Microorganisms 2021, 9, 1822. [Google Scholar] [CrossRef] [PubMed]
- Ariolii, S.; Eraclio, G.; Della Scala, G.; Neri, E.; Colombo, S.; Scaloni, A.; Fortina, M.G.; Mora, D. Role of Temperate Bacteriophage φ20617 on Streptococcus thermophilus DSM 20617 T Autolysis and Biology. Front. Microbiol. 2018, 9, 2719. [Google Scholar] [CrossRef] [PubMed]
- Abraha, H.B.; Kim, K.P. Complete genome sequence analysis, morphology and structural protein identification of two Bacillus subtilis phages, BSTP4 and BSTP6, which may form a new species in the genus Salasvirus. Virus Genes 2023, 59, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, T.Y.; Jin, M.; Zeng, R.Y. Characterization of Bacillus phage Gxv1, a novel lytic Salasvirus phage isolated from deep-sea seamount sediments. Mar. Life Sci. Technol. 2021, 3, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.M.; Zang, Q.B.; Gu, Y.; Niu, G.P.; Ding, C.; Zhang, P.Y. Viral metagenomics analysis of picobirnavirus-positive feces from children with sporadic diarrhea in China. Arch. Virol. 2016, 161, 971–975. [Google Scholar] [CrossRef]
- Ramos, E.D.F.; Ribeiro, G.D.; Villanova, F.; Milagres, F.A.D.; Brustulin, R.; Araujo, E.L.L.; Pandey, R.P.; Raj, V.S.; Deng, X.T.; Delwart, E.; et al. Composition of Eukaryotic Viruses and Bacteriophages in Individuals with Acute Gastroenteritis. Viruses 2021, 13, 2365. [Google Scholar] [CrossRef]
- Yang, S.; He, Y.; Zhang, J.; Zhang, D.; Wang, Y.; Lu, X.; Wang, X.; Shen, Q.; Ji, L.; Lu, H.; et al. Viral metagenomics reveals diverse viruses in the fecal samples of children with diarrhea. Virol. Sin. 2022, 37, 82–93. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Sahoo, G.C.; Nayak, M.K.; Rajendran, K.; Dutta, P.; Mitra, U.; Bhattacharya, M.K.; Naik, T.N.; Bhattacharya, S.K.; Krishnan, T. Detection of genogroup I and II human picobirnaviruses showing small genomic RNA profile causing acute watery diarrhoea among children in Kolkata, India. Infect. Genet. Evol. 2007, 7, 229–238. [Google Scholar] [CrossRef]
- van Leeuwen, M.; Williams, M.M.W.; Koraka, P.; Simon, J.H.; Smits, S.L.; Osterhaus, A.D.M.E. Human Picobirnaviruses Identified by Molecular Screening of Diarrhea Samples. J. Clin. Microbiol. 2010, 48, 1787–1794. [Google Scholar] [CrossRef]
- Giordano, M.O.; Martinez, L.C.; Rinaldi, D.; Espul, C.; Martinez, N.; Isa, M.B.; Depetris, A.R.; Medeot, S.I.; Nates, S.V. Diarrhea and enteric emerging viruses in HIV-infected patients. Aids Res. Hum. Retroviruses 1999, 15, 1427–1432. [Google Scholar] [CrossRef]
- Kashnikov, A.Y.; Epifanova, N.V.; Novikova, N.A. Picobirnaviruses: Prevalence, genetic diversity, detection methods. Vavilovskii Zhurnal Genet. I Sel. 2020, 24, 661–672. [Google Scholar] [CrossRef]
- Febvre, H.P.; Rao, S.; Gindin, M.; Goodwin, N.D.M.; Finer, E.; Vivanco, J.S.; Lu, S.; Manter, D.K.; Wallace, T.C.; Weir, T.L. PHAGE Study: Effects of Supplemental Bacteriophage Intake on Inflammation and Gut Microbiota in Healthy Adults. Nutrients 2019, 11, 666. [Google Scholar] [CrossRef]
- Grubb, D.S.; Wrigley, S.D.; Freedman, K.E.; Wei, Y.R.; Vazquez, A.R.; Trotter, R.E.; Wallace, T.C.; Johnson, S.A.; Weir, T.L. PHAGE-2 Study: Supplemental Bacteriophages Extend Bifidobacterium animalis subsp. Lactis BL04 Benefits on Gut Health and Microbiota in Healthy Adults. Nutrients 2020, 12, 2474. [Google Scholar] [CrossRef]
- Campbell, D.E.; Ly, L.K.; Ridlon, J.M.; Hsiao, A.; Whitaker, R.J.; Degnan, P.H. Infection with Bacteroides Phage BV01 Alters the Host Transcriptome and Bile Acid Metabolism in a Common Human Gut Microbe. Cell Rep. 2020, 32, 108142. [Google Scholar] [CrossRef]
- Oh, J.H.; Alexander, L.M.; Pan, M.; Schueler, K.L.; Keller, M.P.; Attie, A.D.; Walter, J.; van Pijkeren, J.P. Dietary Fructose and Microbiota-Derived Short-Chain Fatty Acids Promote Bacteriophage Production in the Gut Symbiont Lactobacillus reuteri. Cell Host Microbe 2019, 25, 273–284. [Google Scholar] [CrossRef]
- Scanlan, J.G.; Hall, A.R.; Scanlan, P.D. Impact of bile salts on coevolutionary dynamics between the gut bacterium Escherichia coli and its lytic phage PP01. Infect. Genet. Evol. 2019, 73, 425–432. [Google Scholar] [CrossRef]
- Black, C.J.; Ng, C.E.; Goodoory, V.C.; Ford, A.C. Novel Symptom Subgroups in Individuals With Irritable Bowel Syndrome Predict Disease Impact and Burden. Clin. Gastroenterol. Hepatol. 2024, 22, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Black, C.J.; Yiannakou, Y.; Guthrie, E.A.; West, R.; Houghton, L.A.; Ford, A.C. A Novel Method to Classify and Subgroup Patients with IBS Based on Gastrointestinal Symptoms and Psychological Profiles. Am. J. Gastroenterol. 2021, 116, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Jedrusiak, A.; Fortuna, W.; Majewska, J.; Gorski, A.; Jonczyk-Matysiak, E. Phage Interactions with the Nervous System in Health and Disease. Cells 2023, 12, 1720. [Google Scholar] [CrossRef] [PubMed]
- Pargin, E.; Roach, M.J.; Skye, A.; Papudeshi, B.; Inglis, L.K.; Mallawaarachchi, V.; Grigson, S.R.; Harker, C.; Edwards, R.A.; Giles, S.K. The human gut virome: Composition, colonization, interactions, and impacts on human health. Front. Microbiol. 2023, 14, 963173. [Google Scholar] [CrossRef] [PubMed]
- Ritz, N.L.; Draper, L.A.; Bastiaanssen, T.F.S.; Turkington, C.J.R.; Peterson, V.L.; van de Wouw, M.; Vlckova, K.; Fülling, C.; Guzzetta, K.E.; Burokas, A.; et al. The gut virome is associated with stress-induced changes in behaviour and immune responses in mice. Nat. Microbiol. 2024, 9, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Mayneris-Perxachs, J.; Castells-Nobau, A.; Arnoriaga-Rodriguez, M.; Garre-Olmo, J.; Puig, J.; Ramos, R.; Martinez-Hernandez, F.; Burokas, A.; Coll, C.; Maria Moreno-Navarrete, J.; et al. Caudovirales bacteriophages are associated with improved executive function and memory in flies, mice, and humans. Cell Host Microbe 2022, 30, 340–356. [Google Scholar] [CrossRef]
- Xie, P.; Luo, M.; Deng, X.; Fan, J.; Xiong, L. Outcome-Specific Efficacy of Different Probiotic Strains and Mixtures in Irritable Bowel Syndrome: A Systematic Review and Network Meta-Analysis. Nutrients 2023, 15, 3856. [Google Scholar] [CrossRef]
- Mihindukulasuriya, K.A.; Mars, R.A.T.; Johnson, A.J.; Ward, T.; Priya, S.; Lekatz, H.R.; Kalari, K.R.; Droit, L.; Zheng, T.; Blekhman, R.; et al. Multi-Omics Analyses Show Disease, Diet, and Transcriptome Interactions with the Virome. Gastroenterology 2021, 161, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541. [Google Scholar] [CrossRef]
- Priya, S.; Burns, M.B.; Ward, T.; Mars, R.A.T.; Adamowicz, B.; Lock, E.F.; Kashyap, P.C.; Knights, D.; Blekhman, R. Identification of shared and disease-specific host gene-microbiome associations across human diseases using multi-omic integration. Nat. Microbiol. 2022, 7, 780–795. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scale | Performance Index | Virus | Bacteria | Metabolites | Multiomics |
|---|---|---|---|---|---|
| GSS | MAE | 1.60 | 3.75 | 5.99 | 1.35 |
| MAPE | 0.94 | 0.22 | 0.72 | 0.07 | |
| R2 | 0.94 | 0.73 | 0.37 | 0.85 | |
| ML algorithm | MLP-XGB | GRNN | GRU | SVM | |
| SDS | MAE | 3.03 | 7.73 | 1.67 | 1.19 |
| MAPE | 0.08 | 0.15 | 0.04 | 0.02 | |
| R2 | 0.71 | 0.15 | 0.88 | 0.97 | |
| ML algorithm | BiLSTM-RF | RF | LSTM-XGB | LSTM | |
| SAS | MAE | 5.27 | 2.50 | 3.11 | 0.85 |
| MAPE | 0.18 | 0.07 | 0.10 | 0.02 | |
| R2 | 0.55 | 0.84 | 0.34 | 0.89 | |
| ML algorithm | MLP | LSBoost | BiLSTM | CNN-GRU-SE |
| Scale | Performance Index | Virus | Bacteria | Metabolites | Multiomics |
|---|---|---|---|---|---|
| GSS abdominal pain | MAE | 0.70 | 0.45 | 0.58 | 0.26 |
| MAPE | 0.42 | 0.53 | 0.13 | 0.51 | |
| R2 | 0.70 | 0.64 | 0.87 | 0.92 | |
| ML algorithm | LSBoost | GRNN | GRNN | GAM | |
| GSS diarrhea | MAE | 0.97 | 0.68 | 1.07 | 0.25 |
| MAPE | 0.63 | 0.20 | 0.74 | 0.21 | |
| R2 | 0.67 | 0.82 | 0.69 | 0.97 | |
| ML algorithm | LSTM-XGB | MLP-SVM | MLP-RF | CNN-BiLSTM | |
| SDS physiological concomitants | MAE | 1.20 | 0.81 | 0.72 | 0.52 |
| MAPE | 0.07 | 0.08 | 0.06 | 0.03 | |
| R2 | 0.93 | 0.92 | 0.94 | 0.96 | |
| ML algorithm | GAM | XGBoost | LSBoost | ELM | |
| SDS psychological concomitants | MAE | 2.05 | 3.09 | 0.92 | 0.63 |
| MAPE | 0.09 | 0.14 | 0.07 | 0.04 | |
| R2 | 0.69 | 0.53 | 0.97 | 0.98 | |
| ML algorithm | CNN-LSTM-SE | SVM-RF | MLP-SVM | CNN-LSTM-SE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, P.; Luo, M.; Fan, J.; Xiong, L. Multiomics Analysis Reveals Gut Virome–Bacteria–Metabolite Interactions and Their Associations with Symptoms in Patients with IBS-D. Viruses 2024, 16, 1054. https://doi.org/10.3390/v16071054
Xie P, Luo M, Fan J, Xiong L. Multiomics Analysis Reveals Gut Virome–Bacteria–Metabolite Interactions and Their Associations with Symptoms in Patients with IBS-D. Viruses. 2024; 16(7):1054. https://doi.org/10.3390/v16071054
Chicago/Turabian StyleXie, Peiwei, Mei Luo, Jiahui Fan, and Lishou Xiong. 2024. "Multiomics Analysis Reveals Gut Virome–Bacteria–Metabolite Interactions and Their Associations with Symptoms in Patients with IBS-D" Viruses 16, no. 7: 1054. https://doi.org/10.3390/v16071054