

Low-Input, High-Resolution 5′ Terminal Filovirus RNA Sequencing with ViBE-Seq

 ,
,  , ,
, ,  ,
,  , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Biosafety Statement
2.2. Cell Culture
2.3. Virus Propagation
2.4. Animal Tissue
2.5. Viral Sequences
2.6. Analysis of Reverse Transcriptase Terminal Deoxynucleotide Transferase Activity
2.7. RNA Isolation
2.7.1. Virion RNA
2.7.2. Total Cellular RNA from Infected Cells
2.7.3. Total Cellular RNA from Infected NHP Liver Tissue
2.8. Reverse Transcription
2.9. cDNA Marker Synthesis
2.10. cDNA Purification
2.11. cDNA Circularization
2.12. Target Sequence Amplification
2.13. Sequencing
2.14. Sequencing Result Analysis
3. Results
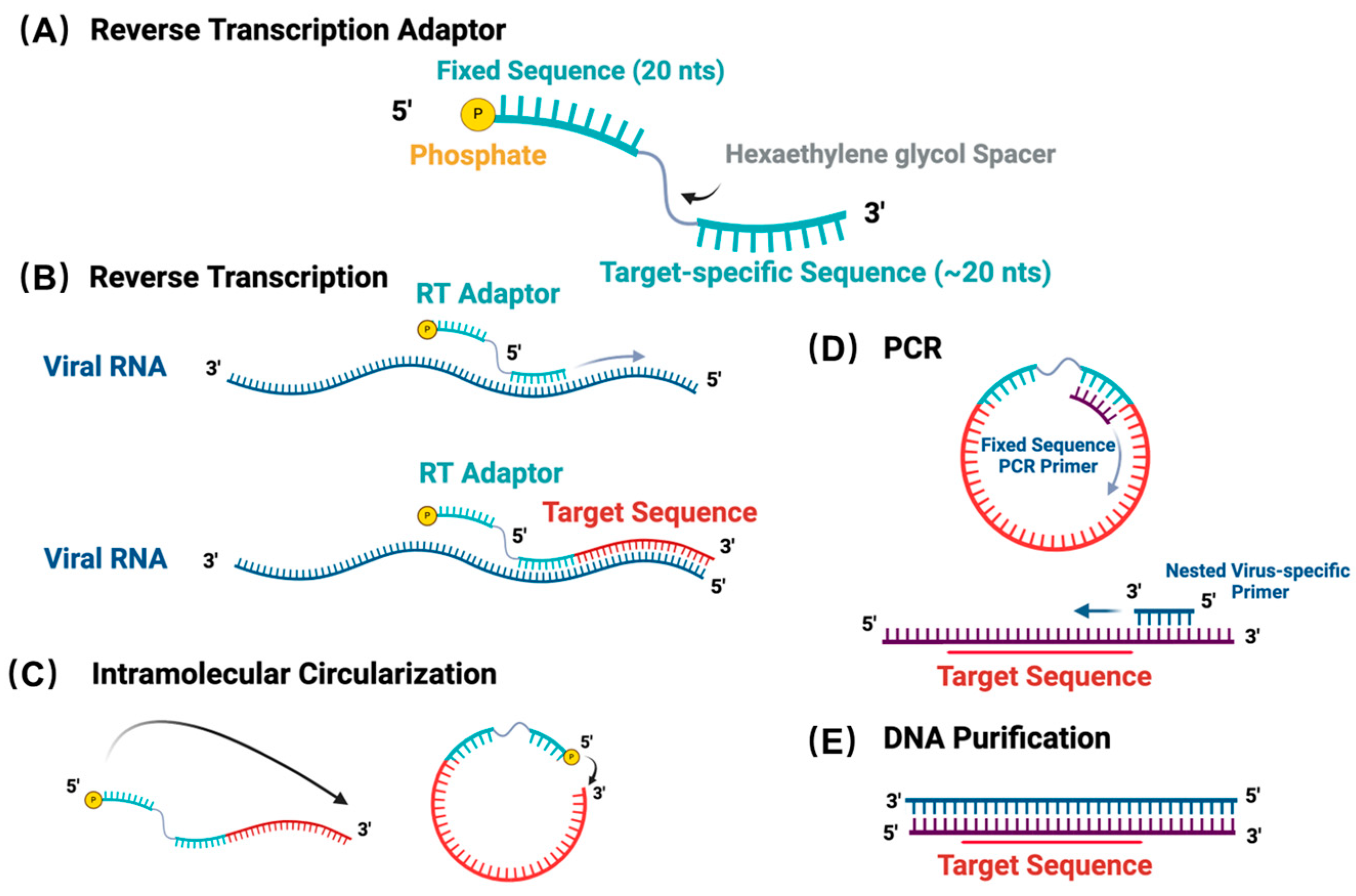
3.1. ViBE-Seq Utilizes cDNA Circularization to Amplify Target Sequences
3.2. An Amount of 50 ng of Purified Virion RNA Is Sufficient for 5′ End Sequence Determination
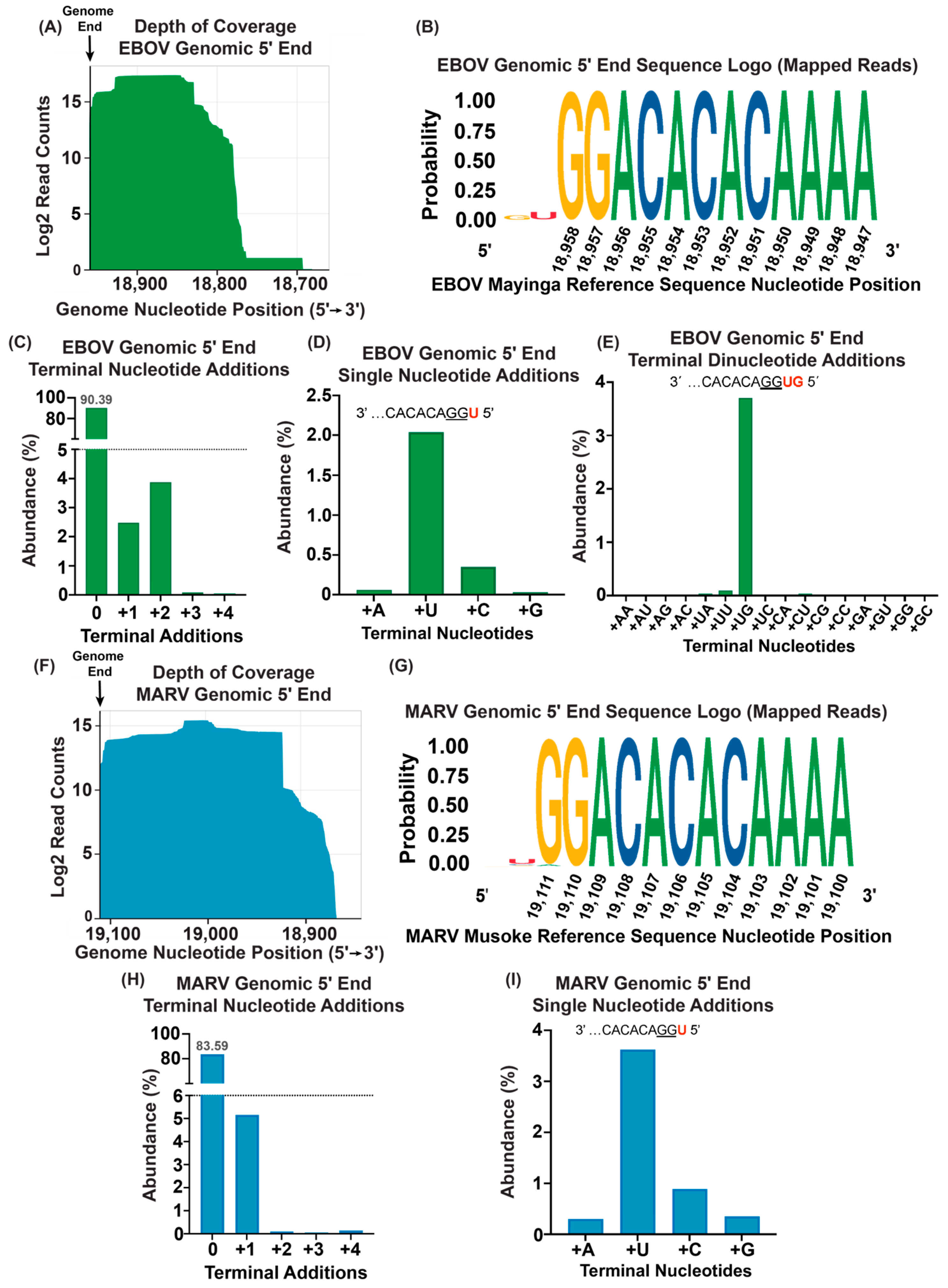
3.3. Viral Genome and mRNA 5′ Termini Captured from RNA Isolated from Infected Cells
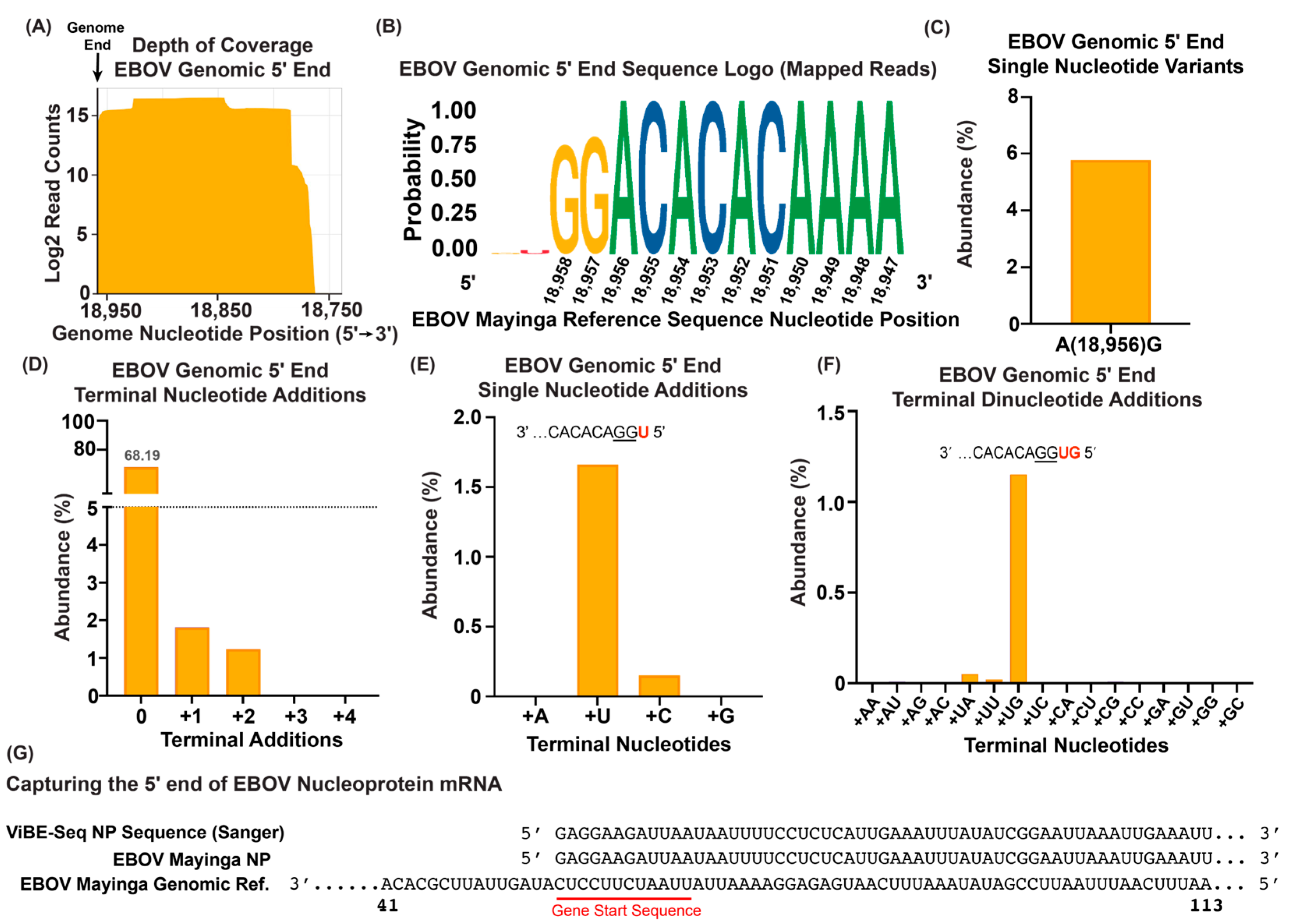
3.4. ViBE-Seq Captures EBOV 5′ Genome Ends from Infected Animal Tissue
3.5. ViBE-Seq Captures Terminal Deoxynucleotidyl Transferase Activity of RT Polymerases
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gatherer, D. The 2014 Ebola virus disease outbreak in West Africa. J. Gen. Virol. 2014, 95, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Hsu, V.P.; Hossain, M.J.; Parashar, U.D.; Ali, M.M.; Ksiazek, T.G.; Kuzmin, I.; Niezgoda, M.; Rupprecht, C.; Bresee, J.; Breiman, R.F. Nipah Virus Encephalitis Reemergence, Bangladesh. Emerg. Infect. Dis. 2004, 10, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.B.; Goh, K.J.; Wong, K.T.; Kamarulzaman, A.; Tan, P.S.K.; Ksiazek, T.G.; Zaki, S.R.; Paul, G.; Lam, S.K.; Tan, C.T. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 1999, 354, 1257–1259. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.E.J.; Howey, R.; Gaunt, E.; Reilly, L.; Chase-Topping, M.; Savill, N. Temporal trends in the discovery of human viruses. Proc. R. Soc. B Biol. Sci. 2008, 275, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; To, K.K.W.; Chan, J.F.W.; Lau, S.K.P.; Woo, P.C.Y.; Yuen, K.-Y. A Systematic Approach to Novel Virus Discovery in Emerging Infectious Disease Outbreaks. J. Mol. Diagn. 2015, 17, 230–241. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Gloza-Rausch, F.; Seebens, A.; Annan, A.; Ipsen, A.; Kruppa, T.; Müller, M.A.; Kalko, E.K.V.; Adu-Sarkodie, Y.; et al. Henipavirus RNA in African bats. PLoS ONE 2009, 4, e6367. [Google Scholar] [CrossRef] [PubMed]
- Kemenesi, G.; Kurucz, K.; Dallos, B.; Zana, B.; Földes, F.; Boldogh, S.; Görföl, T.; Carroll, M.W.; Jakab, F. Re-emergence of Lloviu virus in Miniopterus schreibersii bats, Hungary, 2016. Emerg. Microbes Infect. 2018, 7, 66. [Google Scholar] [CrossRef]
- Yang, X.-L.; Tan, C.W.; Anderson, D.E.; Jiang, R.-D.; Li, B.; Zhang, W.; Zhu, Y.; Lim, X.F.; Zhou, P.; Liu, X.-L.; et al. Characterization of a filovirus (Měnglà virus) from Rousettus bats in China. Nat. Microbiol. 2019, 4, 390–395. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Yang, F.; Ren, X.; Jiang, J.; Dong, J.; Sun, L.; Zhu, Y.; Zhou, H.; Jin, Q. Novel Henipa-like virus, Mojiang Paramyxovirus, in rats, China, 2012. Emerg. Infect. Dis. 2014, 20, 1064–1066. [Google Scholar] [CrossRef]
- Zhang, X.-A.; Li, H.; Jiang, F.-C.; Zhu, F.; Zhang, Y.-F.; Chen, J.-J.; Tan, C.-W.; Anderson, D.E.; Fan, H.; Dong, L.-Y.; et al. A Zoonotic Henipavirus in Febrile Patients in China. N. Engl. J. Med. 2022, 387, 470–472. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Hu, T.; Yan, X.; Pa, Y.; Liu, Y.; Liu, Y.; Li, N.; Yu, J.; Zhang, H.; Liu, Y.; et al. Isolation, characterization, and circulation sphere of a filovirus in fruit bats. Proc. Natl. Acad. Sci. USA 2024, 121, e2313789121. [Google Scholar] [CrossRef] [PubMed]
- Manhart, W.A.; Pacheco, J.R.; Hume, A.J.; Cressey, T.N.; Deflubé, L.R.; Mühlberger, E. A Chimeric Lloviu Virus Minigenome System Reveals that the Bat-Derived Filovirus Replicates More Similarly to Ebolaviruses than Marburgviruses. Cell Rep. 2018, 24, 2573–2580.e4. [Google Scholar] [CrossRef] [PubMed]
- Laing, E.D.; Amaya, M.; Navaratnarajah, C.K.; Feng, Y.-R.; Cattaneo, R.; Wang, L.-F.; Broder, C.C. Rescue and characterization of recombinant cedar virus, a non-pathogenic Henipavirus species. Virol. J. 2018, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Ikegame, S.; Carmichael, J.C.; Wells, H.; Furler O’Brien, R.L.; Acklin, J.A.; Chiu, H.P.; Oguntuyo, K.Y.; Cox, R.M.; Patel, A.R.; Kowdle, S.; et al. Metagenomics-enabled reverse-genetics assembly and characterization of myotis bat morbillivirus. Nat. Microbiol. 2023, 8, 1108–1122. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, T.; Brandt, J.; Caì, Y.; Kuhn, J.H.; Finch, C. Reverse Genetics of Filoviruses. Curr. Top. Microbiol. Immunol. 2017, 411, 421–445. [Google Scholar] [CrossRef] [PubMed]
- Hume, A.J.; Heiden, B.; Olejnik, J.; Suder, E.L.; Ross, S.; Scoon, W.A.; Bullitt, E.; Ericsson, M.; White, M.R.; Turcinovic, J.; et al. Recombinant Lloviu virus as a tool to study viral replication and host responses. PLoS Pathog. 2022, 18, e1010268. [Google Scholar] [CrossRef]
- Martínez-Sobrido, L.; Cheng, B.Y.; de la Torre, J.C. Reverse Genetics Approaches to Control Arenavirus. Methods Mol. Biol. 2016, 1403, 313–351. [Google Scholar] [CrossRef] [PubMed]
- Hume, A.J.; Mühlberger, E. Distinct Genome Replication and Transcription Strategies within the Growing Filovirus Family. J. Mol. Biol. 2019, 431, 4290–4320. [Google Scholar] [CrossRef]
- Halpin, K.; Bankamp, B.; Harcourt, B.H.; Bellini, W.J.; Rota, P.A. Nipah virus conforms to the rule of six in a minigenome replication assay. J. Gen. Virol. 2004, 85, 701–707. [Google Scholar] [CrossRef]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; DePasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Seitz, S. Opportunities and Challenges of Data-Driven Virus Discovery. Biomolecules 2022, 12, 1073. [Google Scholar] [CrossRef] [PubMed]
- Djikeng, A.; Spiro, D. Advancing full length genome sequencing for human RNA viral pathogens. Future Virol. 2008, 4, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fan, H.; An, X.; Fan, H.; Jiang, H.; Chen, Y.; Tong, Y. Scrutinizing Virus Genome Termini by High-Throughput Sequencing. PLoS ONE 2014, 9, e85806. [Google Scholar] [CrossRef] [PubMed]
- Marston, D.A.; McElhinney, L.M.; Ellis, R.J.; Horton, D.L.; Wise, E.L.; Leech, S.L.; David, D.; de Lamballerie, X.; Fooks, A.R. Next generation sequencing of viral RNA genomes. BMC Genom. 2013, 14, 444. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, P.G.; Tsiakanikas, P.; Stolidi, I.; Scorilas, A. A versatile 5′ RACE-Seq methodology for the accurate identification of the 5′ termini of mRNAs. BMC Genom. 2022, 23, 163. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, J.; Hume, A.J.; Ross, S.J.; Scoon, W.A.; Seitz, S.; White, M.R.; Slutzky, B.; Yun, N.E.; Mühlberger, E. Art of the Kill: Designing and Testing Viral Inactivation Procedures for Highly Pathogenic Negative Sense RNA Viruses. Pathogens 2023, 12, 952. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, J.; Forero, A.; Deflube, L.R.; Hume, A.J.; Manhart, W.A.; Nishida, A.; Marzi, A.; Katze, M.G.; Ebihara, H.; Rasmussen, A.L.; et al. Ebolaviruses Associated with Differential Pathogenicity Induce Distinct Host Responses in Human Macrophages. J. Virol. 2017, 91, e00179-17. [Google Scholar] [CrossRef] [PubMed]
- Mühlberger, E.; Weik, M.; Volchkov, V.E.; Klenk, H.-D.; Becker, S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Virol. 1999, 73, 2333–2342. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 March 2023).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Bioinform. Action 2011, 17, 3. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Broad Institute, GitHub Repository. Picard Tools. Available online: http://broadinstitute.github.io/picard/ (accessed on 19 March 2024).
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2014. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.; Bertrand, D.; Yeo, G.H.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Huppertz, I.; Attig, J.; D’Ambrogio, A.; Easton, L.E.; Sibley, C.R.; Sugimoto, Y.; Tajnik, M.; König, J.; Ule, J. iCLIP: Protein-RNA interactions at nucleotide resolution. Methods 2014, 65, 274–287. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Deflubé, L.R.; Cressey, T.N.; Hume, A.J.; Olejnik, J.; Haddock, E.; Feldmann, F.; Ebihara, H.; Fearns, R.; Mühlberger, E. Ebolavirus polymerase uses an unconventional genome replication mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 8535–8543. [Google Scholar] [CrossRef]
- Chen, D.; Patton, J.T. Reverse transcriptase adds nontemplated nucleotides to cDNAs during 5′-RACE and primer extension. Biotechniques 2001, 30, 574–580, 582. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Volchkova, V.A.; Chepurnov, A.A.; Blinov, V.M.; Dolnik, O.; Netesov, S.V.; Feldmann, H. Characterization of the L gene and 5′ trailer region of Ebola virus. J. Gen. Virol. 1999, 80, 355–362. [Google Scholar] [CrossRef]
- Prescott, J.; Bushmaker, T.; Fischer, R.; Miazgowicz, K.; Judson, S.; Munster, V.J. Postmortem stability of Ebola virus. Emerg. Infect. Dis. 2015, 21, 856–859. [Google Scholar] [CrossRef] [PubMed]
- QIAGEN. Reverse Transcription. Available online: https://www.qiagen.com/us/applications/oem-and-custom-solutions/master-mixes/reverse-transcription-test-pack (accessed on 4 March 2023).
- Braun, M.R.; Deflubé, L.R.; Noton, S.L.; Mawhorter, M.E.; Tremaglio, C.Z.; Fearns, R. RNA elongation by respiratory syncytial virus polymerase is calibrated by conserved region V. PLoS Pathog. 2017, 13, e1006803. [Google Scholar] [CrossRef]
- Yoneda, M.; Guillaume, V.; Ikeda, F.; Sakuma, Y.; Sato, H.; Wild, T.F.; Kai, C. Establishment of a Nipah virus rescue system. Proc. Natl. Acad. Sci. USA 2006, 103, 16508–16513. [Google Scholar] [CrossRef]
- Yun, T.; Park, A.; Hill, T.E.; Pernet, O.; Beaty, S.M.; Juelich, T.L.; Smith, J.K.; Zhang, L.; Wang, Y.E.; Vigant, F.; et al. Efficient reverse genetics reveals genetic determinants of budding and fusogenic differences between Nipah and Hendra viruses and enables real-time monitoring of viral spread in small animal models of henipavirus infection. J. Virol. 2015, 89, 1242–1253. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ross, S.J.; Hume, A.J.; Olejnik, J.; Turcinovic, J.; Honko, A.N.; McKay, L.G.A.; Connor, J.H.; Griffiths, A.; Mühlberger, E.; Cifuentes, D. Low-Input, High-Resolution 5′ Terminal Filovirus RNA Sequencing with ViBE-Seq. Viruses 2024, 16, 1064. https://doi.org/10.3390/v16071064
Ross SJ, Hume AJ, Olejnik J, Turcinovic J, Honko AN, McKay LGA, Connor JH, Griffiths A, Mühlberger E, Cifuentes D. Low-Input, High-Resolution 5′ Terminal Filovirus RNA Sequencing with ViBE-Seq. Viruses. 2024; 16(7):1064. https://doi.org/10.3390/v16071064
Chicago/Turabian StyleRoss, Stephen J., Adam J. Hume, Judith Olejnik, Jacquelyn Turcinovic, Anna N. Honko, Lindsay G. A. McKay, John H. Connor, Anthony Griffiths, Elke Mühlberger, and Daniel Cifuentes. 2024. "Low-Input, High-Resolution 5′ Terminal Filovirus RNA Sequencing with ViBE-Seq" Viruses 16, no. 7: 1064. https://doi.org/10.3390/v16071064
APA StyleRoss, S. J., Hume, A. J., Olejnik, J., Turcinovic, J., Honko, A. N., McKay, L. G. A., Connor, J. H., Griffiths, A., Mühlberger, E., & Cifuentes, D. (2024). Low-Input, High-Resolution 5′ Terminal Filovirus RNA Sequencing with ViBE-Seq. Viruses, 16(7), 1064. https://doi.org/10.3390/v16071064