

Transposase-Assisted RNA/DNA Hybrid Co-Tagmentation for Target Meta-Virome of Foodborne Viruses
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Design of Reverse Transcription Primer Pool
2.3. Nucleic Acid Extraction
2.4. TRACE-seq Library Preparation Based on Specific Primer Pool
2.5. Standard Illumina RNA-seq Library Preparation
2.6. Bioinformatics Analysis
3. Results
3.1. Updated TRACE-seq Experimental Methodology
3.2. Sample Information
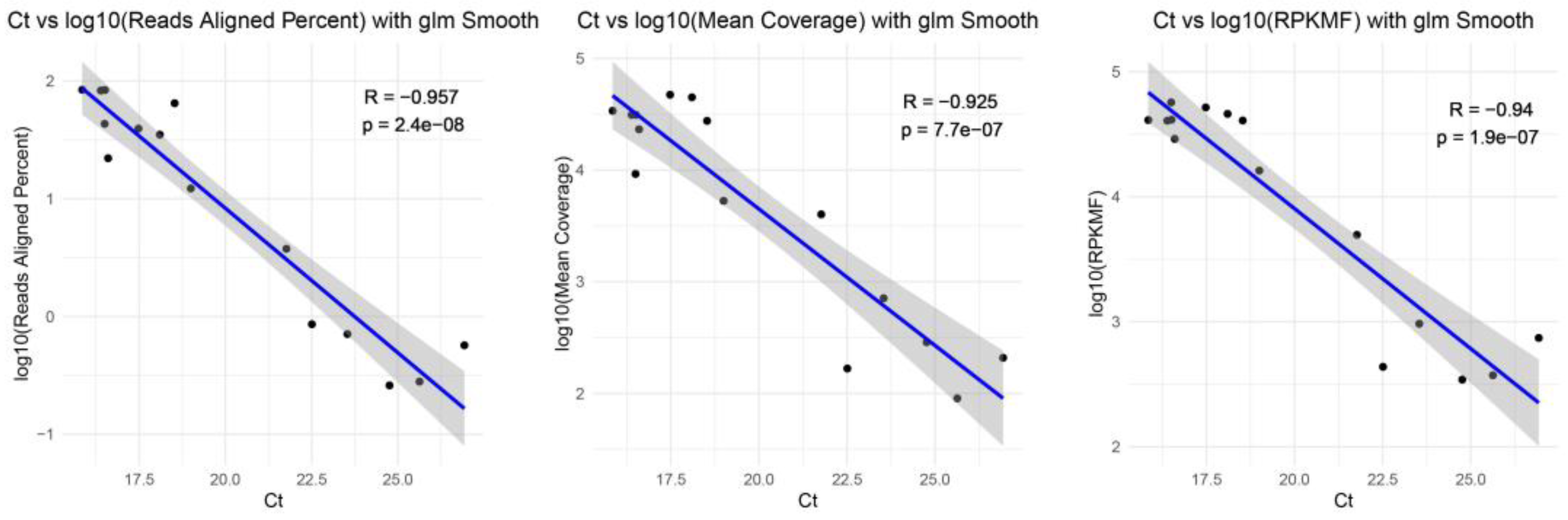
3.3. Whole-Genome Sequences Results for Updated TRACE-seq
3.4. Coinfection Screening of Various Foodborne Viruses
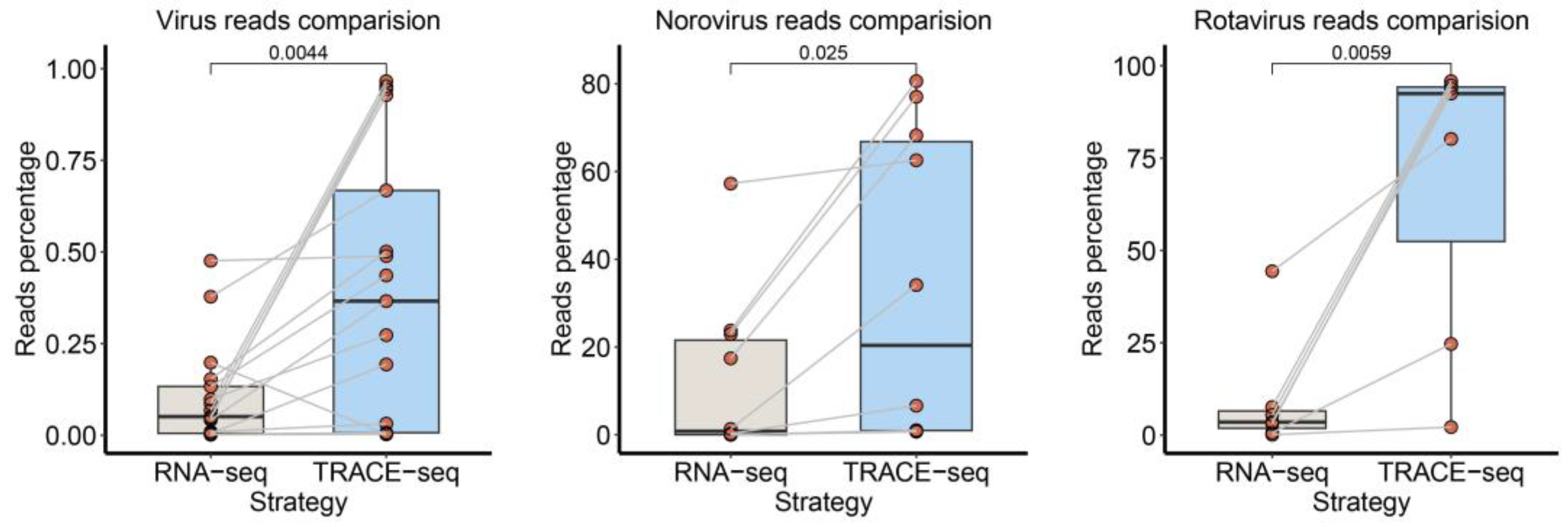
3.5. Comparison of Updated TRACE-seq with Standard RNA-seq Method
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agüeria, D.A.; Terni, C.; Baldovino, V.M.; Civit, D. Food safety knowledge, practices and attitudes of fishery workers in Mar del Plata, Argentina. Food Control 2018, 91, 5–11. [Google Scholar] [CrossRef]
- Havelaar, A.H.; Cawthorne, A.; Angulo, F.; Bellinger, D.; Corrigan, T.; Cravioto, A.; Gibb, H.; Hald, T.; Ehiri, J.; Kirk, M.; et al. WHO Initiative to Estimate the Global Burden of Foodborne Diseases. Lancet 2013, 381, S59. [Google Scholar] [CrossRef]
- Luo, H. Study on Epidemic Characteristics and Changing Trend of Other Infectious Diarrhoeal Diseases in China from 2005 to 2019; Chinese Center for Disease Control and Prevention: Beijing, China, 2020. [Google Scholar]
- Koopmans, M.; Duizer, E. Foodborne viruses: An emerging problem. Int. J. Food Microbiol. 2004, 90, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Alidjinou, E.K.; Sane, F.; Firquet, S.; Lobert, P.-E.; Hober, D. Resistance of Enteric Viruses on Fomites. Intervirology 2019, 61, 205–213. [Google Scholar] [CrossRef]
- Wilhelmi, I.; Roman, E.; Sanchez-Fauquier, A. Viruses causing gastroenteritis. Clin. Microbiol. Infect. 2003, 9, 247–262. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Li, S.; Zhang, Z.; Hou, J.; Wang, D.; Wu, Q.; Jiang, Y.; Tian, Z. Imported human norovirus in travelers, Shanghai port, China 2018: An epidemiological and whole genome sequencing study. Travel Med. Infect. Dis. 2021, 43, 102140. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Ye, L.; Shi, X.; Yan, K.; Huang, J.; Lin, K.; Xing, D.; Ye, S.; Wu, Y.; Li, B.; et al. Dysbiosis in Peripheral Blood Mononuclear Cell Virome Associated with Systemic Lupus Erythematosus. Front. Cell. Infect. Microbiol. 2020, 10, 131. [Google Scholar] [CrossRef]
- López-Labrador, F.X.; Brown, J.R.; Fischer, N.; Harvala, H.; Van Boheemen, S.; Cinek, O.; Sayiner, A.; Madsen, T.V.; Auvinen, E.; Kufner, V.; et al. Recommendations for the introduction of metagenomic high-throughput sequencing in clinical virology, part I: Wet lab procedure. J. Clin. Virol. 2021, 134, 104691. [Google Scholar] [CrossRef]
- Lu, B.; Dong, L.; Yi, D.; Zhang, M.; Zhu, C.; Li, X.; Yi, C. Transposase-assisted tagmentation of RNA/DNA hybrid duplexes. eLife 2020, 9, e54919. [Google Scholar] [CrossRef]
- Lu, B.; Yan, Y.; Dong, L.; Han, L.; Liu, Y.; Yu, J.; Chen, J.; Yi, D.; Zhang, M.; Deng, X.; et al. Integrated characterization of SARS-CoV-2 genome, microbiome, antibiotic resistance and host response from single throat swabs. Cell Discov. 2021, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, D.; Wang, D.; Wu, Q. Library Preparation Based on Transposase Assisted RNA/DNA Hybrid Co-Tagmentation for Next-Generation Sequencing of Human Noroviruses. Viruses 2021, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Tisza, M.; Javornik Cregeen, S.; Avadhanula, V.; Zhang, P.; Ayvaz, T.; Feliz, K.; Hoffman, K.L.; Clark, J.R.; Terwilliger, A.; Ross, M.C.; et al. Wastewater sequencing reveals community and variant dynamics of the collective human virome. Nat. Commun. 2023, 14, 6878. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.V.D.; Avoort, H.V.D.; Peñaranda, S.; Oberste, M.S.; Vinjé, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Z.; Li, S.; Wu, Q.; Tian, P.; Zhang, Z.; Wang, D. Fingerprinting of human noroviruses co-infections in a possible foodborne outbreak by metagenomics. Int. J. Food Microbiol. 2020, 333, 108787. [Google Scholar] [CrossRef]
- Bintsis, T. Foodborne pathogens. Aims Microbiol. 2017, 3, 529–563. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; Erchia, A.M.D.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Desdouits, M.; de Graaf, M.; Strubbia, S.; Munnink, B.B.O.; Kroneman, A.; Le Guyader, F.S.; Koopmans, M.P.G. Novel opportunities for NGS-based one health surveillance of foodborne viruses. ONE Health Outlook 2020, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.A.; Alwood, A.; Thaipisuttikul, I.; Spencer, D.; Haugen, E.; Ernst, S.; Will, O.; Kaul, R.; Raymond, C.; Levy, R.; et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 14339–14344. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Sample ID | RT-qPCR | Ct | Total Filtered Reads | Reads Aligned | Reads Aligned% | Covered Bases | Mean Coverage | RPKMF * | Virus Species | Virus Genotype |
|---|---|---|---|---|---|---|---|---|---|---|
| TR306110255 | Norovirus positive | 25.63 | 2,270,814 | 6416 | 0.28% | 7558 | 90.40 | 373.39 | Norovirus | GII.4 Sydney[P16] |
| TR308030002 | Norovirus positive | 24.76 | 7,029,256 | 18,305 | 0.26% | 7278 | 287.17 | 344.64 | Norovirus | GII.17[P17] |
| 7,029,256 | 8157 | 0.12% | 1233 | 3.37 | 7.16 | Human betaherpesvirus 6B | ||||
| TR310260051 | Norovirus positive | 23.54 | 7,352,203 | 52,078 | 0.71% | 7452 | 711.93 | 964.05 | Norovirus | GII.20[P7] |
| TR311040037 | Norovirus positive | 19.00 | 2,986,176 | 365,863 | 12.25% | 7567 | 5295.76 | 16,191.20 | Norovirus | GII.4 Sydney[P16] |
| 29,86,176 | 603,968 | 20.23% | 6171 | 10,681.10 | 32,775.00 | Astrovirus MLB1 | ||||
| TR310210044 | Norovirus positive | 21.77 | 7,978,050 | 299,652 | 3.76% | 7577 | 4014.02 | 4957.05 | Norovirus | GII.13[P21] |
| TR311130018 | Norovirus positive | 26.93 | 2,749,256 | 15,786 | 0.57% | 7732 | 208.54 | 742.60 | Norovirus | GI.3[P10] |
| JN230418N1 | Norovirus positive | 16.50 | 1,641,314 | 710,458 | 43.29% | 7605 | 9226.61 | 56,917.70 | Norovirus | GII.4 Sydney[P31] |
| JN230418N2 | Norovirus positive | 18.10 | 7,795,456 | 2,731,336 | 35.04% | 7605 | 44,760.58 | 46,071.70 | Norovirus | GII.4 Sydney[P31] |
| JN230418N3 | Norovirus positive | 16.60 | 7,488,859 | 1,654,221 | 22.09% | 7605 | 23,139.51 | 29,045.50 | Norovirus | GII.4 Sydney[P31] |
| JN230418N4 | Norovirus positive | 17.48 | 7,258,419 | 2,861,419 | 39.42% | 7605 | 47,187.67 | 51,837.00 | Norovirus | GII.4 Sydney[P31] |
| TR307130012 | Rotavirus positive | 22.51 | 2,917,878 | 25,102 | 0.86% | 18,214 | 167.21 | 435.94 | Rotavirus | R1-C1-M1-P[8]-A1-I1-T1-N1-G3-E1-H1 |
| TR309140031 | Rotavirus positive | - | 9,764,213 | 6,389,046 | 65.43% | 19,016 | 34,646.50 | 30,022.98 | Rotavirus | R1-C1-M1-P[8]-A1-I1-T1-N1-G3-E1-H1 |
| TR310080053 | Rotavirus positive | - | 110,570 | 24,478 | 22.14% | 18,283 | 161.44 | 11,101.98 | Rotavirus | R2-C2-M2-P[8]-A2-I2-T2-N2-G3-E2-H2 |
| JN230418L1 | Rotavirus positive | 16.39 | 6,125,493 | 5,088,710 | 83.07% | 18,996 | 31,128.48 | 40,607.76 | Rotavirus | R1-C1-M1-P[8]-A1-I1-T1-N1-G3-E1-H1 |
| JN230418L2 | Rotavirus positive | 16.51 | 5,735,717 | 4,811,697 | 83.89% | 19,013 | 31,172.48 | 41,334.89 | Rotavirus | R1-C1-M1-P[8]-A1-I1-T1-N1-G3-E1-H1 |
| JN230418L3 | Rotavirus positive | 15.85 | 6,318,787 | 5,324,226 | 84.26% | 19,043 | 33,894.67 | 41,087.07 | Rotavirus | R1-C1-M1-P[8]-A1-I1-T1-N1-G3-E1-H1 |
| JN230418L4 | Rotavirus positive | 18.53 | 5,021,832 | 3,254,866 | 64.81% | 19,015 | 27,548.76 | 40,760.53 | Rotavirus | R1-C1-M1-P[8]-A1-I1-T1-N1-G3-E1-H1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Zhang, Z.; Wang, Z.; Xue, L.; Liu, F.; Lu, Y.; Yu, S.; Li, S.; Zheng, H.; Zhang, Z.; et al. Transposase-Assisted RNA/DNA Hybrid Co-Tagmentation for Target Meta-Virome of Foodborne Viruses. Viruses 2024, 16, 1068. https://doi.org/10.3390/v16071068
Liu D, Zhang Z, Wang Z, Xue L, Liu F, Lu Y, Yu S, Li S, Zheng H, Zhang Z, et al. Transposase-Assisted RNA/DNA Hybrid Co-Tagmentation for Target Meta-Virome of Foodborne Viruses. Viruses. 2024; 16(7):1068. https://doi.org/10.3390/v16071068
Chicago/Turabian StyleLiu, Danlei, Zilei Zhang, Zhiyi Wang, Liang Xue, Fei Liu, Ye Lu, Shiwei Yu, Shumin Li, Huajun Zheng, Zilong Zhang, and et al. 2024. "Transposase-Assisted RNA/DNA Hybrid Co-Tagmentation for Target Meta-Virome of Foodborne Viruses" Viruses 16, no. 7: 1068. https://doi.org/10.3390/v16071068