
Diverse Circular DNA Viral Communities in Blood, Oral, and Fecal Samples of Captive Lemurs

 , , ,
, , , 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. Extraction, Library Preparation, and Sequencing
2.3. Virus De Novo Assembly and Genome Identification
2.4. Distribution of Virus Genomes across Samples
2.5. Phylogenetic Analyses
2.5.1. Anelloviruses
2.5.2. Cressdnaviruses
2.5.3. Microviruses
2.5.4. Large Bacteriophages and Inoviruses
3. Results and Discussion
3.1. Novel Anelloviruses in Blood, Oral, and Fecal Samples
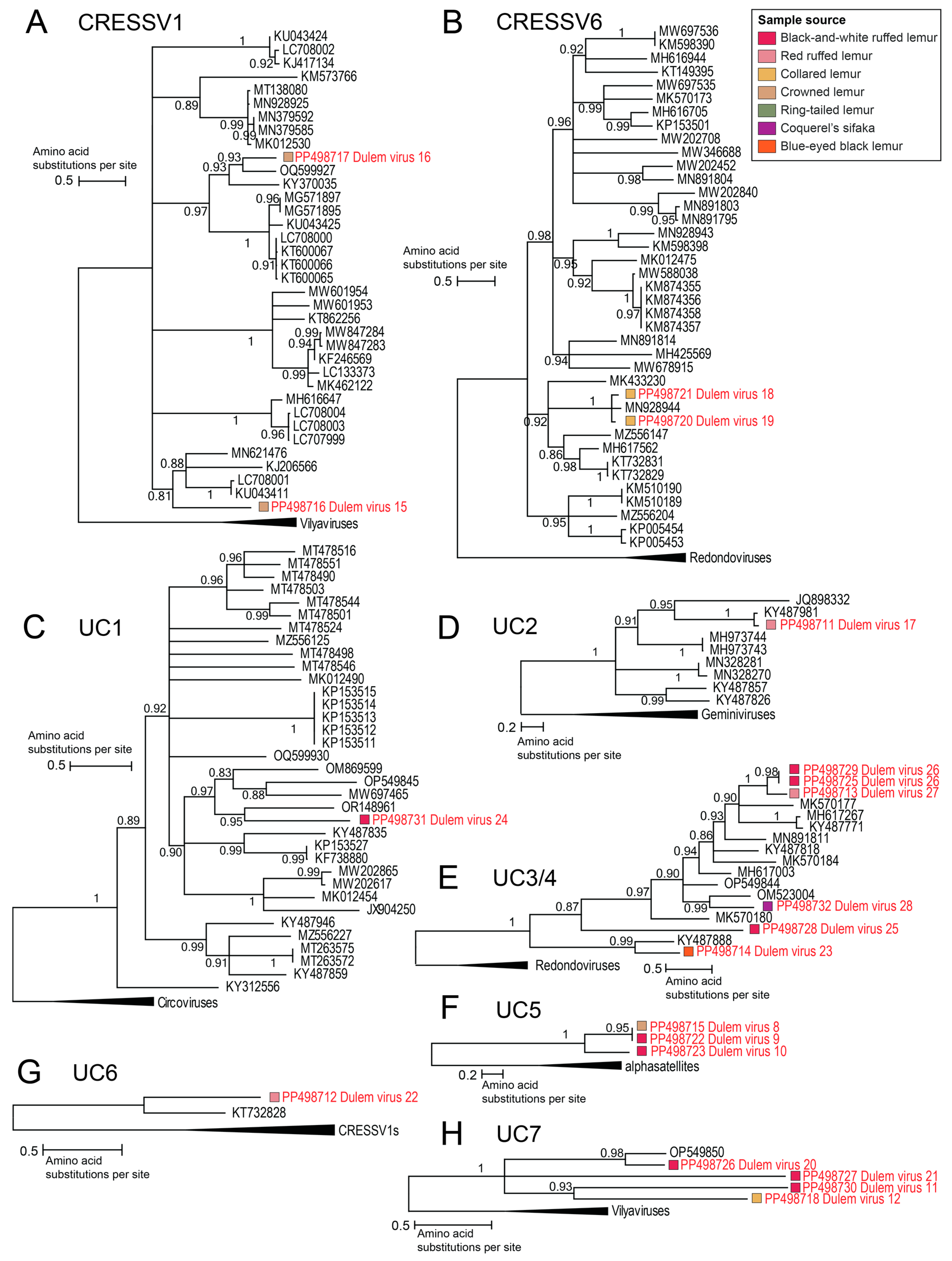
3.2. Diverse Cressdnaviruses in Fecal and Oral Samples
3.2.1. Smacoviruses
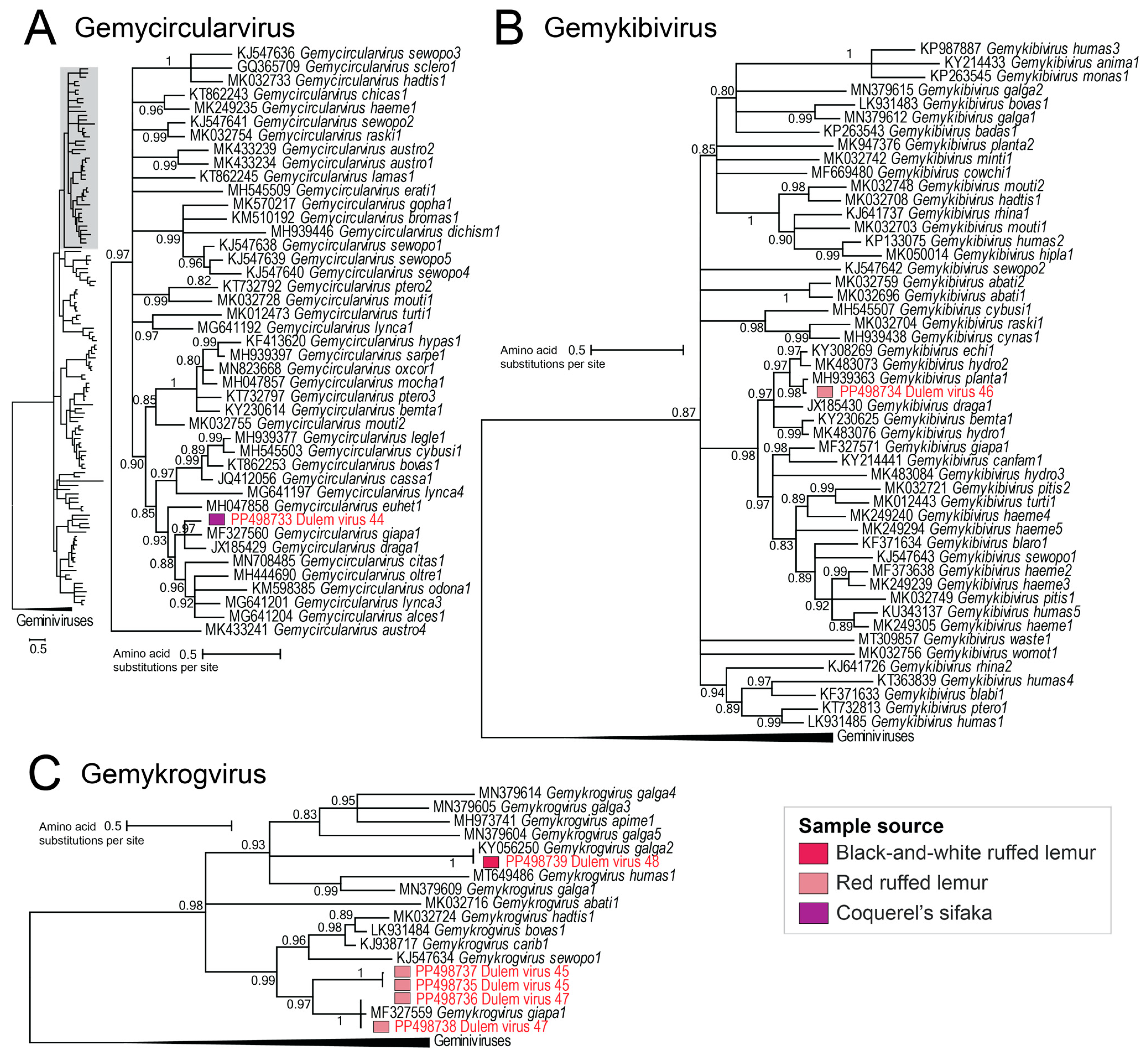
3.2.2. Genomoviruses
3.2.3. Geminivirus
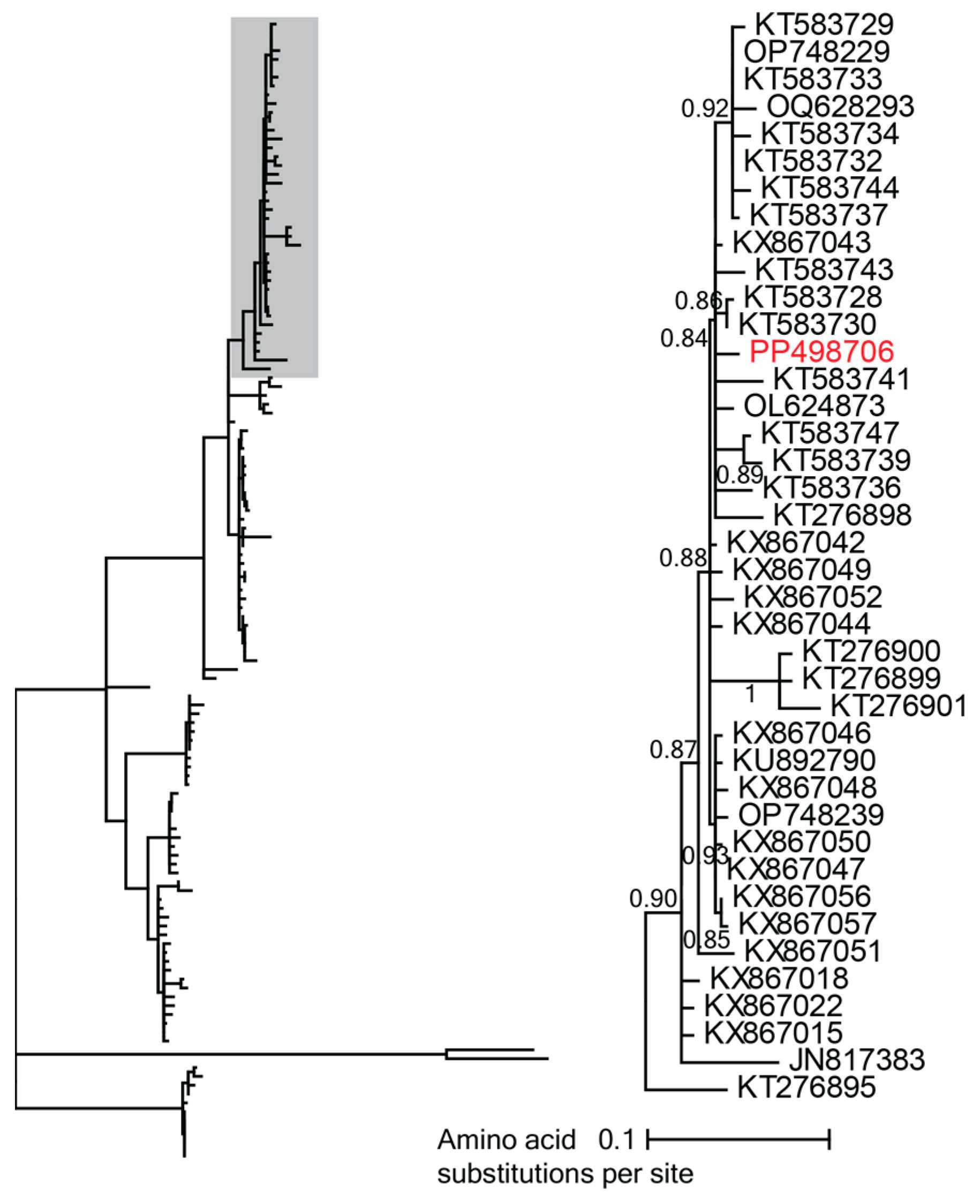
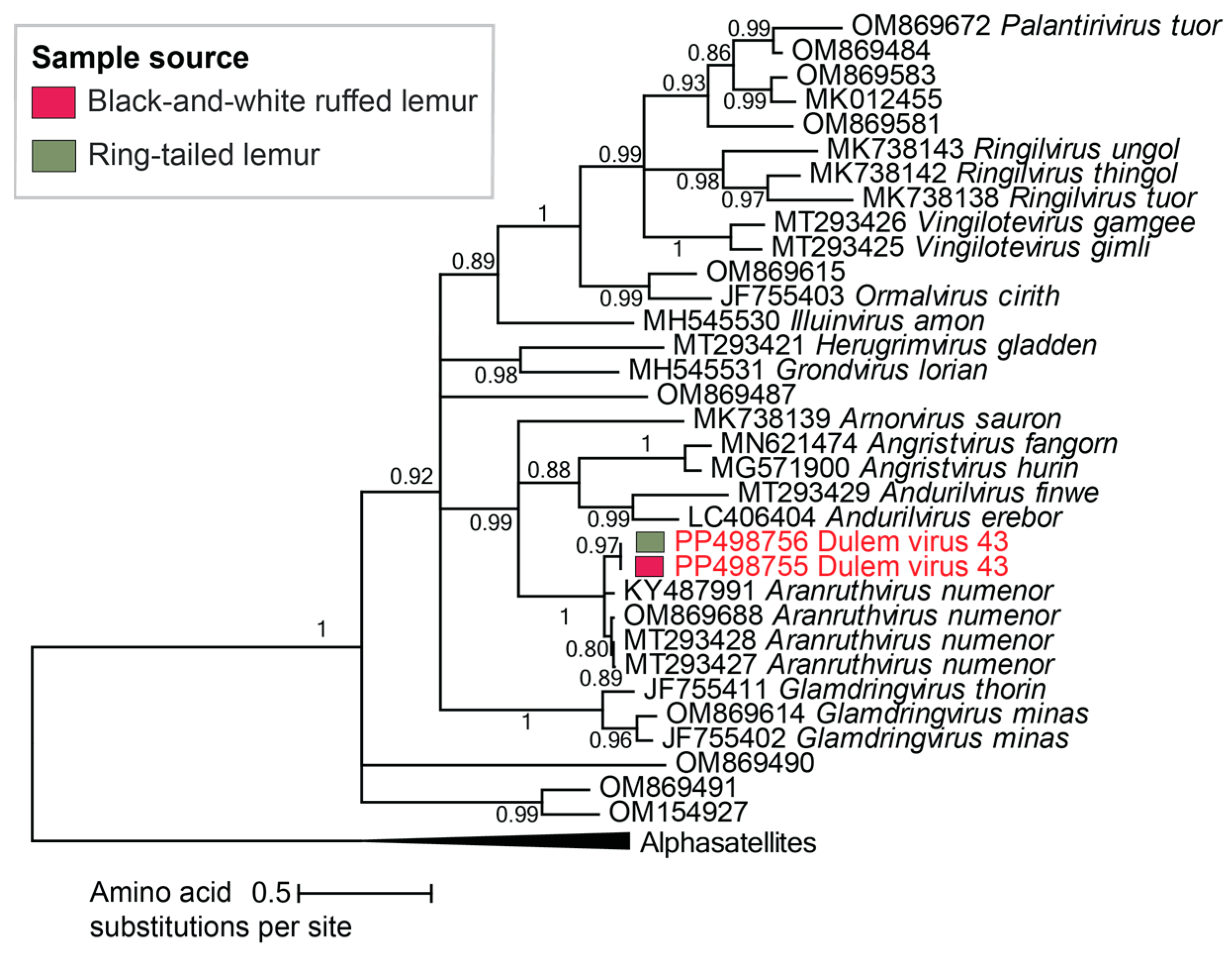
3.2.4. Vilyaviruses
3.2.5. Unclassified Cressdnaviruses
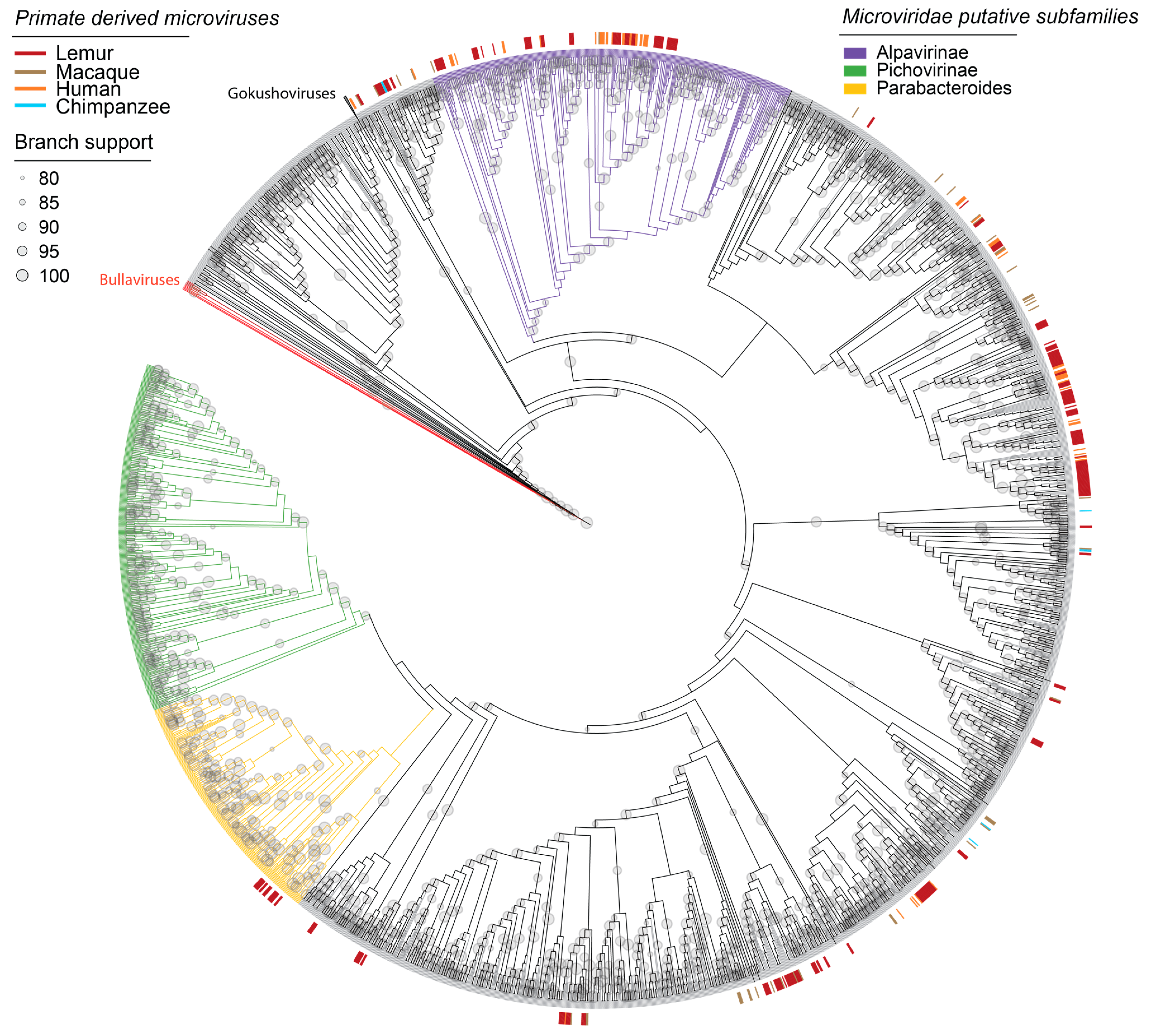
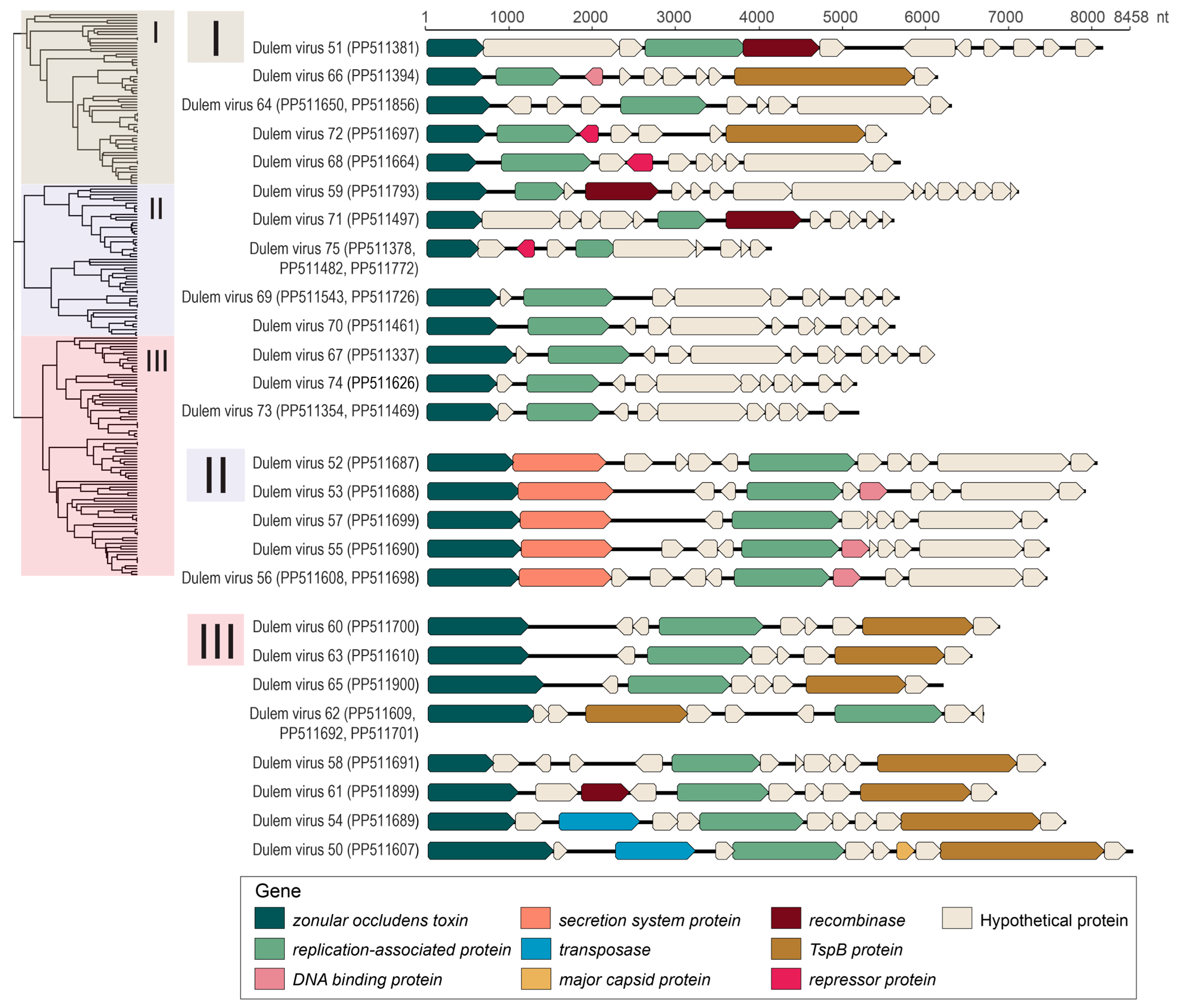
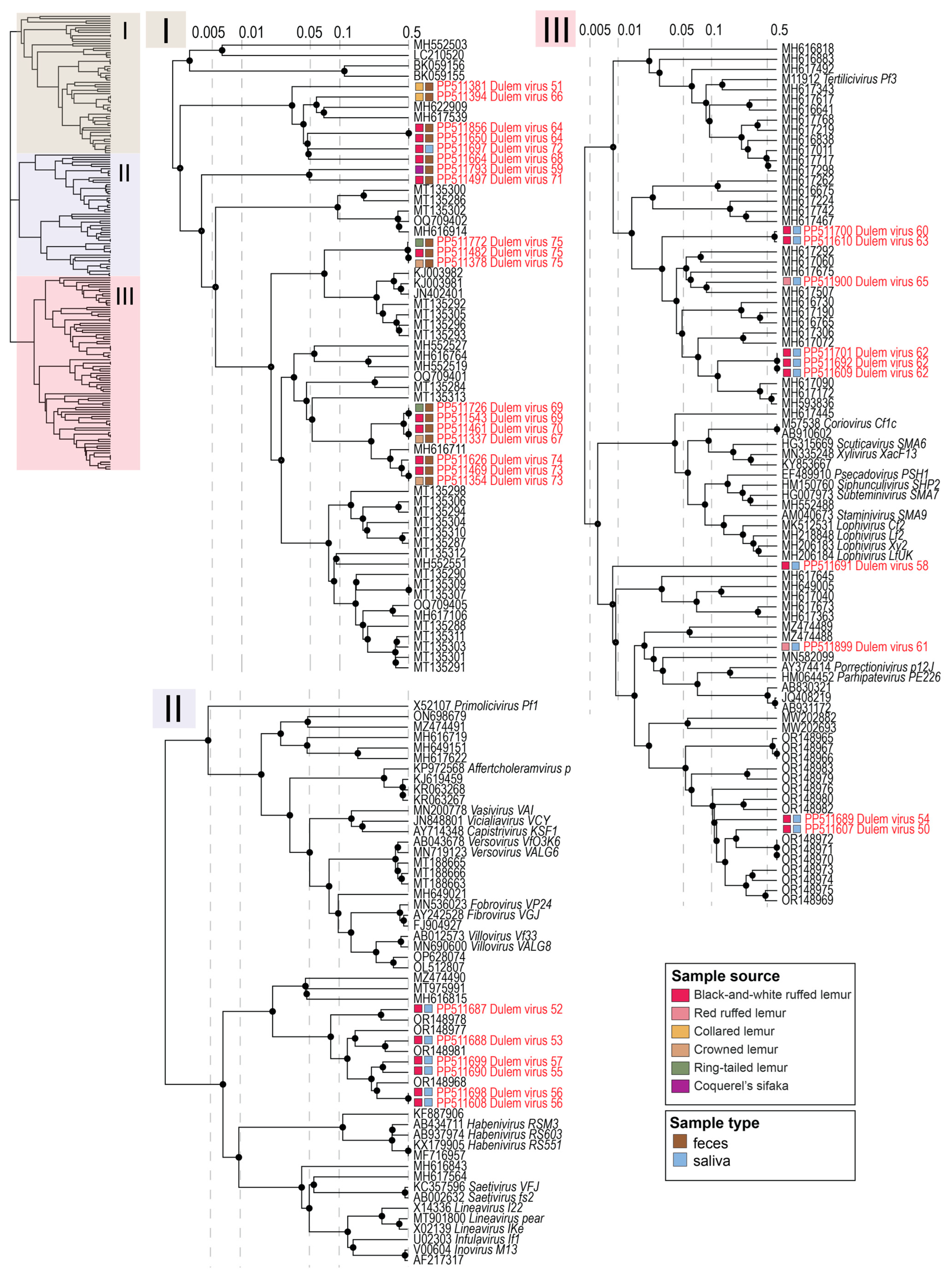
3.3. Microviruses in Fecal and Oral Samples
3.4. Inoviruses in Fecal and Oral Samples
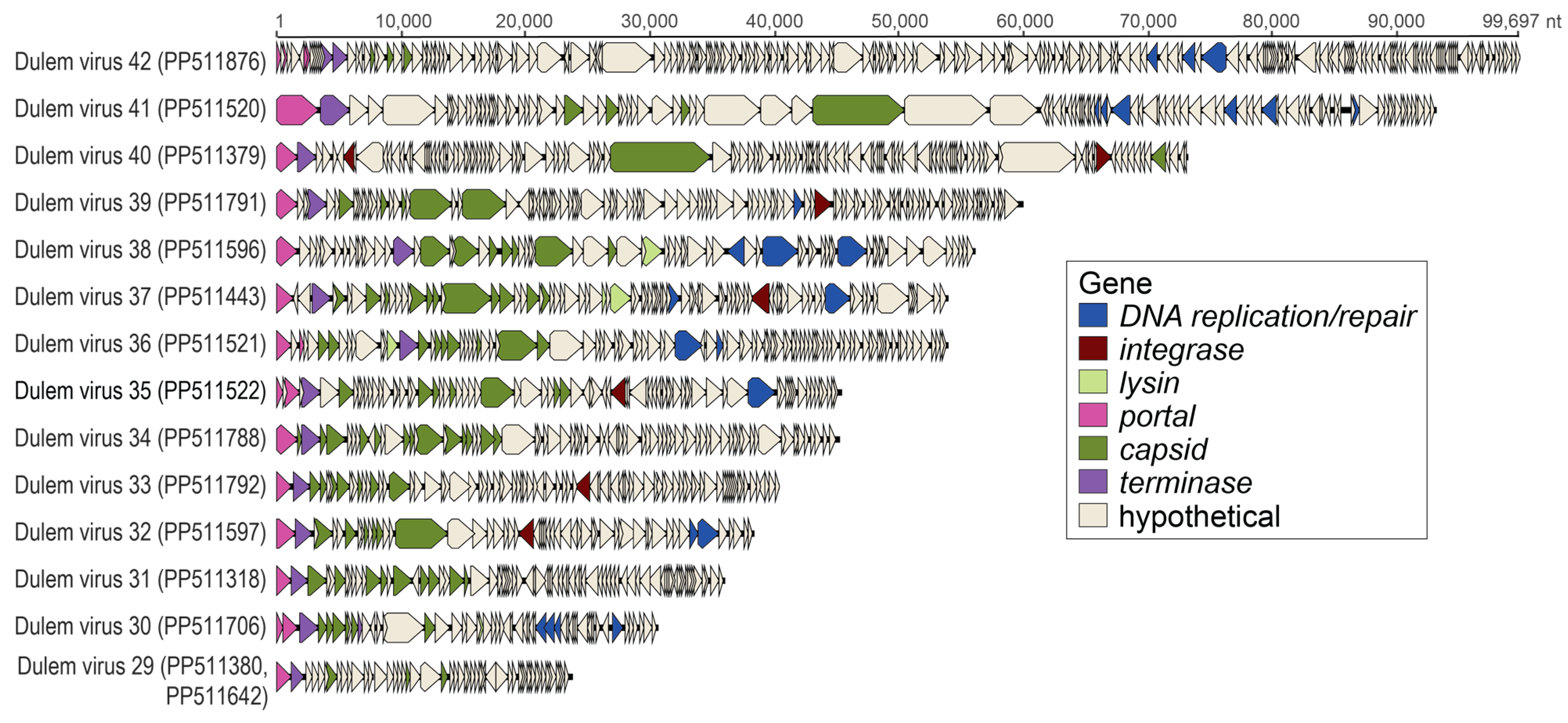
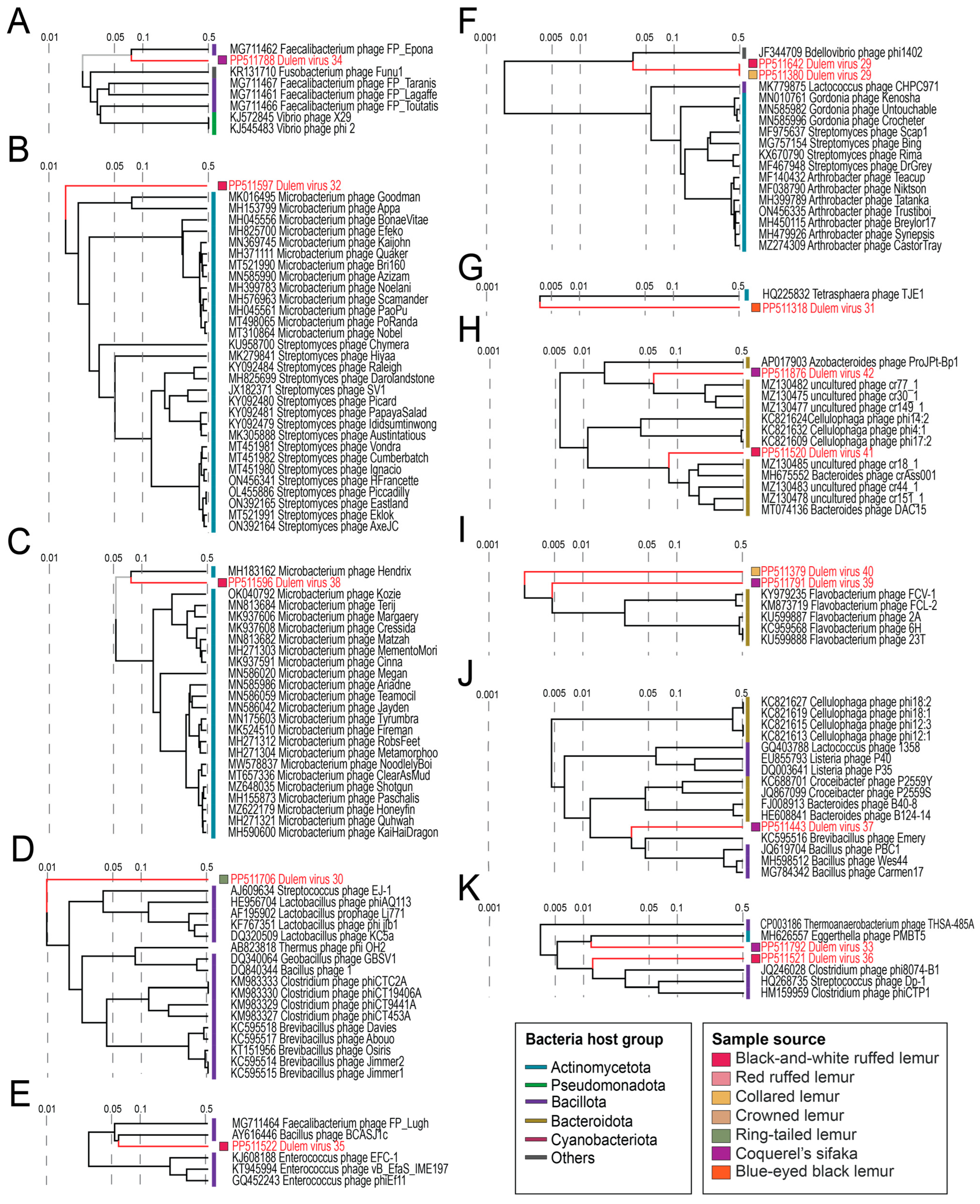
3.5. Caudoviruses in Fecal and Oral Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiao, W.; Ma, Z.S. Inter-Individual Diversity Scaling Analysis of the Human Virome with Classic Diversity-Area Relationship (DAR) Modeling. Front. Genet. 2021, 12, 627128. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Paietta, E.N.; Kraberger, S.; Custer, J.M.; Vargas, K.L.; Espy, C.; Ehmke, E.; Yoder, A.D.; Varsani, A. Characterization of Diverse Anelloviruses, Cressdnaviruses, and Bacteriophages in the Human Oral DNA Virome from North Carolina (USA). Viruses 2023, 15, 1821. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, H.; Feng, L.; Pan, J.; Chen, Z.; Wang, H.; Yang, S.; Shen, Q.; Wang, X.; Shan, T.; et al. Fecal, oral, blood and skin virome of laboratory rabbits. Arch. Virol. 2020, 165, 2847–2856. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Tian, Z.; Yin, C.; Zou, J.; Wu, S.; Luo, Y.; Chen, X.; Dai, Y.; Yang, S.; Li, Y.; et al. The Analysis of Oral and Fecal Virome Detects Multiple Novel Emerging Viruses in Snakes. Transbound. Emerg. Dis. 2023, 2023, 4214812. [Google Scholar] [CrossRef]
- Li, L.; Giannitti, F.; Low, J.; Keyes, C.; Ullmann, L.S.; Deng, X.; Aleman, M.; Pesavento, P.A.; Pusterla, N.; Delwart, E. Exploring the virome of diseased horses. J. Gen. Virol. 2015, 96, 2721–2733. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Droit, L.; Gilbert, M.H.; Schiro, F.R.; Didier, P.J.; Si, X.; Paredes, A.; Handley, S.A.; Virgin, H.W.; Bohm, R.P.; et al. Virome biogeography in the lower gastrointestinal tract of rhesus macaques with chronic diarrhea. Virology 2019, 527, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Liu, B.; Cheng, M.; Dong, J.; Hu, Y.; Jin, Q.; Yang, F. An atlas of the blood virome in healthy individuals. Virus Res. 2023, 323, 199004. [Google Scholar] [CrossRef]
- Rascovan, N.; Duraisamy, R.; Desnues, C. Metagenomics and the Human Virome in Asymptomatic Individuals. Annu. Rev. Microbiol. 2016, 70, 125–141. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Anindita, P.D.; Sasaki, M.; Gonzalez, G.; Phongphaew, W.; Carr, M.; Hang’Ombe, B.M.; Mweene, A.S.; Ito, K.; Orba, Y.; Sawa, H. Discovery and genetic characterization of diverse smacoviruses in Zambian non-human primates. Sci. Rep. 2019, 9, 5045. [Google Scholar] [CrossRef] [PubMed]
- Kapusinszky, B.; Ardeshir, A.; Mulvaney, U.; Deng, X.; Delwart, E. Case-Control Comparison of Enteric Viromes in Captive Rhesus Macaques with Acute or Idiopathic Chronic Diarrhea. J. Virol. 2017, 91, 18. [Google Scholar] [CrossRef]
- Sawaswong, V.; Fahsbender, E.; Altan, E.; Kemthong, T.; Deng, X.; Malaivijitnond, S.; Payungporn, S.; Delwart, E. High Diversity and Novel Enteric Viruses in Fecal Viromes of Healthy Wild and Captive Thai Cynomolgus Macaques (Macaca fascicularis). Viruses 2019, 11, 971. [Google Scholar] [CrossRef] [PubMed]
- D’arc, M.; Furtado, C.; Siqueira, J.D.; Seuánez, H.N.; Ayouba, A.; Peeters, M.; Soares, M.A. Assessment of the gorilla gut virome in association with natural simian immunodeficiency virus infection. Retrovirology 2018, 15, 19. [Google Scholar] [CrossRef]
- Ng, T.F.F.; Zhang, W.; Sachsenröder, J.; Kondov, N.O.; Da Costa, A.C.; Vega, E.; Holtz, L.R.; Wu, G.; Wang, D.; Stine, C.O.; et al. A diverse group of small circular ssDNA viral genomes in human and non-human primate stools. Virus Evol. 2015, 1, vev017. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef] [PubMed]
- Paietta, E.N.; Kraberger, S.; Custer, J.M.; Vargas, K.L.; Van Doorslaer, K.; Yoder, A.D.; Varsani, A. Identification of diverse papillomaviruses in captive black-and-white ruffed lemurs (Varecia variegata). Arch. Virol. 2022, 168, 13. [Google Scholar] [CrossRef]
- Paietta, E.N.; Kraberger, S.; Regney, M.; Custer, J.M.; Ehmke, E.; Yoder, A.D.; Varsani, A. Interspecies Papillomavirus Type Infection and a Novel Papillomavirus Type in Red Ruffed Lemurs (Varecia rubra). Viruses 2024, 16, 37. [Google Scholar] [CrossRef]
- Herrera, J.P. Interactions between plants and primates shape community diversity in a rainforest in Madagascar. J. Anim. Ecol. 2016, 85, 982–993. [Google Scholar] [CrossRef]
- Herrera, J.P. The effects of biogeography and biotic interactions on lemur community assembly. Int. J. Primatol. 2017, 38, 692–716. [Google Scholar] [CrossRef]
- Yoder, A.D. Lemurs. Curr. Biol. 2007, 17, R866–R868. [Google Scholar] [CrossRef] [PubMed]
- IUCN. The IUCN Red List of Threatened Species. Available online: https://www.iucnredlist.org (accessed on 19 September 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Belford, A.K.; Domínguez-Huerta, G.; Bolduc, B.; Buck, C.B. Cenote-Taker 2 democratizes virus discovery and sequence annotation. Virus Evol. 2021, 7, veaa100. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Short-Read Aligner, and Other Bioinformatics Tools. 2015. Available online: http://sourceforge.net/projects/bbmap/ (accessed on 15 August 2016).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, D.; Varsani, A.; Krupovic, M. Pervasive Chimerism in the Replication-Associated Proteins of Uncultured Single-Stranded DNA Viruses. Viruses 2018, 10, 187. [Google Scholar] [CrossRef] [PubMed]
- Zallot, R.; Oberg, N.; Gerlt, J.A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. [Google Scholar] [CrossRef] [PubMed]
- Chrzastek, K.; Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Kulkarni, A.; Chappell, L.; Dufour-Zavala, L.; Kapczynski, D.R.; Varsani, A. Diverse Single-Stranded DNA Viruses Identified in Chicken Buccal Swabs. Microorganisms 2021, 9, 2602. [Google Scholar] [CrossRef] [PubMed]
- Custer, J.M.; White, R.; Taylor, H.; Schmidlin, K.; Fontenele, R.S.; Stainton, D.; Kraberger, S.; Briskie, J.V.; Varsani, A. Diverse single-stranded DNA viruses identified in New Zealand (Aotearoa) South Island robin (Petroica australis) fecal samples. Virology 2022, 565, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Fontenele, R.S.; Lacorte, C.; Lamas, N.S.; Schmidlin, K.; Varsani, A.; Ribeiro, S.G. Single Stranded DNA Viruses Associated with Capybara Faeces Sampled in Brazil. Viruses 2019, 11, 710. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.; Larsen, B.B.; Otto, H.W.; Potticary, A.L.; Kraberger, S.; Custer, J.M.; Suazo, C.; Upham, N.S.; Worobey, M.; Van Doorslaer, K.; et al. Diverse DNA virus genomes identified in fecal samples of Mexican free-tailed bats (Tadarida brasiliensis) captured in Chiricahua Mountains of southeast Arizona (USA). Virology 2023, 580, 98–111. [Google Scholar] [CrossRef]
- Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Walters, M.; Varsani, A. Unravelling the Single-Stranded DNA Virome of the New Zealand Blackfly. Viruses 2019, 11, 532. [Google Scholar] [CrossRef]
- Levy, H.; Fontenele, R.S.; Harding, C.; Suazo, C.; Kraberger, S.; Schmidlin, K.; Djurhuus, A.; Black, C.E.; Hart, T.; Smith, A.L.; et al. Identification and Distribution of Novel Cressdnaviruses and Circular molecules in Four Penguin Species in South Georgia and the Antarctic Peninsula. Viruses 2020, 12, 1029. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.C.; Larsen, B.B.; Rowsey, D.M.; Otto, H.W.; Gryseels, S.; Kraberger, S.; Custer, J.M.; Steger, L.; Yule, K.M.; Harris, R.E.; et al. Using archived and biocollection samples towards deciphering the DNA virus diversity associated with rodent species in the families cricetidae and heteromyidae. Virology 2023, 585, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Orton, J.P.; Morales, M.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Leavitt, D.J.; Webster, T.H.; Wilson, M.A.; Kusumi, K.; Dolby, G.A.; et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses 2020, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef]
- Roux, S.; Camargo, A.P.; Coutinho, F.H.; Dabdoub, S.M.; Dutilh, B.E.; Nayfach, S.; Tritt, A. iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria. PLoS Biol. 2023, 21, e3002083. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Varsani, A.; Opriessnig, T.; Celer, V.; Maggi, F.; Okamoto, H.; Blomström, A.-L.; Cadar, D.; Harrach, B.; Biagini, P.; Kraberger, S. Taxonomic update for mammalian anelloviruses (family Anelloviridae). Arch. Virol. 2021, 166, 2943–2953. [Google Scholar] [CrossRef]
- Amatya, R.; Deem, S.L.; Porton, I.J.; Wang, D.; Lim, E.S. Complete Genome Sequence of Torque teno indri virus 1, a Novel Anellovirus in Blood from a Free-Living Lemur. Genome Announc. 2017, 5, e00698-17. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowska, J.; Deijs, M.; Klein, M.; Bakker, M.; Jebbink, M.F.; Sparreboom, M.; Kinsella, C.M.; Timmerman, A.L.; van der Hoek, L. Diversity and Long-Term Dynamics of Human Blood Anelloviruses. J. Virol. 2022, 96, e00109-22. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.J.; Keeler, E.L.; Bushman, F.D.; Collman, R.G. The enigmatic roles of Anelloviridae and Redondoviridae in humans. Curr. Opin. Virol. 2022, 55, 101248. [Google Scholar] [CrossRef] [PubMed]
- Butkovic, A.; Kraberger, S.; Smeele, Z.; Martin, D.P.; Schmidlin, K.; Fontenele, R.S.; Shero, M.R.; Beltran, R.S.; Kirkham, A.L.; Aleamotu’a, M.; et al. Evolution of anelloviruses from a circovirus-like ancestor through gradual augmentation of the jelly-roll capsid protein. Virus Evol. 2023, 9, vead035. [Google Scholar] [CrossRef]
- Varsani, A.; Kraberger, S.; Opriessnig, T.; Maggi, F.; Celer, V.; Okamoto, H.; Biagini, P. Anelloviridae taxonomy update 2023. Arch. Virol. 2023, 168, 277. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H. TT Viruses in Animals; Springer: Berlin/Heidelberg, Germany, 2009; pp. 35–52. [Google Scholar]
- Bigarré, L.; Beven, V.; De Boisséson, C.; Grasland, B.; Rose, N.; Biagini, P.; Jestin, A. Pig anelloviruses are highly prevalent in swine herds in France. J. Gen. Virol. 2005, 86, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.L.; Kraberger, S.; Fontenele, R.S.; Faleye, T.O.C.; Adams, D.; Adhikari, S.; Sandrolini, H.; Finnerty, S.; Halden, R.U.; Scotch, M.; et al. Genome Sequences of Anelloviruses, Genomovirus, and Papillomavirus Isolated from Nasal Pharyngeal Swabs. Microbiol. Resour. Announc. 2022, 11, e00681-22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, S.; Shan, T.; Hou, R.; Liu, Z.; Li, W.; Guo, L.; Wang, Y.; Chen, P.; Wang, X.; et al. Virome comparisons in wild-diseased and healthy captive giant pandas. Microbiome 2017, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef]
- Hrazdilová, K.; Slaninková, E.; Brožová, K.; Modrý, D.; Vodička, R.; Celer, V. New species of Torque Teno miniviruses infecting gorillas and chimpanzees. Virology 2016, 487, 207–214. [Google Scholar] [CrossRef]
- Kapusinszky, B.; Mulvaney, U.; Jasinska, A.J.; Deng, X.; Freimer, N.; Delwart, E. Local Virus Extinctions following a Host Population Bottleneck. J. Virol. 2015, 89, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Welch, N.; Belford, A.K.; Varsani, A.; Pastrana, D.V.; Tisza, M.J.; Starrett, G.J. Widespread Horizontal Gene Transfer among Animal Viruses. bioRxiv 2024. [Google Scholar] [CrossRef]
- Breitbart, M.; Delwart, E.; Rosario, K.; Segalés, J.; Varsani, A.; ICTV Report Consortium. ICTV virus taxonomy profile: Circoviridae. J. Gen. Virol. 2017, 98, 1997–1998. [Google Scholar] [CrossRef] [PubMed]
- Fiallo-Olivé, E.; Lett, J.-M.; Martin, D.P.; Roumagnac, P.; Varsani, A.; Zerbini, F.M.; Navas-Castillo, J. ICTV Virus Taxonomy Profile: Geminiviridae 2021. J. Gen. Virol. 2021, 102, 001696. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.E.; Gronenborn, B.; Harding, R.M.; Mandal, B.; Grigoras, I.; Randles, J.W.; Sano, Y.; Timchenko, T.; Vetten, H.J.; Yeh, H.H.; et al. ICTV Virus Taxonomy Profile: Nanoviridae. J. Gen. Virol. 2021, 102, 001544. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.P.H.; De Resende, F.M.P.; Da Silva, J.C.F.; De Breuil, S.; Nome, C.; Bejerman, N.; Zerbini, F.M. Amesuviridae: A new family of plant-infecting viruses in the phylum Cressdnaviricota, realm Monodnaviria. Arch. Virol. 2023, 168, 223. [Google Scholar] [CrossRef] [PubMed]
- Gronenborn, B.; Randles, J.W.; Knierim, D.; Barrière, Q.; Vetten, H.J.; Warthmann, N.; Cornu, D.; Sileye, T.; Winter, S.; Timchenko, T. Analysis of DNAs associated with coconut foliar decay disease implicates a unique single-stranded DNA virus representing a new taxon. Sci. Rep. 2018, 8, 5698. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Family Genomoviridae: 2021 taxonomy update. Arch. Virol. 2021, 166, 2911–2926. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A. Naryaviridae, Nenyaviridae, and Vilyaviridae: Three new families of single-stranded DNA viruses in the phylum Cressdnaviricota. Arch. Virol. 2022, 167, 2907–2921. [Google Scholar] [CrossRef]
- Keeler, E.L.; Merenstein, C.; Reddy, S.; Taylor, L.J.; Cobián-Güemes, A.G.; Zankharia, U.; Collman, R.G.; Bushman, F.D. Widespread, human-associated redondoviruses infect the commensal protozoan Entamoeba gingivalis. Cell Host Microbe 2023, 31, 58–68.e5. [Google Scholar] [CrossRef]
- Díez-Villaseñor, C.; Rodriguez-Valera, F. CRISPR analysis suggests that small circular single-stranded DNA smacoviruses infect Archaea instead of humans. Nat. Commun. 2019, 10, 294. [Google Scholar] [CrossRef]
- Ilyina, T.V.; Koonin, E.V. Conserved sequence motifs in the initiator proteins for rolling circle DNA replication encoded by diverse replicons from eubacteria, eucaryotes and archaebacteria. Nucleic Acids Res. 1992, 20, 3279–3285. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Koonin, E.V.; Wolf, Y.I. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 1990, 262, 145–148. [Google Scholar] [CrossRef]
- Nash, T.E.; Dallas, M.B.; Reyes, M.I.; Buhrman, G.K.; Ascencio-Ibañez, J.T.; Hanley-Bowdoin, L. Functional Analysis of a Novel Motif Conserved across Geminivirus Rep Proteins. J. Virol. 2011, 85, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Varsani, A. A 2021 taxonomy update for the family Smacoviridae. Arch. Virol. 2021, 166, 3245–3253. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, S.; Borrel, G.; Krupovic, M.; Gribaldo, S. A compendium of viruses from methanogenic archaea reveals their diversity and adaptations to the gut environment. Nat. Microbiol. 2023, 8, 2170–2182. [Google Scholar] [CrossRef]
- Villanova, F.; Milagres, F.A.D.P.; Brustulin, R.; Araújo, E.L.L.; Pandey, R.P.; Raj, V.S.; Deng, X.; Delwart, E.; Luchs, A.; Costa, A.C.D.; et al. A New Circular Single-Stranded DNA Virus Related with Howler Monkey Associated Porprismacovirus 1 Detected in Children with Acute Gastroenteritis. Viruses 2022, 14, 1472. [Google Scholar] [CrossRef]
- Yinda, C.K.; Vanhulle, E.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Shi, C.; Ghogomu, S.M.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Gut Virome Analysis of Cameroonians Reveals High Diversity of Enteric Viruses, Including Potential Interspecies Transmitted Viruses. mSphere 2019, 4, e00585-18. [Google Scholar] [CrossRef]
- Britt, A. Diet and Feeding Behaviour of the Black-and-White Ruffed Lemur (Varecia variegata variegata) in the Betampona Reserve, Eastern Madagascar. Folia Primatol. 2000, 71, 133–141. [Google Scholar] [CrossRef]
- FJ, W. Social organization, feeding ecology, and reproductive strategy of ruffed lemurs, Varecia variegata. In Primatology Today; Elsevier: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Ganzhorn, J.U. Soil Consumption of Two Groups of Semi-free-ranging Lemurs {Lemur catta and Lemur fulvus). Ethology 1987, 74, 146–154. [Google Scholar] [CrossRef]
- Setz, E.; Enzweiller, J. Geophagy by golden faced sakis (Pithecia pithecia chrysocephala) in central Amazon. In Proceedings of the XIVth Congress of the International Primatological Society, Strasbourg, France, 16–21 August 1992. [Google Scholar]
- Amoroso, C.R.; Frink, A.G.; Nunn, C.L. Water choice as a counterstrategy to faecally transmitted disease: An experimental study in captive lemurs. Behaviour 2017, 154, 1239–1258. [Google Scholar] [CrossRef]
- Hao, F.; Wu, M.; Li, G. Characterization of a novel genomovirus in the phytopathogenic fungus Botrytis cinerea. Virology 2021, 553, 111–116. [Google Scholar] [CrossRef]
- Chabi-Jesus, C.; Najar, A.; Fontenele, R.S.; Kumari, S.G.; Ramos-González, P.L.; Freitas-Astúa, J.; Kraberger, S.; Varsani, A. Viruses representing two new genomovirus species identified in citrus from Tunisia. Arch. Virol. 2020, 165, 1225–1229. [Google Scholar] [CrossRef]
- Vieira, A.C.; Lopes, Í.S.; Fonseca, P.L.C.; Olmo, R.P.; Bittencourt, F.; de Vasconcelos, L.M.; Pirovani, C.P.; Gaiotto, F.A.; Aguiar, E.R.G.R. Expanding the environmental virome: Infection profile in a native rainforest tree species. Front. Microbiol. 2022, 13, 874319. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, L.d.N.A.; Boiteux, L.S.; de Noronha Fonseca, M.E.; Batista, J.G.; Nery, F.M.B.; de Cássia Pereira–Carvalho, R. Complete genomic sequence of an isolate of plant-associated genomovirus 12 (genus Gemycircularvirus) from open–field tomatoes in Brazil. J. Plant Pathol. 2022, 104, 1129–1134. [Google Scholar] [CrossRef]
- Jimoh, A.O.; Balle, C.; Brown, B.; Feng, C.; Havyarimana, E.; Konstantinus, I.N.; Gill, K.; Bekker, L.-G.; Passmore, J.-A.S.; Jaspan, H.B. Genome sequences of anelloviruses, a genomovirus, microviruses, polyomaviruses, and an unclassified caudovirus identified in vaginal secretions from South African adolescents. Microbiol. Resour. Announc. 2023, 12, e01143-22. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.N.; Campos, F.S.; Finoketti, F.; Dos Santos, A.C.; Campos, A.A.S.; Wagner, P.G.C.; Roehe, P.M.; De Carvalho Ruthner Batista, H.B.; Franco, A.C. Viral diversity in oral cavity from Sapajus nigritus by metagenomic analyses. Braz. J. Microbiol. 2020, 51, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Negrey, J.D.; Emery Thompson, M.; Dunn, C.D.; Otali, E.; Wrangham, R.W.; Mitani, J.C.; Machanda, Z.P.; Muller, M.N.; Langergraber, K.E.; Goldberg, T.L. Female reproduction and viral infection in a long-lived mammal. J. Anim. Ecol. 2022, 91, 1999–2009. [Google Scholar] [CrossRef]
- Bolatti, E.M.; Zorec, T.M.; Montani, M.E.; Hošnjak, L.; Chouhy, D.; Viarengo, G.; Casal, P.E.; Barquez, R.M.; Poljak, M.; Giri, A.A. A preliminary study of the virome of the South American free-tailed bats (Tadarida brasiliensis) and identification of two novel mammalian viruses. Viruses 2020, 12, 422. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.A.; Alves, R.S.; Do Canto Olegário, J.; De Camargo, L.J.; Weber, M.N.; Da Silva, M.S.; Canova, R.; Mosena, A.C.S.; Cibulski, S.P.; Varela, A.P.M.; et al. The virome of the white-winged vampire bat Diaemus youngi is rich in circular DNA viruses. Virus Genes 2022, 58, 214–226. [Google Scholar] [CrossRef]
- Yang, S.; Shan, T.; Xiao, Y.; Zhang, H.; Wang, X.; Shen, Q.; Wang, Y.; Yao, Y.; Liu, Q.; Wang, H.; et al. Digging metagenomic data of pangolins revealed SARS-CoV-2 related viruses and other significant viruses. J. Med. Virol. 2021, 93, 1786–1791. [Google Scholar] [CrossRef]
- Smith, K.; Fielding, R.; Schiavone, K.; Hall, K.R.; Reid, V.S.; Boyea, D.; Smith, E.L.; Schmidlin, K.; Fontenele, R.S.; Kraberger, S.; et al. Circular DNA viruses identified in short-finned pilot whale and orca tissue samples. Virology 2021, 559, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Cook, C.N.; Schmidlin, K.; Fontenele, R.S.; Bautista, J.; Smith, B.; Varsani, A. Diverse single-stranded DNA viruses associated with honey bees (Apis mellifera). Infect. Genet. Evol. 2019, 71, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, B.; Fu, Y.; Jiang, D.; Ghabrial, S.A.; Li, G.; Peng, Y.; Xie, J.; Cheng, J.; Huang, J.; et al. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Natl. Acad. Sci. USA 2010, 107, 8387–8392. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, S.; Zhang, L.; Qiu, D.; Zhou, X.; Guo, L. A tripartite ssDNA mycovirus from a plant pathogenic fungus is infectious as cloned DNA and purified virions. Sci. Adv. 2020, 6, eaay9634. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Dayaram, A.; Marinov, M.; Ware, J.; Kraberger, S.; Stainton, D.; Breitbart, M.; Varsani, A. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 2012, 93, 2668–2681. [Google Scholar] [CrossRef]
- Kraberger, S.; Polston, J.E.; Capobianco, H.M.; Alcalá-Briseño, R.I.; Fontenele, R.S.; Varsani, A. Genomovirus Genomes Recovered from Echinothrips americanus Sampled in Florida, USA. Genome Announc. 2017, 5, e00445-17. [Google Scholar] [CrossRef]
- Fontenele, R.S.; Roumagnac, P.; Richet, C.; Kraberger, S.; Stainton, D.; Aleamotu’a, M.; Filloux, D.; Bernardo, P.; Harkins, G.W.; McCarthy, J.; et al. Diverse genomoviruses representing twenty-nine species identified associated with plants. Arch. Virol. 2020, 165, 2891–2901. [Google Scholar] [CrossRef]
- Lima, D.A.; Cibulski, S.P.; Finkler, F.; Teixeira, T.F.; Varela, A.P.M.; Cerva, C.; Loiko, M.R.; Scheffer, C.M.; Dos Santos, H.F.; Mayer, F.Q.; et al. Faecal virome of healthy chickens reveals a large diversity of the eukaryote viral community, including novel circular ssDNA viruses. J. Gen. Virol. 2017, 98, 690–703. [Google Scholar] [CrossRef]
- Victoria, J.G.; Kapoor, A.; Li, L.; Blinkova, O.; Slikas, B.; Wang, C.; Naeem, A.; Zaidi, S.; Delwart, E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J. Virol. 2009, 83, 4642–4651. [Google Scholar] [CrossRef]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.Q.; Wei, C.L.; Soh, S.W.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y. RNA viral community in human feces: Prevalence of plant pathogenic viruses. PLoS Biol. 2006, 4, e3. [Google Scholar] [CrossRef] [PubMed]
- Blinkova, O.; Victoria, J.; Li, Y.; Keele, B.F.; Sanz, C.; Ndjango, J.B.; Peeters, M.; Travis, D.; Lonsdorf, E.V.; Wilson, M.L.; et al. Novel circular DNA viruses in stool samples of wild-living chimpanzees. J. Gen. Virol. 2010, 91, 74–86. [Google Scholar] [CrossRef]
- Esposito, A.M.; Esposito, M.M.; Ptashnik, A. Phylogenetic Diversity of Animal Oral and Gastrointestinal Viromes Useful in Surveillance of Zoonoses. Microorganisms 2022, 10, 1815. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tao, J.; Li, B.; Shen, X.; Cheng, J.; Liu, H. The Gut Viral Metagenome Analysis of Domestic Dogs Captures Snapshot of Viral Diversity and Potential Risk of Coronavirus. Front. Vet. Sci. 2021, 8, 695088. [Google Scholar] [CrossRef]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The Fecal Viral Flora of Wild Rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat Guano Virome: Predominance of Dietary Viruses from Insects and Plants plus Novel Mammalian Viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef]
- Briddon, R.W. Geminiviridae. In Encyclopedia of Life Sciences; Wiley: Hoboken, NJ, USA, 2009; pp. 1–12. [Google Scholar]
- Montazeri, R.; Shams-Bakhsh, M.; Mahmoudi, S.B.; Rajabi, A. Evaluation of sugar beet lines for resistance to beet curly top viruses. Euphytica 2016, 210, 31–40. [Google Scholar] [CrossRef]
- Kinsella, C.M.; Bart, A.; Deijs, M.; Broekhuizen, P.; Kaczorowska, J.; Jebbink, M.F.; van Gool, T.; Cotten, M.; van der Hoek, L. Entamoeba and Giardia parasites implicated as hosts of CRESS viruses. Nat. Commun. 2020, 11, 4620. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Lan, X.; Dan, J.; Ren, Z.; Cao, S.; Shen, L.; Deng, J.; Zuo, Z.; Yu, S.; et al. Occurrence and multilocus genotyping of Giardia duodenalis in captive non-human primates from 12 zoos in China. PLoS ONE 2020, 15, e0228673. [Google Scholar] [CrossRef]
- Fomsgaard, A.; Worsøe Rosenstierne, M.; Rasmussen, M.; Wright, P.; Bueno, G.; Noromalala, E.; Bornbusch, S.; Stensvold, C.R.; Thomas-Poulsen, M.; Hvilsom, C. Prevalence, infection intensity and genotyping of Giardia duodenalis in ring-tailed lemurs Lemur catta from European zoos and wild populations. J. Zoo Aquar. Res. 2020, 8, 253–258. [Google Scholar] [CrossRef]
- Villers, L.M.; Jang, S.S.; Lent, C.L.; Lewin-Koh, S.C.; Norosoarinaivo, J.A. Survey and comparison of major intestinal flora in captive and wild ring-tailed lemur (Lemur catta) populations. Am. J. Primatol. 2008, 70, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Dixon, B.R. Giardia duodenalis in humans and animals—Transmission and disease. Res. Vet. Sci. 2021, 135, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Hamlen, H.J.; Lawrence, J.M. Giardiasis in laboratory-housed squirrel monkeys: A retrospective study. Lab. Anim. Sci. 1994, 44, 235–239. [Google Scholar] [PubMed]
- Kalishman, J.; Paul-Murphy, J.; Scheffler, J.; Thomson, J.A. Survey of Cryptosporidium and Giardia spp. in a captive population of common marmosets. Lab. Anim. Sci. 1996, 46, 116–119. [Google Scholar] [PubMed]
- Feng, Y.; Xiao, L. Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin. Microbiol. Rev. 2011, 24, 110–140. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, C.M.; Deijs, M.; Becker, C.; Broekhuizen, P.; van Gool, T.; Bart, A.; Schaefer, A.S.; van der Hoek, L. Host prediction for disease-associated gastrointestinal cressdnaviruses. Virus Evol. 2022, 8, veac087. [Google Scholar] [CrossRef]
- Abe, N.; Nagoshi, M.; Takami, K.; Sawano, Y.; Yoshikawa, H. A survey of Blastocystis sp. in livestock, pets, and zoo animals in Japan. Vet. Parasitol. 2002, 106, 203–212. [Google Scholar] [CrossRef]
- Hess, S.C.; Weiss, K.C.B.; Custer, J.M.; Lewis, J.S.; Kraberger, S.; Varsani, A. Identification of small circular DNA viruses in coyote fecal samples from Arizona (USA). Arch. Virol. 2023, 169, 12. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Yang, S.; Wang, H.; Wang, H.; Zhang, J.; Gong, G.; Xiao, Y.; Yang, J.; Wang, X.; Lu, J.; et al. Virome in the cloaca of wild and breeding birds revealed a diversity of significant viruses. Microbiome 2022, 10, 60. [Google Scholar] [CrossRef]
- Pearson, V.M.; Caudle, S.B.; Rokyta, D.R. Viral recombination blurs taxonomic lines: Examination of single-stranded DNA viruses in a wastewater treatment plant. PeerJ 2016, 4, e2585. [Google Scholar] [CrossRef]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H.; et al. Discovery of several thousand highly diverse circular DNA viruses. eLife 2020, 9, e51971. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Mao, Q.; Wang, Y.; He, J.; Yang, J.; Chen, X.; Xiao, Y.; He, Y.; Zhao, M.; Lu, J.; et al. Expanding known viral diversity in plants: Virome of 161 species alongside an ancient canal. Environ. Microbiome 2022, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Male, M.F.; Kraberger, S.; Stainton, D.; Kami, V.; Varsani, A. Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infect. Genet. Evol. 2016, 39, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Olivo, D.; Khalifeh, A.; Custer, J.M.; Kraberger, S.; Varsani, A. Diverse Small Circular DNA Viruses Identified in an American Wigeon Fecal Sample. Microorganisms 2024, 12, 196. [Google Scholar] [CrossRef] [PubMed]
- Trubl, G.; Roux, S.; Borton, M.A.; Varsani, A.; Li, Y.-F.; Sun, C.; Jang, H.B.; Woodcroft, B.J.; Tyson, G.W.; Wrighton, K.C.; et al. Population ecology and potential biogeochemical impacts of ssDNA and dsDNA soil viruses along a permafrost thaw gradient. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zhang, Y.; Sharma, S.; Tom, L.; Liao, Y.-T.; Wu, V.C.H. Gut Phageome—An Insight into the Role and Impact of Gut Microbiome and Their Correlation with Mammal Health and Diseases. Microorganisms 2023, 11, 2454. [Google Scholar] [CrossRef] [PubMed]
- Townsend, E.M.; Kelly, L.; Muscatt, G.; Box, J.D.; Hargraves, N.; Lilley, D.; Jameson, E. The human gut phageome: Origins and roles in the human gut microbiome. Front. Cell. Infect. Microbiol. 2021, 11, 643214. [Google Scholar] [CrossRef] [PubMed]
- Gogarten, J.F.; Rühlemann, M.; Archie, E.; Tung, J.; Akoua-Koffi, C.; Bang, C.; Deschner, T.; Muyembe-Tamfun, J.-J.; Robbins, M.M.; Schubert, G.; et al. Primate phageomes are structured by superhost phylogeny and environment. Proc. Natl. Acad. Sci. USA 2021, 118, e2013535118. [Google Scholar] [CrossRef] [PubMed]
- Kirchberger, P.C.; Martinez, Z.A.; Ochman, H. Organizing the Global Diversity of Microviruses. mBio 2022, 13, e00588-22. [Google Scholar] [CrossRef]
- Uchiyama, A.; Chen, M.; Fane, B.A. Characterization and function of putative substrate specificity domain in microvirus external scaffolding proteins. J. Virol. 2007, 81, 8587–8592. [Google Scholar] [CrossRef]
- Doore, S.M.; Fane, B.A. The microviridae: Diversity, assembly, and experimental evolution. Virology 2016, 491, 45–55. [Google Scholar] [CrossRef]
- Wang, H.; Ling, Y.; Shan, T.; Yang, S.; Xu, H.; Deng, X.; Delwart, E.; Zhang, W. Gut virome of mammals and birds reveals high genetic diversity of the family Microviridae. Virus Evol. 2019, 5, vez013. [Google Scholar] [CrossRef] [PubMed]
- Penner, J.C.; Ferreira, J.A.; Secor, P.R.; Sweere, J.M.; Birukova, M.K.; Joubert, L.-M.; Haagensen, J.A.; Garcia, O.; Malkovskiy, A.V.; Kaber, G. Pf4 bacteriophage produced by Pseudomonas aeruginosa inhibits Aspergillus fumigatus metabolism via iron sequestration. Microbiology 2016, 162, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Nazik, H.; Joubert, L.-M.; Secor, P.R.; Sweere, J.M.; Bollyky, P.L.; Sass, G.; Cegelski, L.; Stevens, D.A. Pseudomonas phage inhibition of Candida albicans. Microbiology 2017, 163, 1568–1577. [Google Scholar] [CrossRef]
- Knezevic, P.; Adriaenssens, E.M.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Inoviridae. J. Gen. Virol. 2021, 102, 001614. [Google Scholar] [CrossRef]
- Ilyina, T.S. Filamentous bacteriophages and their role in the virulence and evolution of pathogenic bacteria. Mol. Genet. Microbiol. Virol. 2015, 30, 1–9. [Google Scholar] [CrossRef]
- Li, H.; Li, H.; Wang, J.; Guo, L.; Fan, H.; Zheng, H.; Yang, Z.; Huang, X.; Chu, M.; Yang, F.; et al. The altered gut virome community in rhesus monkeys is correlated with the gut bacterial microbiome and associated metabolites. Virol. J. 2019, 16, 105. [Google Scholar] [CrossRef]
- Greene, L.K.; Andriambeloson, J.-B.; Blanco, M.B.; Ehmke, E.E. Forest access restores foraging and ranging behavior in captive sifakas. Zoo Biol. 2023, 42, 209–222. [Google Scholar] [CrossRef]
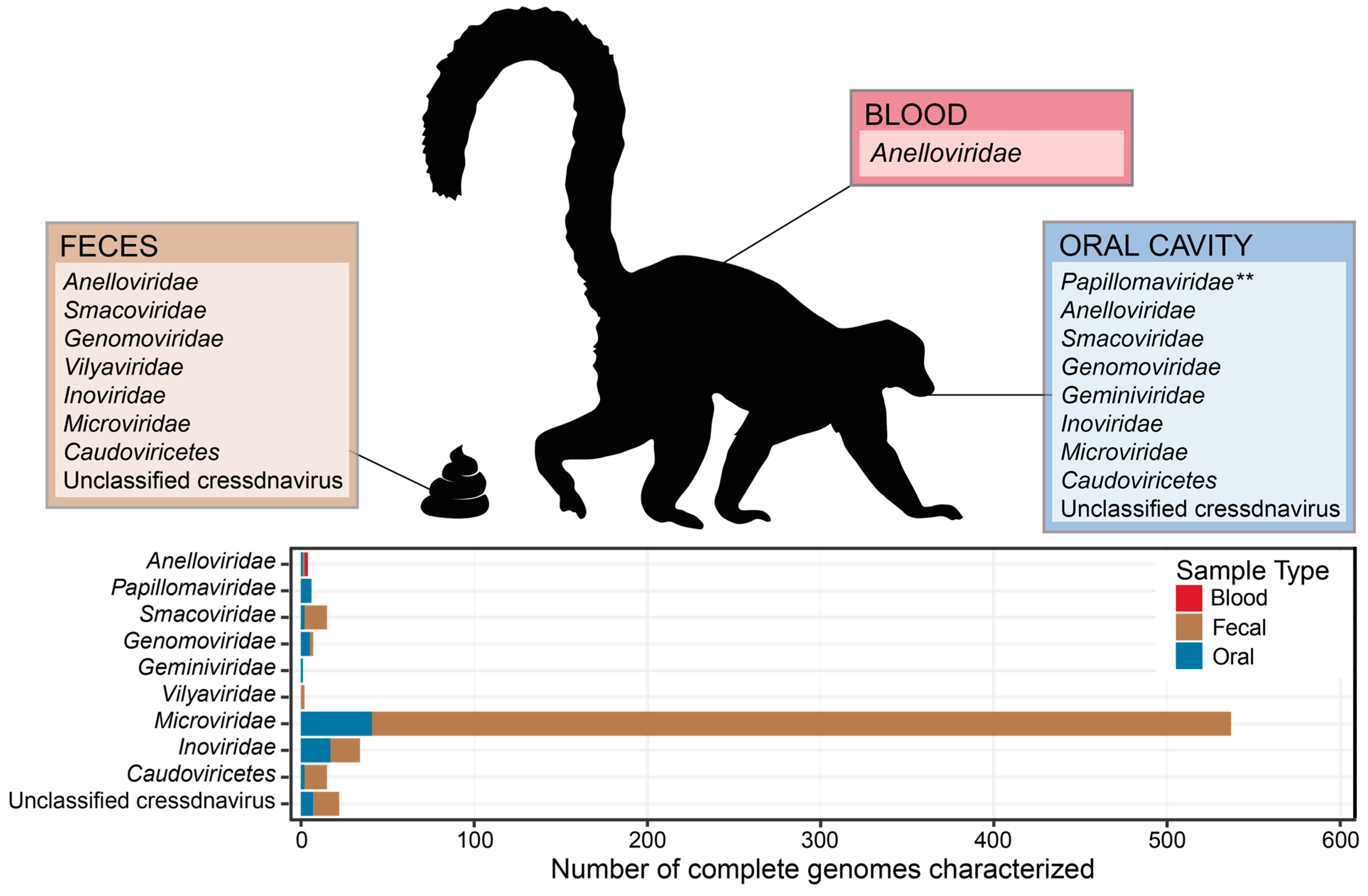
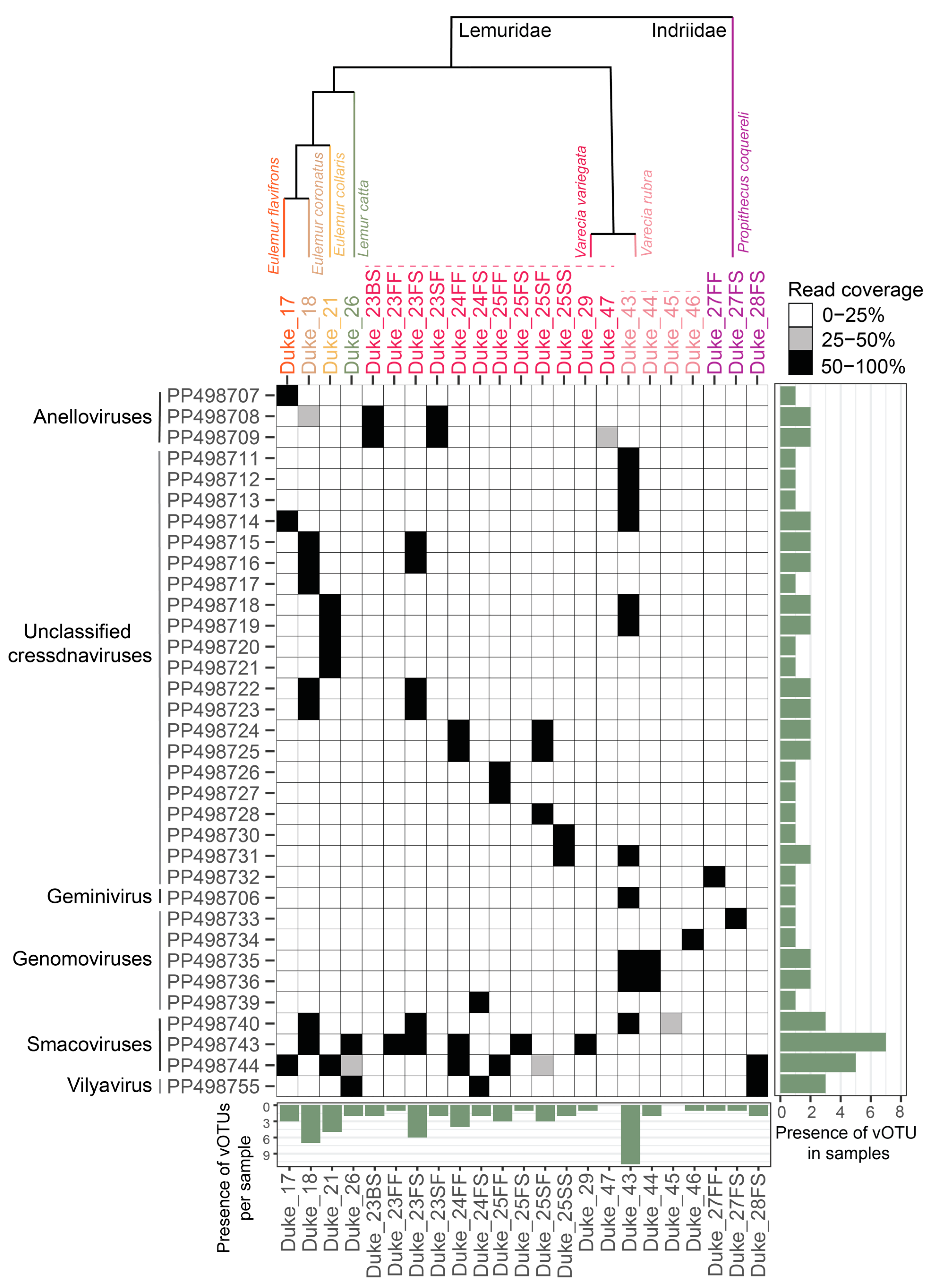

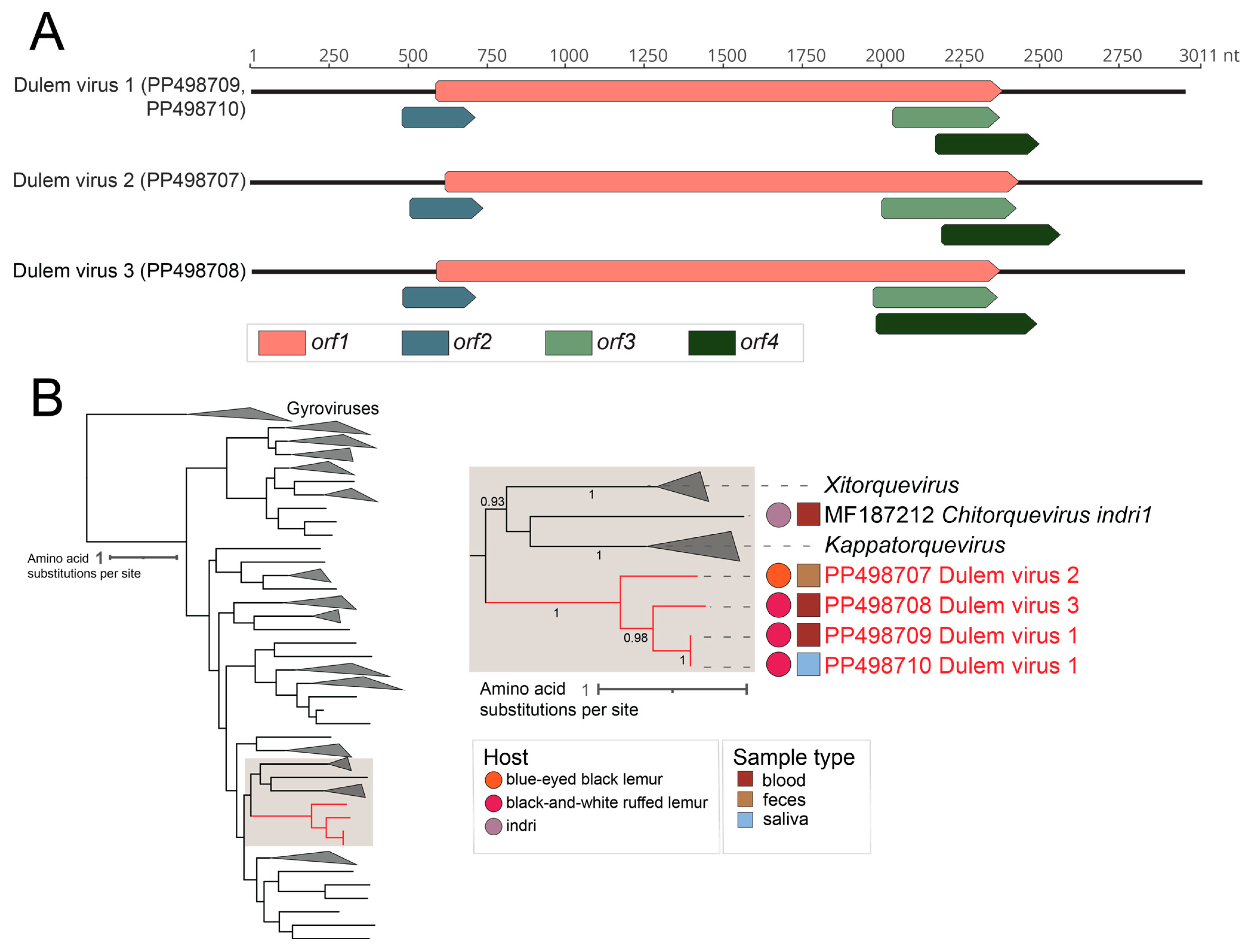


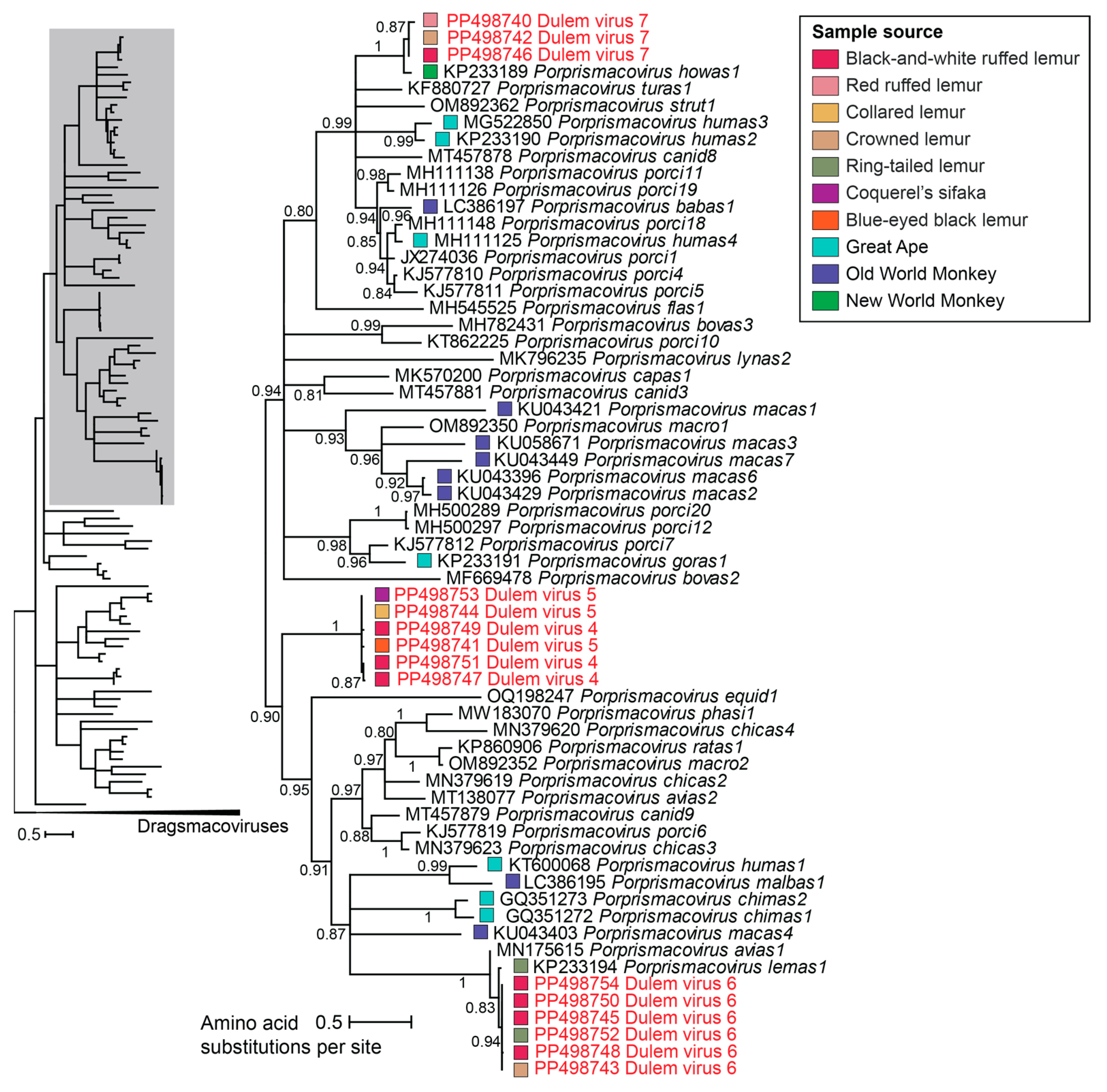


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paietta, E.N.; Kraberger, S.; Lund, M.C.; Vargas, K.L.; Custer, J.M.; Ehmke, E.; Yoder, A.D.; Varsani, A. Diverse Circular DNA Viral Communities in Blood, Oral, and Fecal Samples of Captive Lemurs. Viruses 2024, 16, 1099. https://doi.org/10.3390/v16071099
Paietta EN, Kraberger S, Lund MC, Vargas KL, Custer JM, Ehmke E, Yoder AD, Varsani A. Diverse Circular DNA Viral Communities in Blood, Oral, and Fecal Samples of Captive Lemurs. Viruses. 2024; 16(7):1099. https://doi.org/10.3390/v16071099
Chicago/Turabian StylePaietta, Elise N., Simona Kraberger, Michael C. Lund, Karla L. Vargas, Joy M. Custer, Erin Ehmke, Anne D. Yoder, and Arvind Varsani. 2024. "Diverse Circular DNA Viral Communities in Blood, Oral, and Fecal Samples of Captive Lemurs" Viruses 16, no. 7: 1099. https://doi.org/10.3390/v16071099
APA StylePaietta, E. N., Kraberger, S., Lund, M. C., Vargas, K. L., Custer, J. M., Ehmke, E., Yoder, A. D., & Varsani, A. (2024). Diverse Circular DNA Viral Communities in Blood, Oral, and Fecal Samples of Captive Lemurs. Viruses, 16(7), 1099. https://doi.org/10.3390/v16071099