
Characterization of Six Ampeloviruses Infecting Pineapple in Reunion Island Using a Combination of High-Throughput Sequencing Approaches
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RNA Extraction
2.3. High-Throughput Sequencing
2.4. Data Analysis and Assembly of Viral Genomes
2.5. Detection of PMWaVs by RT-PCR
2.6. RACE PCR
2.7. Search for Recombination and Phylogenetic Analyses
3. Results
3.1. Identification of Known and Novel Ampeloviruses
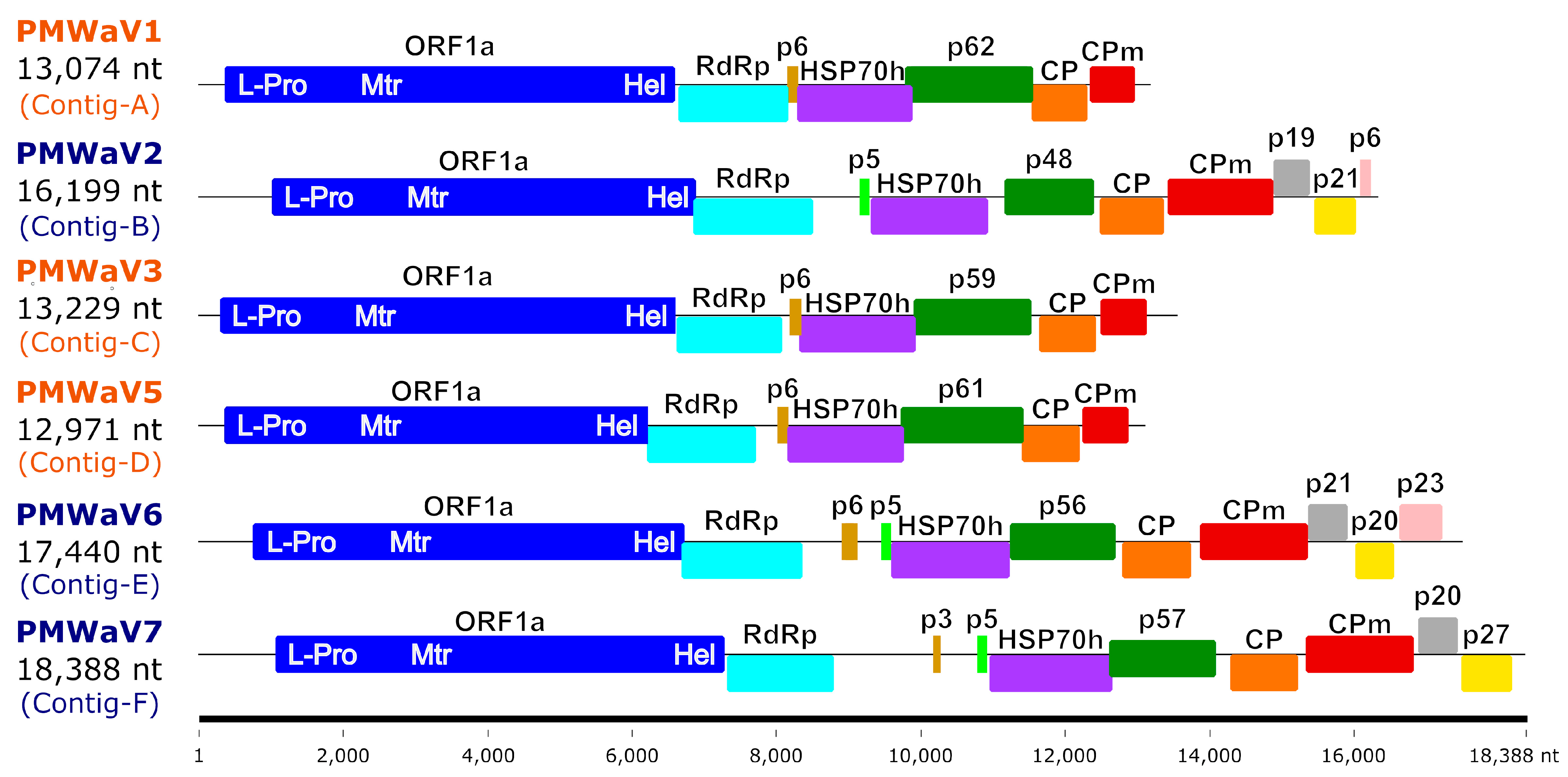
3.2. Species Identification and Genome Organization
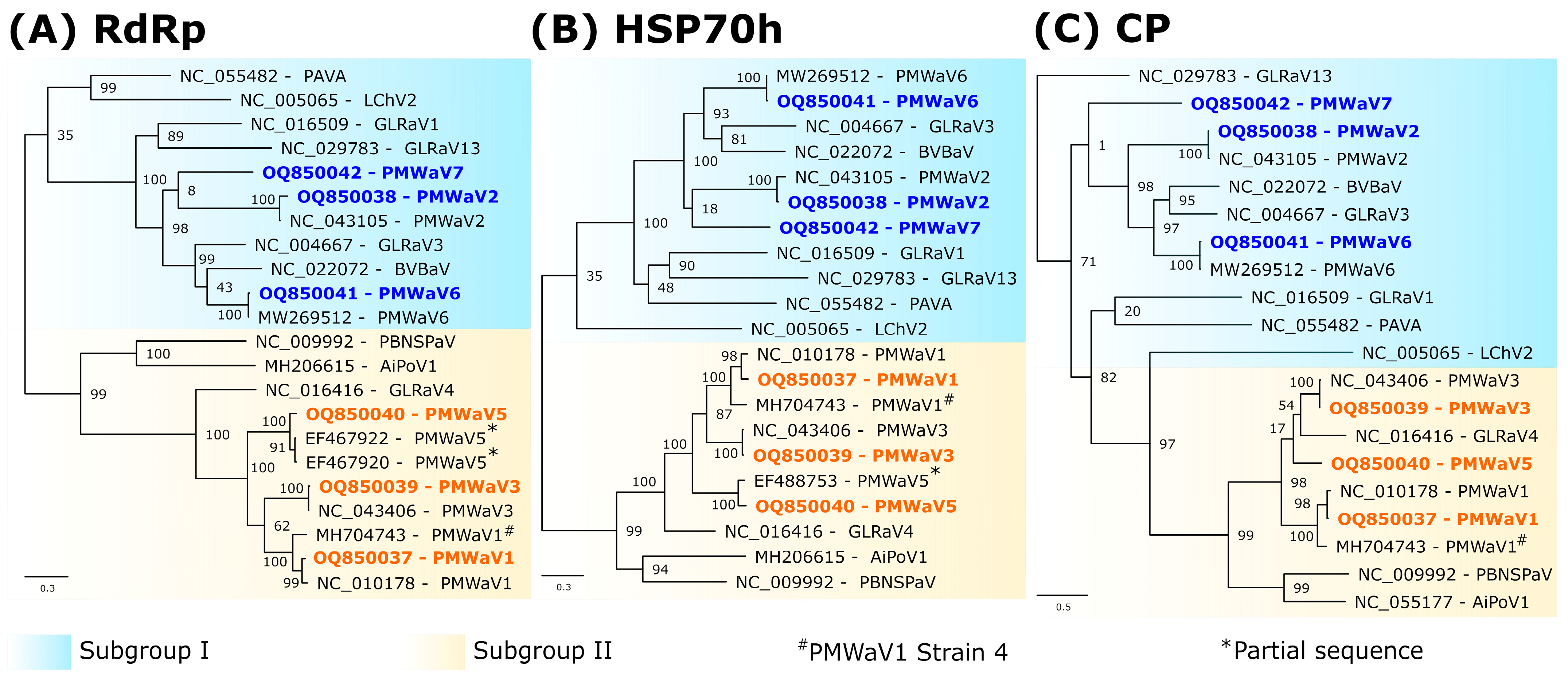
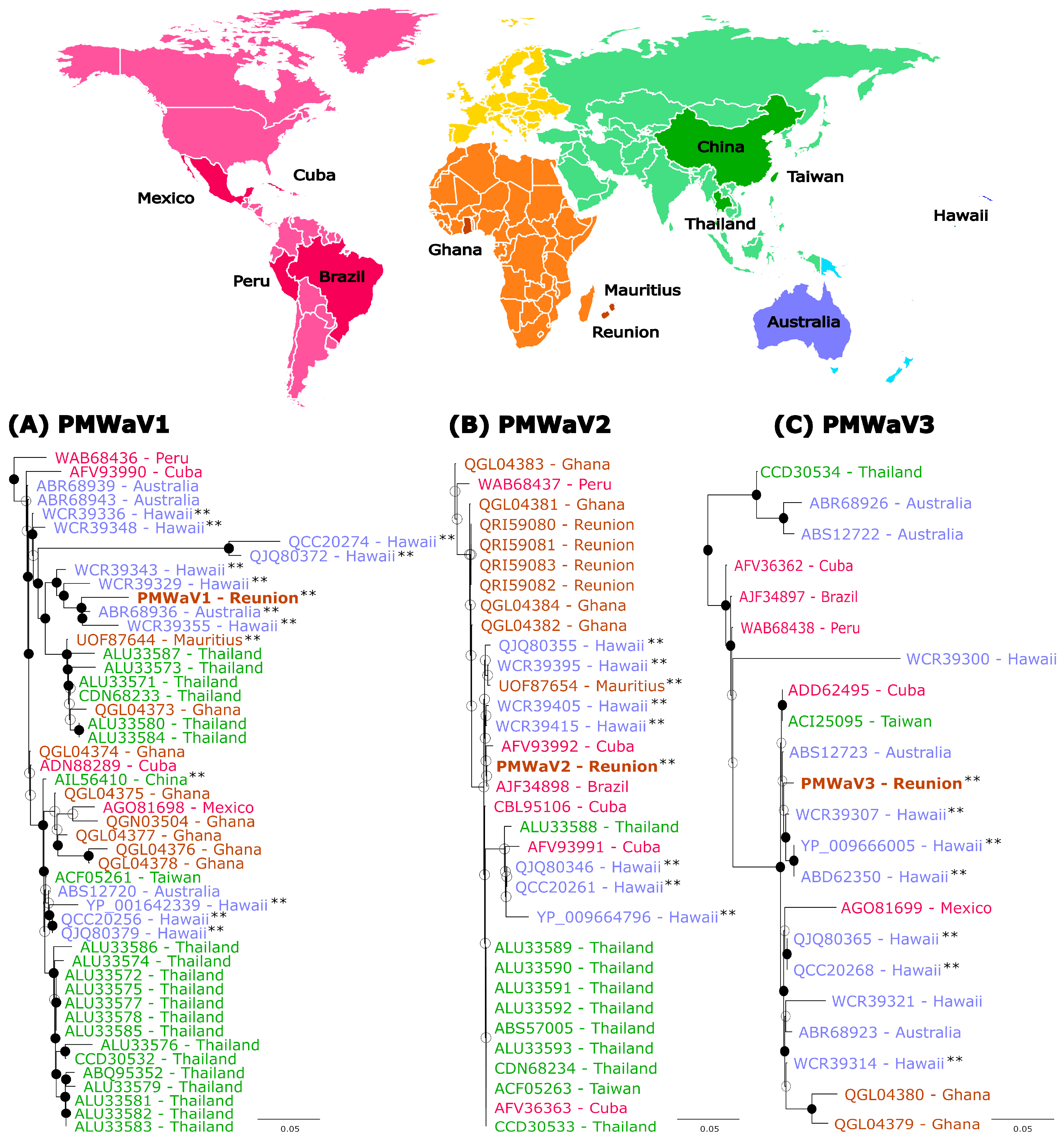
3.3. Phylogenetic Analyses
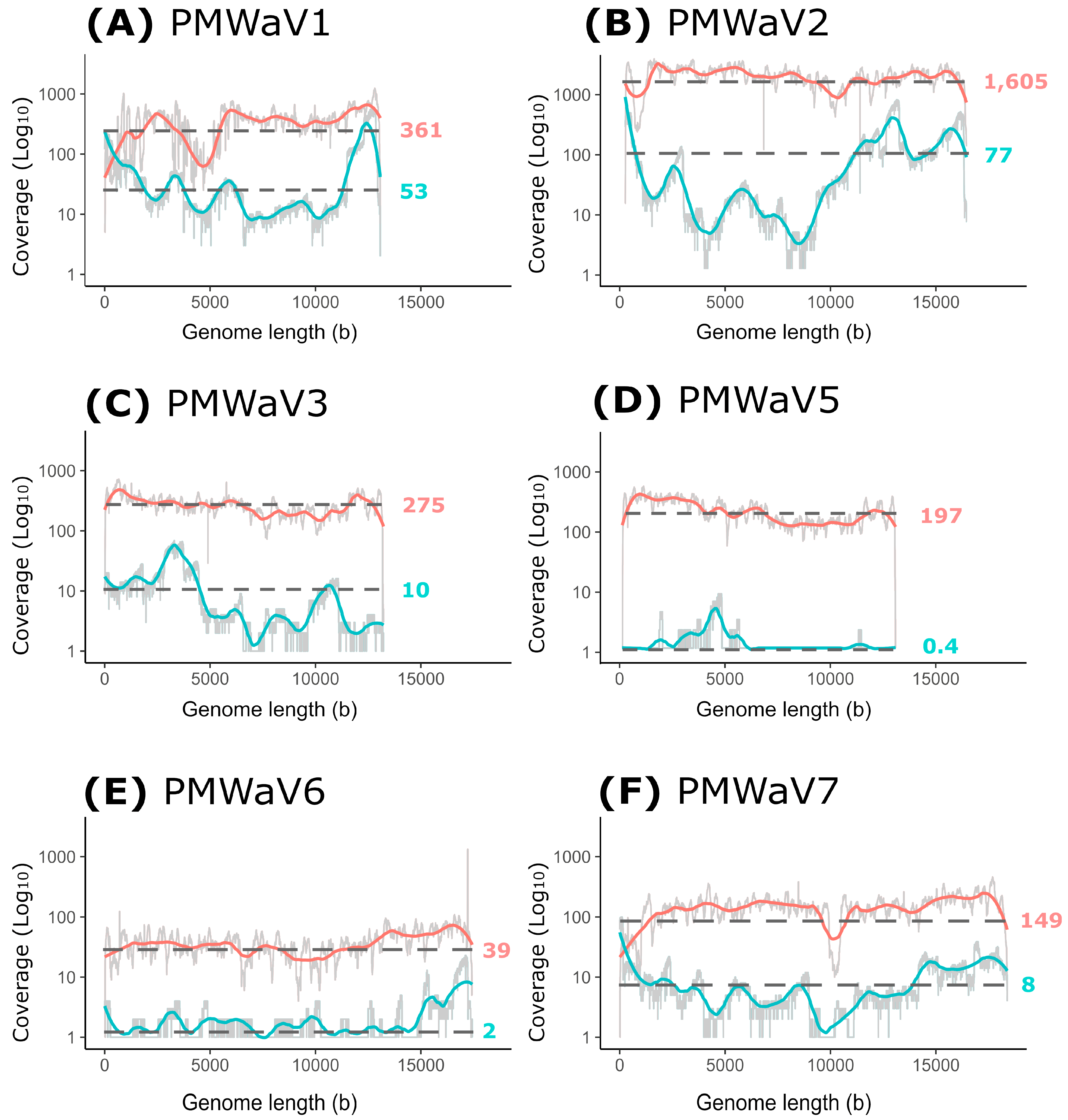
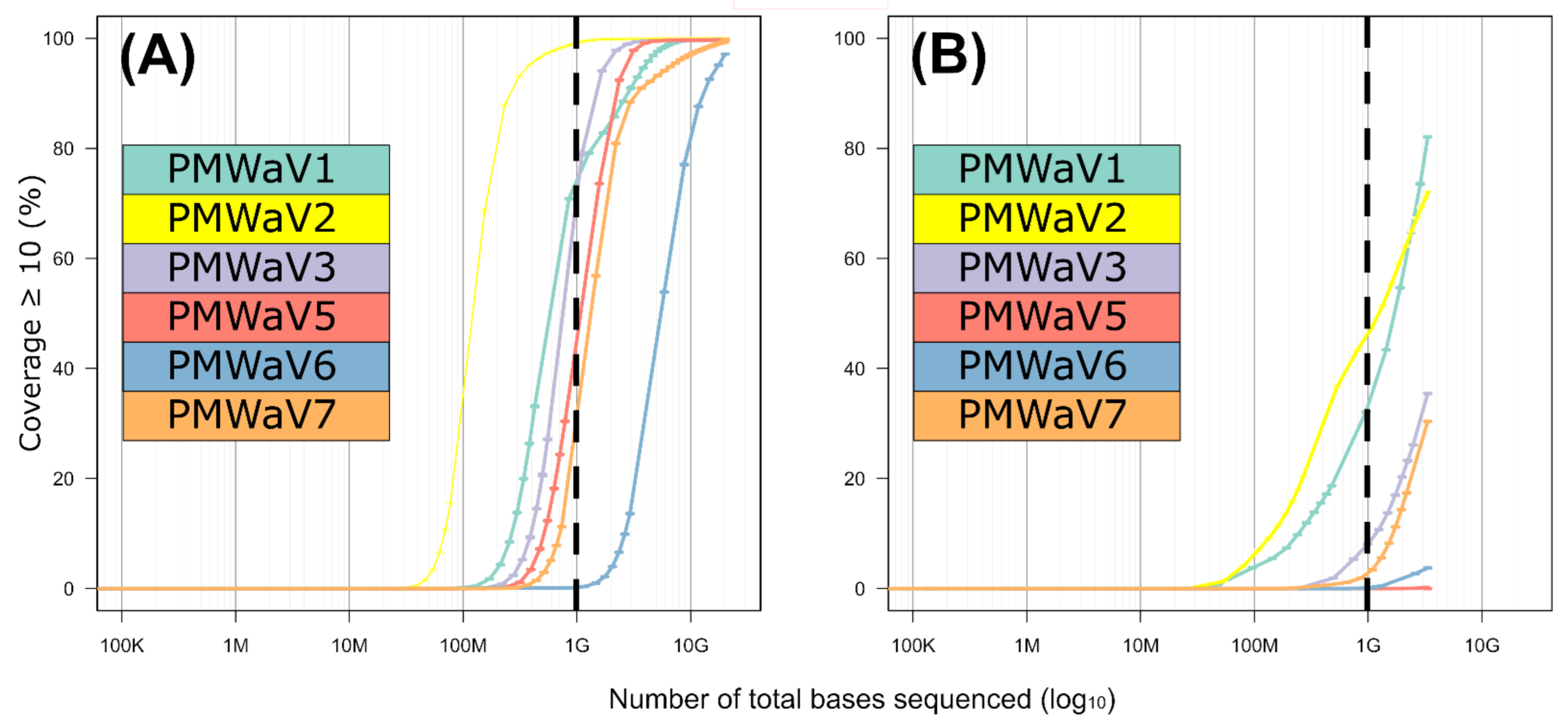
3.4. Comparison between the Illumina Short Reads and Nanopore Long Reads Approaches
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAOSTAT. Food and Agriculture Organization of the United Nations. In FAOSTAT Statistics Database; FAO: Rome, Italy, 2023. [Google Scholar]
- Sether, D.M.; Melzer, M.J.; Busto, J.; Zee, F.; Hu, J.S. Diversity and mealybug transmissibility of ampeloviruses in pineapple. Plant Dis. 2005, 89, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Carter, W. The influence of plant nutrition on susceptibility of pineapple plants to mealybug wilt. Phytopathology 1945, 35, 316–323. [Google Scholar]
- Carter, W. (Ed.) The systemic phytotoxemias: Mealybug wilt of pineapple. In Insects in Relation to Plant Disease; Interscience: New York, NY, USA, 1962; pp. 238–265. [Google Scholar]
- Carter, W. Mealybug wilt of pineapple; a reapraisal. Ann. N. Y. Acad. Sci. 1963, 105, 741–764. [Google Scholar] [CrossRef]
- Larsen, L.D. Diseases of pineapple: Hawaii Sugar Planters’ Association. Pathol. Physiol. Ser. Exp. Stn. Bull. 1910, 10, 1–72. [Google Scholar]
- Gambley, C.F.; Steele, V.; Geering, A.D.W.; Thomas, J.E. The genetic diversity of ampeloviruses in Australian pineapples and their association with mealybug wilt disease. Australas. Plant Pathol. 2008, 37, 95–105. [Google Scholar] [CrossRef]
- Sether, D.M.; Hu, J.S. Yield Impact and Spread of Pineapple mealybug wilt associated virus-2 and Mealybug Wilt of Pineapple in Hawaii. Plant Dis. 2002, 86, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Ito, K. Additional immunological evidence supporting the virus nature of mealybug wilt. Pineapple Res. Inst. News 1962, 10, 158–162. [Google Scholar]
- Gunasinghe, U.; German, T. Association of virus-particles with mealybug-wilt of pineapple. Phytopathology 1986, 76, 1073. [Google Scholar]
- Gunasinghe, U.; German, T. Further chacterization of virus associated with mealybug-wilt of pineapple. Phytopathology 1987, 77, 1776. [Google Scholar]
- Melzer, M.J.; Karasev, A.V.; Sether, D.M.; Hu, J.S. Nucleotide sequence, genome organization and phylogenetic analysis of pineapple mealybug wilt-associated virus-2. J. Gen. Virol. 2001, 82, 1–7. [Google Scholar] [CrossRef]
- Gunasinghe, U.; German, T. Purification and partial characterization of a virus from pineapple. Phytopathology 1989, 79, 1337–1341. [Google Scholar] [CrossRef]
- Martelli, G.P.; Agranovsky, A.A.; Bar-Joseph, M.B.D.; Boscia, D.; Candresse, T.; Coutts, R.H.A.; Dolja, V.V.; Falk, B.W.; Gonsalves, D.; Jelkmann, W.; et al. ICTV Study Group on closteroviruses and allied viruses. The family Closteroviridae revised. Arch. Virol. 2002, 147, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Bar-Joseph, M.; Candresse, T.; Maree, H.J.; Martelli, G.P.; Melzer, M.J.; Menzel, W.; Minafra, A.; Sabanadzovic, S.; Report Consortium, I. ICTV Virus Taxonomy Profile: Closteroviridae. J. Gen. Virol. 2020, 101, 364–365. [Google Scholar] [CrossRef]
- Maree, H.J.; Almeida, R.P.P.; Bester, R.; Chooi, K.M.; Cohen, D.; Dolja, V.V.; Fuchs, M.F.; Golino, D.A.; Jooste, A.E.C.; Martelli, G.P.; et al. Grapevine leafroll-associated virus 3. Front. Microbiol. 2013, 4, 82. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.K.; Green, J.C.; Melzer, M.; Borth, W.; Hu, J.S. Mealybug Wilt of Pineapple and Associated Viruses. Horticulturae 2018, 4, 52. [Google Scholar] [CrossRef]
- Larrea-Sarmiento, A.; Olmedo-Velarde, A.; Wang, X.; Borth, W.; Matsumoto, T.K.; Suzuki, J.Y.; Wall, M.M.; Melzer, M.; Hu, J. A novel ampelovirus associated with mealybug wilt of pineapple (Ananas comosus). Virus Genes 2021, 57, 464–468. [Google Scholar] [CrossRef]
- Herrbach, E.; Le Maguet, J.; Hommay, G. CHAPTER 11: Virus Transmission by Mealybugs and Soft Scales (Hemiptera: Coccoidea). In Vector-Mediated Transmission of Plant Pathogens; American Phytopathological Society (APS): Saint Paul, MN, USA, 2016; pp. 147–161. [Google Scholar] [CrossRef]
- Sether, D.M.; Borth, W.B.; Melzer, M.J.; Hu, J. Spatial and Temporal Incidences of Pineapple mealybug wilt-associated viruses in Pineapple Planting Blocks. Plant Dis. 2010, 94, 196–200. [Google Scholar] [CrossRef]
- Sether, D.M.; Hu, J.S. Closterovirus infection and mealybug exposure are necessary for the development of mealybug wilt of pineapple disease. Phytopathology 2002, 92, 928–935. [Google Scholar] [CrossRef]
- Larrea-Sarmiento, A.; Geering, A.D.W.; Olmedo-Velarde, A.; Wang, X.; Borth, W.; Matsumoto, T.K.; Suzuki, J.Y.; Wall, M.M.; Melzer, M.; Moyle, R.; et al. Genome sequence of pineapple secovirus B, a second sadwavirus reported infecting Ananas comosus. Arch. Virol. 2022, 167, 2801–2804. [Google Scholar] [CrossRef]
- Larrea-Sarmiento, A.; Olmedo-Velarde, A.; Green, J.C.; Al Rwahnih, M.; Wang, X.; Li, Y.H.; Wu, W.; Zhang, J.; Matsumoto, T.K.; Suzuki, J.Y.; et al. Identification and complete genomic sequence of a novel sadwavirus discovered in pineapple (Ananas comosus). Arch. Virol. 2020, 165, 1245–1248. [Google Scholar] [CrossRef]
- Larrea-Sarmiento, A.E.; Olmedo-Velarde, A.; Wang, X.; Borth, W.; Domingo, R.; Matsumoto, T.K.; Suzuki, J.Y.; Wall, M.M.; Melzer, M.J.; Hu, J. Genetic Diversity of Viral Populations Associated with Ananas Germplasm and Improvement of Virus Diagnostic Protocols. Pathogens 2022, 11, 1470. [Google Scholar] [CrossRef]
- Massé, D.; Filloux, D.; Candresse, T.; Massart, S.; Marais, A.; Verdin, E.; Cassam, N.; Fernandez, E.; Roumagnac, P.; Teycheney, P.Y.; et al. Identification of a novel vitivirus from pineapple in Reunion Island. Arch. Virol. 2022, 167, 2355–2357. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Roossinck, M.J. Deep Sequencing for Discovery and Evolutionary Analysis of Plant Viruses. Virus Res. 2017, 239, 82–86. [Google Scholar] [CrossRef]
- Wu, Q.; Ding, S.-W.; Zhang, Y.; Zhu, S. Identification of Viruses and Viroids by Next-Generation Sequencing and Homology-Dependent and Homology-Independent Algorithms. Annu. Rev. Phytopathol. 2015, 53, 425–444. [Google Scholar] [CrossRef]
- Green, J.C.; Rwahnih, M.A.; Olmedo-Velarde, A.; Melzer, M.J.; Hamim, I.; Borth, W.B.; Brower, T.M.; Wall, M.; Hu, J.S. Further genomic characterization of pineapple mealybug wilt-associated viruses using high-throughput sequencing. Trop. Plant Pathol. 2020, 45, 64–72. [Google Scholar] [CrossRef]
- Marais, A.; Murolo, S.; Faure, C.; Brans, Y.; Larue, C.; Maclot, F.; Massart, S.; Chiumenti, M.; Minafra, A.; Romanazzi, G.; et al. Sixty Years from the First Disease Description, a Novel Badnavirus Associated with Chestnut Mosaic Disease. Phytopathology 2021, 111, 1051–1058. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Rouard, M.D.A.; Droc, G.; Sarah, G.; Dufayard, J.F.; Guignon, V.; Hamelin, C.; Homa, F.; de Lamotte, F.; Larmande, P.; Lariviere, D.; et al. The South Green Bioinformatics platform. In Proceedings of the Plant and Animal Genome, PAG XXII, San Diego, CA, USA, 11–15 January 2014. [Google Scholar]
- Masoudi-Nejad, A.; Tonomura, K.; Kawashima, S.; Moriya, Y.; Suzuki, M.; Itoh, M.; Kanehisa, M.; Endo, T.; Goto, S. EGassembler: Online bioinformatics service for large-scale processing, clustering and assembling ESTs and genomic DNA fragments. Nucleic Acids Res. 2006, 34, W459–W462. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible Emergence of New Geminiviruses by Frequent Recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Mika, O.; Salminen, J.K.C.; Burke, D.S.; Mccutchan, F.E. Identification of Breakpoints in Intergenotypic Recombinants of HIV Type 1 by Bootscanning. AIDS Res. Hum. Retroviruses 1995, 11, 1423–1425. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An Exact Nonparametric Method for Inferring Mosaic Structure in Sequence Triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Dolja, V.V.; Kreuze, J.F.; Valkonen, J.P.T. Comparative and functional genomics of closteroviruses. Virus Res. 2006, 117, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Peremyslov, V.V.; Pan, Y.W.; Dolja, V.V. Movement Protein of a Closterovirus Is a Type III Integral Transmembrane Protein Localized to the Endoplasmic Reticulum. J. Virol. 2004, 78, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Massé, D.; Cassam, N.; Hostachy, B.; Iskra-Caruana, M.-L.; Darnaudery, M.; Lefeuvre, P.; Lett, J.-M. First Report of Three Pineapple Mealybug Wilt-Associated Viruses in Queen Victoria Pineapples in Reunion Island. Plant Dis. 2021, 105, 715. [Google Scholar] [CrossRef]
- Moreno, I.; Rodríguez-Arévalo, K.A.; Tarazona-Velásquez, R.; Kondo, T. Occurrence and distribution of pineapple mealybug wilt-associated viruses (PMWaVs) in MD2 pineapple fields in the Valle del Cauca Department, Colombia. Trop. Plant Pathol. 2023, 48, 217–225. [Google Scholar] [CrossRef]
- Bradamante, G.; Mittelsten Scheid, O.; Incarbone, M. Under siege: Virus control in plant meristems and progeny. Plant Cell 2021, 33, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Paull, R.E.; Duarte, O. Tropical Fruits Volume I. Crop Production Science, Horticulture; CABI Publishing: Wallingford, UK, 2011. [Google Scholar]
- Zanella, C.M.; Janke, A.; Palma-Silva, C.; Kaltchuk-Santos, E.; Pinheiro, F.G.; Paggi, G.M.; Soares, L.E.S.; Goetze, M.; Büttow, M.V.; Bered, F. Genetics, evolution and conservation of Bromeliaceae. Genet. Mol. Biol. 2012, 35, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Joy, P.; Anjana, R. Evolution of Pineapple; Pineapple Research Station: Muvattupuzha, India, 2015; pp. 670–686. [Google Scholar]
- Malmstrom, C.M.; Shu, R.; Linton, E.W.; Newton, L.A.; Cook, M.A. Barley yellow dwarf viruses (BYDVs) preserved in herbarium specimens illuminate historical disease ecology of invasive and native grasses. J. Ecol. 2007, 95, 1153–1166. [Google Scholar] [CrossRef]
- Rwahnih, M.A.; Rowhani, A.; Golino, D.A.; Islas, C.M.; Preece, J.E.; Sudarshana, M.R. Detection and genetic diversity of Grapevine red blotch-associated virus isolates in table grape accessions in the National Clonal Germplasm Repository in California. Can. J. Plant Pathol. 2015, 37, 130–135. [Google Scholar] [CrossRef]
- Rieux, A.; Campos, P.; Duvermy, A.; Scussel, S.; Martin, D.; Gaudeul, M.; Lefeuvre, P.; Becker, N.; Lett, J.-M. Contribution of historical herbarium small RNAs to the reconstruction of a cassava mosaic geminivirus evolutionary history. Sci. Rep. 2021, 11, 21280. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.J.; Sether, D.M.; Karasev, A.V.; Borth, W.; Hu, J.S. Complete nucleotide sequence and genome organization of pineapple mealybug wilt-associated virus-1. Arch. Virol. 2008, 153, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Sether, D.M.; Melzer, M.J.; Borth, W.B.; Hu, J.S. Genome organization and phylogenetic relationship of Pineapple mealybug wilt associated virus-3 with family Closteroviridae members. Virus Genes 2009, 38, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Adams, I.; Al Rwahnih, M.; Baeyen, S.; Bilodeau, G.J.; Blouin, A.G.; Boonham, N.; Candresse, T.; Chandellier, A.; De Jonghe, K. Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pests. Peer Community J. 2022, 2, e62. [Google Scholar] [CrossRef]
- Fontdevila Pareta, N.; Khalili, M.; Maachi, A.; Rivarez, M.P.S.; Rollin, J.; Salavert, F.; Temple, C.; Aranda, M.A.; Boonham, N.; Botermans, M.; et al. Managing the deluge of newly discovered plant viruses and viroids: An optimized scientific and regulatory framework for their characterization and risk analysis. Front. Microbiol. 2023, 14, 1181562. [Google Scholar] [CrossRef]
- Gambley, C.F.; Geering, A.D.W.; Thomas, J.E. Development of an immunomagnetic capture-reverse transcriptase-PCR assay for three pineapple ampeloviruses. J. Virol. Methods 2009, 155, 187–192. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Supercontigs ID (Length bp) | Number of Contigs | Contigs Length (bp) | Number of Reads | Cov ≥ 10 1 (%) | Cov ≥ 100 1 (%) | Mean Depth | Most Similar Virus on NCBI (BLASTn) | Accession Numbers | Identity (%) 2 | Query Cover (%) | Virus Specises |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Contig-A (13,074) | 67 | 86–13,073 | 42,491 | 99.9 | 98.3 | 415 | Pineapple mealybug wilt-associated virus 1 | OP860292 | 86.3 | 99.3 | PMWaV1 |
| Contig-B (16,199) | 38 | 81–5816 | 200,516 | 100.0 | 99.9 | 1683 | Pineapple mealybug wilt-associated virus 2 | OP860299 | 99.4 | 99.2 | PMWaV2 |
| Contig-C (13,229) | 59 | 80–5040 | 28,779 | 99.8 | 98.3 | 285 | Pineapple mealybug wilt-associated virus 3 | MN539274 | 96.3 | 99.4 | PMWaV3 |
| Contig-D (12,971) | 8 | 159–6146 | 19,534 | 99.8 | 90.6 | 197 | Pineapple mealybug wilt-associated virus 5 | EF467920 | 84.1 | 13.1 | PMWaV5 |
| Contig-E (17,440) | 4 | 466–7117 | 7304 | 97.5 | 1.1 | 41 | Pineapple mealybug wilt-associated virus 6 | OP86029 | 99.1 | 99.1 | PMWaV6 |
| Contig-F (18,388) | 7 | 389–18,092 | 23,269 | 100.0 | 85.1 | 157 | Grapevine leafroll-associated virus 3 | KY073324 | 79.3 | 0.5 | PMWaV7 |
| Protein | Supercontig | Virus Species | Most Closely Related Virus (BLASTp) | Nucleotides Identities (%) | Amino Acid Identities (%) ¹ |
|---|---|---|---|---|---|
| RdRp | Contig-A | PMWaV1 | NC_010178—Pineapple mealybug wilt-associated virus 1 | 90.6 | 90.7 |
| Contig-B | PMWaV2 | NC_043105—Pineapple mealybug wilt-associated virus 2 | 98.9 | 98.4 | |
| Contig-C | PMWaV3 | NC_043406—Pineapple mealybug wilt-associated virus 3 | 97.6 | 98.6 | |
| Contig-D | PMWaV5 | EF467922—Pineapple mealybug wilt-associated virus 5 | 85.6 | 85.7 | |
| Contig-E | PMWaV6 | MW269512—Pineapple mealybug wilt-associated virus 6 | 99.5 | 98.8 | |
| Contig-F | PMWaV7 | NC_004667—Grapevine leafroll-associated virus 3 | 55.2 | 43.0 | |
| HSP70h | Contig-A | PMWaV1 | NC_010178—Pineapple mealybug wilt-associated virus 1 | 88.9 | 86.4 |
| Contig-B | PMWaV2 | NC_043105—Pineapple mealybug wilt-associated virus 2 | 97.5 | 95.9 | |
| Contig-C | PMWaV3 | NC_043406—Pineapple mealybug wilt-associated virus 3 | 97.2 | 94.8 | |
| Contig-D | PMWaV5 | EF467920—Pineapple mealybug wilt-associated virus 5 | 84.7 | 91.8 | |
| Contig-E | PMWaV6 | MW269512—Pineapple mealybug wilt-associated virus 6 | 99.3 | 99.1 | |
| Contig-F | PMWaV7 | NC_004667—Grapevine leafroll-associated virus 3 | 55.2 | 41.2 | |
| CP | Contig-A | PMWaV1 | NC_010178—Pineapple mealybug wilt-associated virus 1 | 91.1 | 94.9 |
| Contig-B | PMWaV2 | NC_043105—Pineapple mealybug wilt-associated virus 2 | 99.5 | 99.2 | |
| Contig-C | PMWaV3 | NC_043406—Pineapple mealybug wilt-associated virus 3 | 97.2 | 97.3 | |
| Contig-D | PMWaV5 | NC_043406—Pineapple mealybug wilt-associated virus 3 | 66.6 | 60.6 | |
| Contig-E | PMWaV6 | MW269512—Pineapple mealybug wilt-associated virus 6 | 99.4 | 98.6 | |
| Contig-F | PMWaV7 | NC_022072—Blackberry vein banding-associated virus | 46.9 | 34.3 |
| Virus Species | Approach | Number of Reads | Cov ≥ 1 1 (%) | Cov ≥ 10 1 (%) | Mean Depth | Number Viral Bases/ Million Sequenced Bases |
|---|---|---|---|---|---|---|
| PMWaV1 | Illumina short reads Nanopore long reads | 41,417 1074 | 100 100 | 99.8 82.1 | 361.0 53.3 | 310.6 234.7 |
| PMWaV2 | Illumina short reads Nanopore long reads | 198,147 2374 | 100 99.7 | 100 72.0 | 1606 77.0 | 1710.1 420 |
| PMWaV3 | Illumina short reads Nanopore long reads | 28,561 218 | 100 94.8 | 99.7 35.5 | 275.0 10.1 | 238.9 45.3 |
| PMWaV5 | Illumina short reads Nanopore long reads | 19,517 17 | 99.9 18.8 | 99.8 0.0 | 197.0 0.4 | 168.1 1.8 |
| PMWaV6 | Illumina short reads Nanopore long reads | 7235 69 | 99.9 53.9 | 97.1 3.7 | 39.4 1.5 | 45.2 8.7 |
| PMWaV7 | Illumina short reads Nanopore long reads | 22,937 332 | 99.9 94.0 | 99.7 30.4 | 148.8 8.3 | 179.9 51.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massé, D.; Candresse, T.; Filloux, D.; Massart, S.; Cassam, N.; Hostachy, B.; Marais, A.; Fernandez, E.; Roumagnac, P.; Verdin, E.; et al. Characterization of Six Ampeloviruses Infecting Pineapple in Reunion Island Using a Combination of High-Throughput Sequencing Approaches. Viruses 2024, 16, 1146. https://doi.org/10.3390/v16071146
Massé D, Candresse T, Filloux D, Massart S, Cassam N, Hostachy B, Marais A, Fernandez E, Roumagnac P, Verdin E, et al. Characterization of Six Ampeloviruses Infecting Pineapple in Reunion Island Using a Combination of High-Throughput Sequencing Approaches. Viruses. 2024; 16(7):1146. https://doi.org/10.3390/v16071146
Chicago/Turabian StyleMassé, Delphine, Thierry Candresse, Denis Filloux, Sébastien Massart, Nathalie Cassam, Bruno Hostachy, Armelle Marais, Emmanuel Fernandez, Philippe Roumagnac, Eric Verdin, and et al. 2024. "Characterization of Six Ampeloviruses Infecting Pineapple in Reunion Island Using a Combination of High-Throughput Sequencing Approaches" Viruses 16, no. 7: 1146. https://doi.org/10.3390/v16071146