
VOGDB—Database of Virus Orthologous Groups
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Concept
2.2. Preprocessing of Input Data
2.2.1. Input from RefSeq
2.2.2. Polyproteins
2.3. Creation of the First-Layer Clusters—VOGs
2.3.1. Functional Annotation
2.3.2. Naming
2.4. Creation of the Second Layer Clusters—VFAMs
Clustering Using MCL
2.5. Creation of the Third-Layer Clusters—VFOLDs
2.6. Quality Assessment of the Clustering Results
3. Results
3.1. Database
3.2. Content
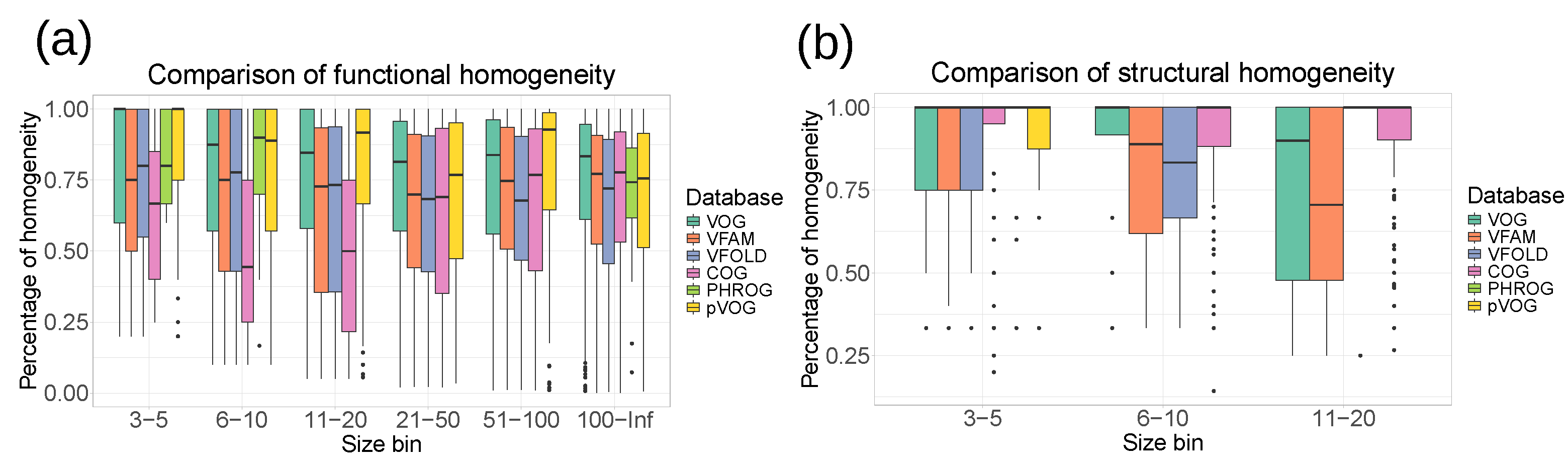
3.3. Quality Assessment
3.4. Comparison with Similar Databases
3.5. Availability
3.5.1. VOGDB Webpage
3.5.2. VOGDB Release Files
4. Discussion
4.1. Limitations
4.2. Support for Bioinformatic Workflows
4.3. Usage for Metagenome Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Villarreal, L. Evolution of Viruses. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 174–184. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.R. Are There 1031 Virus Particles on Earth, or More, or Fewer? J. Bacteriol. 2020, 202, e00052-20. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Krupovic, M.; Dolja, V.V. The global virome: How much diversity and how many independent origins? Environ. Microbiol. 2023, 25, 40–44. [Google Scholar] [CrossRef]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhatla, D.B.; Sherman, W.A.; Chung, B.Y.W.; Cook, S.; Schneider, G.; Eisenhaber, B.; Karlin, D.G. Powerful Sequence Similarity Search Methods and In-Depth Manual Analyses Can Identify Remote Homologs in Many Apparently “Orphan” Viral Proteins. J. Virol. 2014, 88, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Andino, R. Viral Evolution. In Viral Pathogenesis; Elsevier: Amsterdam, The Netherlands, 2016; pp. 233–240. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. The logic of virus evolution. Cell Host Microbe 2022, 30, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Orthologs, Paralogs, and Evolutionary Genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. An Introduction to Sequence Similarity (“Homology”) Searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.J. Hidden Markov Models and their Applications in Biological Sequence Analysis. Curr. Genom. 2009, 10, 402–415. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Terzian, P.; Olo Ndela, E.; Galiez, C.; Lossouarn, J.; Pérez Bucio, R.; Mom, R.; Toussaint, A.; Petit, M.A.; Enault, F. PHROG: Families of prokaryotic virus proteins clustered using remote homology. NAR Genom. Bioinform. 2021, 3, lqab067. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; Badretdin, A.; Coulouris, G.; DiCuccio, M.; Durkin, A.; Jovenitti, E.; Li, W.; Mersha, M.; O’Neill, K.; Virothaisakun, J.; et al. RefSeq and the prokaryotic genome annotation pipeline in the age of metagenomes. Nucleic Acids Res. 2024, 52, D762–D769. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; O’Neill, K.R.; Haft, D.H.; DiCuccio, M.; Chetvernin, V.; Badretdin, A.; Coulouris, G.; Chitsaz, F.; Derbyshire, M.; Durkin, A.S.; et al. RefSeq: Expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res. 2021, 49, D1020–D1028. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Ostell, J.; Pruitt, K.D.; Sayers, E.W. GenBank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef] [PubMed]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods Mol. Biol. 2016, 1374, 23–54. [Google Scholar] [CrossRef] [PubMed]
- Chandonia, J.M.; Guan, L.; Lin, S.; Yu, C.; Fox, N.; Brenner, S. SCOPe: Improvements to the structural classification of proteins—Extended database to facilitate variant interpretation and machine learning. Nucleic Acids Res. 2022, 50, D553–D559. [Google Scholar] [CrossRef] [PubMed]
- Yost, S.A.; Marcotrigiano, J. Viral precursor polyproteins: Keys of regulation from replication to maturation. Curr. Opin. Virol. 2013, 3, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, A.A.; Sigorskih, A.I.; Ocheredko, E.S.; Samborskiy, D.V.; Gorbalenya, A.E. LAMPA, LArge Multidomain Protein Annotator, and its application to RNA virus polyproteins. Bioinformatics 2020, 36, 2731–2739. [Google Scholar] [CrossRef]
- Kristensen, D.M.; Kannan, L.; Coleman, M.K.; Wolf, Y.I.; Sorokin, A.; Koonin, E.V.; Mushegian, A. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 2010, 26, 1481–1487. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Wong, T.K.F.; Kalyaanamoorthy, S.; Meusemann, K.; Yeates, D.K.; Misof, B.; Jermiin, L.S. A minimum reporting standard for multiple sequence alignments. NAR Genom. Bioinform. 2020, 2, lqaa024. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef]
- Van Dongen, S. Graph Clustering Via a Discrete Uncoupling Process. SIAM J. Matrix Anal. Appl. 2008, 30, 121–141. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, D488–D508. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.M.; Söding, J.; Steinegger, M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2023, 42, 243–246. [Google Scholar] [CrossRef]
- Barrio-Hernandez, I.; Yeo, J.; Jänes, J.; Mirdita, M.; Gilchrist, C.L.M.; Wein, T.; Varadi, M.; Velankar, S.; Beltrao, P.; Steinegger, M. Clustering predicted structures at the scale of the known protein universe. Nature 2023, 622, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Vera Alvarez, R.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Plaza, A.; Szklarczyk, D.; Botas, J.; Cantalapiedra, C.; Giner-Lamia, J.; Mende, D.R.; Kirsch, R.; Rattei, T.; Letunic, I.; Jensen, L.; et al. eggNOG 6.0: Enabling comparative genomics across 12 535 organisms. Nucleic Acids Res. 2023, 51, D389–D394. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient Virus World and evolution of cells. Biol. Direct 2006, 1, 29. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Edlund, A.; Yang, Y.; McLean, J.S.; Yooseph, S. Metagenome and Metatranscriptome Analyses Using Protein Family Profiles. PLoS Comput. Biol. 2016, 12, e1004991. [Google Scholar] [CrossRef]
- Laffy, P.W.; Wood-Charlson, E.M.; Turaev, D.; Jutz, S.; Pascelli, C.; Botté, E.S.; Bell, S.C.; Peirce, T.E.; Weynberg, K.D.; Van Oppen, M.J.H.; et al. Reef invertebrate viromics: Diversity, host specificity and functional capacity. Environ. Microbiol. 2018, 20, 2125–2141. [Google Scholar] [CrossRef]
- Yu, R.; Huang, Z.; Lam, T.Y.C.; Sun, Y. Utilizing profile hidden Markov model databases for discovering viruses from metagenomic data: A comprehensive review. Briefings Bioinform. 2024, 25, bbae292. [Google Scholar] [CrossRef]
- Turner, D.; Shkoporov, A.N.; Lood, C.; Millard, A.D.; Dutilh, B.E.; Alfenas-Zerbini, P.; Van Zyl, L.J.; Aziz, R.K.; Oksanen, H.M.; Poranen, M.M.; et al. Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee. Arch. Virol. 2023, 168, 74. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer | Strict | Medium | Low |
|---|---|---|---|
| vOG | 38,562 | 45,613 | 48,627 |
| vFAM | 28,500 | 32,546 | 33,951 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trgovec-Greif, L.; Hellinger, H.-J.; Mainguy, J.; Pfundner, A.; Frishman, D.; Kiening, M.; Webster, N.S.; Laffy, P.W.; Feichtinger, M.; Rattei, T. VOGDB—Database of Virus Orthologous Groups. Viruses 2024, 16, 1191. https://doi.org/10.3390/v16081191
Trgovec-Greif L, Hellinger H-J, Mainguy J, Pfundner A, Frishman D, Kiening M, Webster NS, Laffy PW, Feichtinger M, Rattei T. VOGDB—Database of Virus Orthologous Groups. Viruses. 2024; 16(8):1191. https://doi.org/10.3390/v16081191
Chicago/Turabian StyleTrgovec-Greif, Lovro, Hans-Jörg Hellinger, Jean Mainguy, Alexander Pfundner, Dmitrij Frishman, Michael Kiening, Nicole Suzanne Webster, Patrick William Laffy, Michael Feichtinger, and Thomas Rattei. 2024. "VOGDB—Database of Virus Orthologous Groups" Viruses 16, no. 8: 1191. https://doi.org/10.3390/v16081191