
Comparative Transcriptomic Analyses of Peripheral Blood Mononuclear Cells of COVID-19 Patients without Pneumonia and with Severe Pneumonia in the First Year of Follow-Up
 , , , , , , , , , , , , add
Show full author list
, , , , , , , , , , , , add
Show full author list
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethical Statement
2.2. Data Availability
2.3. Study Population
2.4. Blood Sample Collection and Isolation of Peripheral Blood Mononuclear Cells (PBMCs)
2.5. Transcriptome Library Construction and Next-Generation RNA Sequencing
2.6. RNA-Seq Data Analysis and Differential Expression Analysis
2.7. Functional Annotation and Enrichment Analysis
Identification of 52 Genes Associated with Idiopathic Pulmonary Fibrosis Risk in the PBMCs’ Transcriptome
2.8. Statistical Analysis
3. Results
3.1. Identification of DEGs and DElncRNAs among the Healthy Control, No Pneumonia, and Severe Pneumonia Groups
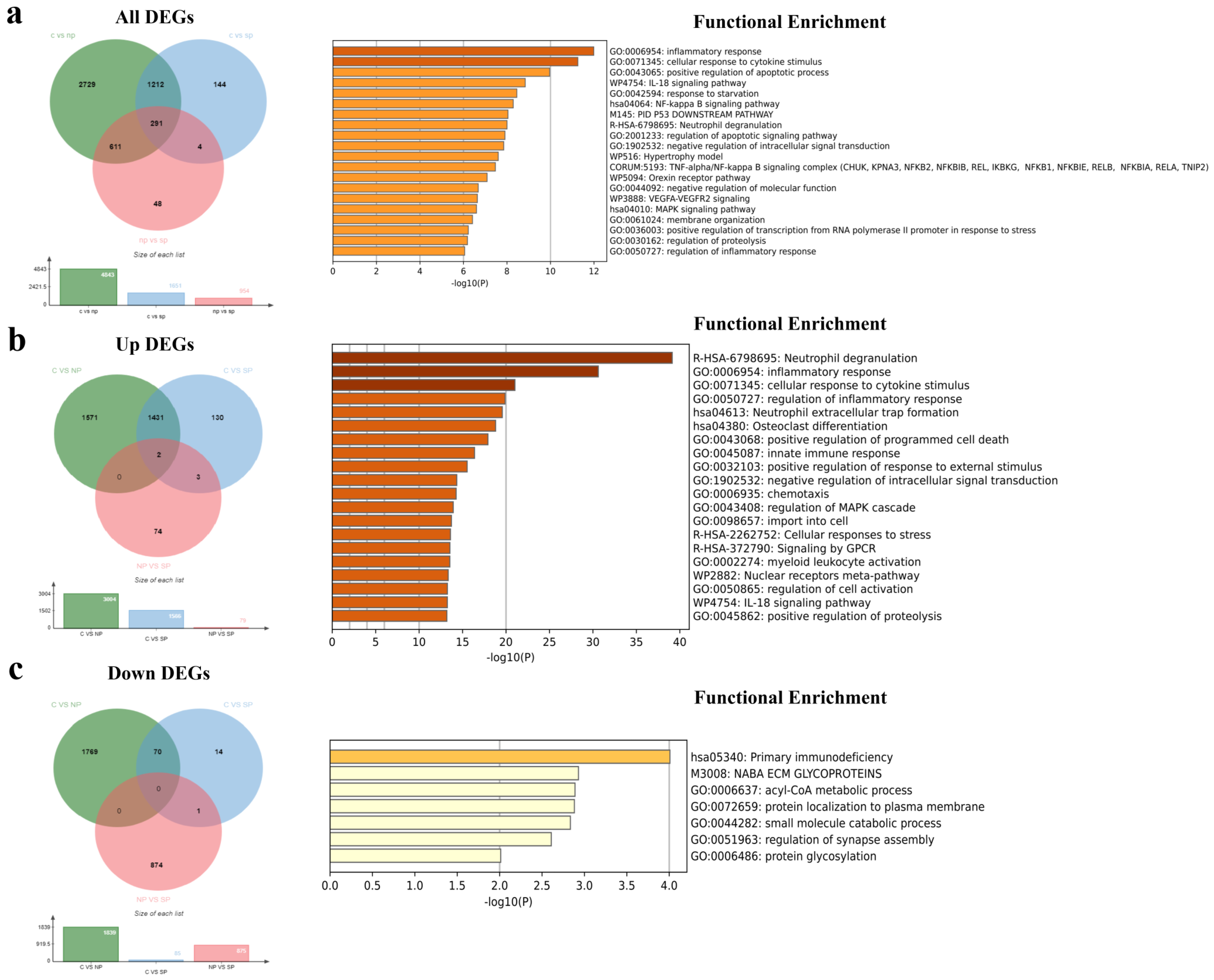
3.2. Functional Enrichment Analysis of All DEGs
3.3. Functional Enrichment Analysis of Common DEGs and DElncRNAs
3.4. Identification of Hub Genes via the Protein–Protein Interaction (PPI) Network
3.5. Identification of Gene–Disease, Gene–Transcription Factor, and Gene–Transcription Factor Target Interactions
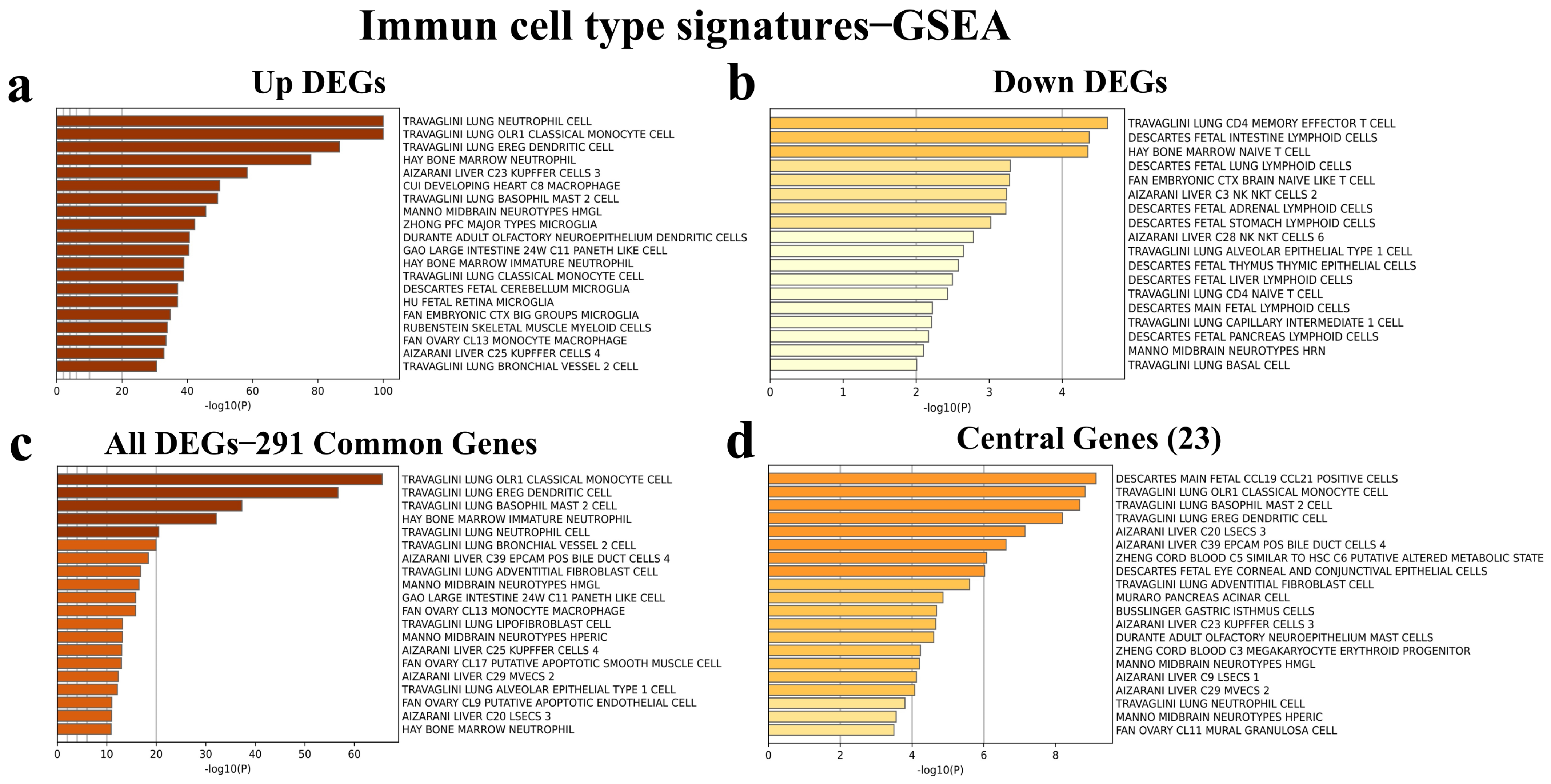
3.6. Identification of Immune Cell Type Signatures of the Common and Central DEGs
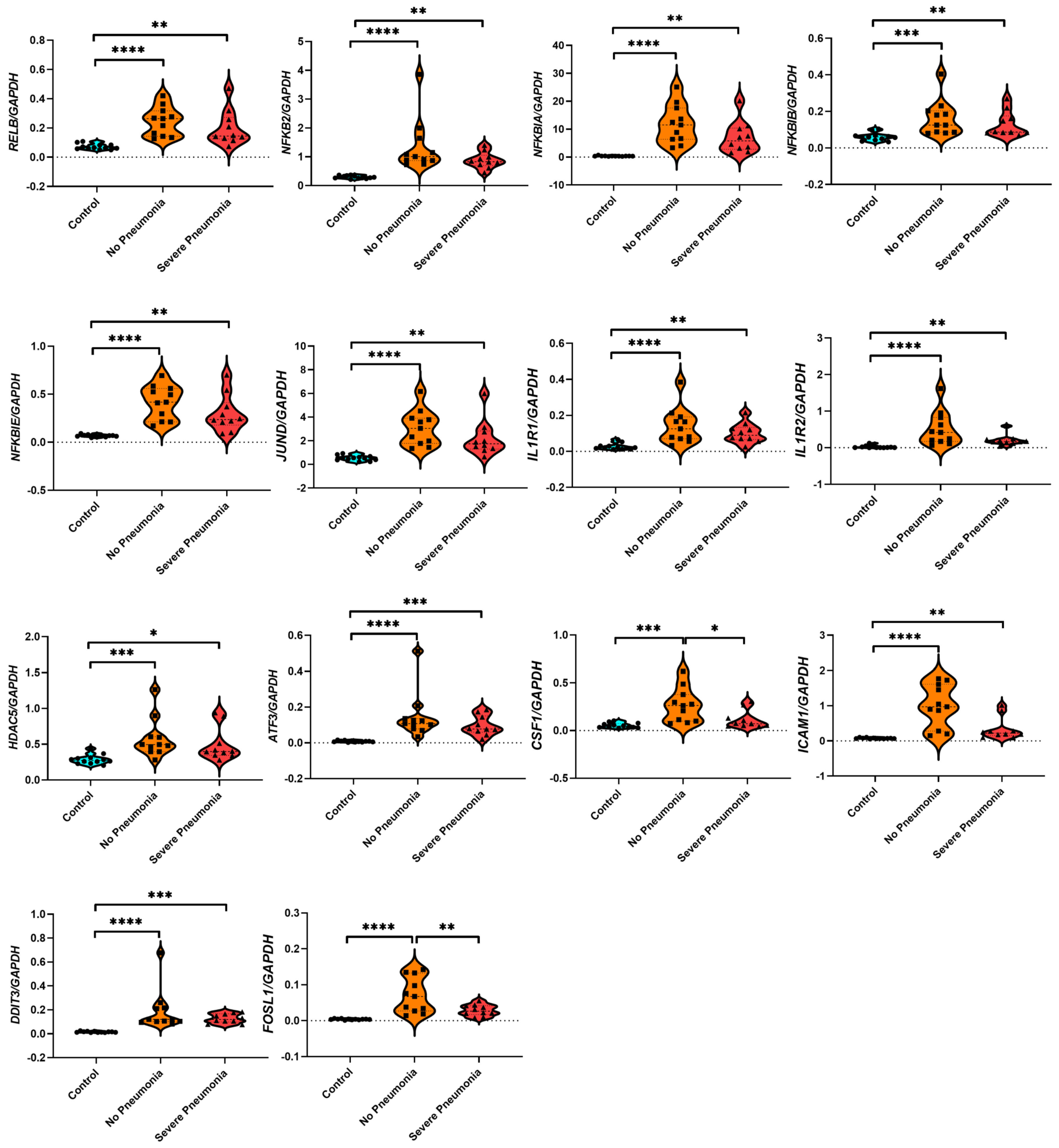
3.7. Gene Expression Analysis Based on RNA-seq Count Data of 14 Hub Genes
3.8. Investigation of 52 Idiopathic Pulmonary Fibrosis (IPF)-Related Gene Signatures in the PBMC Transcriptome of Patients with Non-Pneumonia and Severe Pneumonia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO COVID-19 Dashboard. 2024. Available online: https://data.who.int/dashboards/covid19/cases?n=c (accessed on 23 June 2024).
- Da Rosa Mesquita, R.; Junior, L.C.F.S.; Santana, F.M.S.; de Oliveira, T.F.; Alcântara, R.C.; Arnozo, G.M.; Filho, E.R.d.S.; dos Santos, A.G.G.; da Cunha, E.J.O.; de Aquino, S.H.S.; et al. Clinical manifestations of COVID-19 in the general population: Systematic review. Wien. Klin. Wochenschr. 2021, 133, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, B.; Shilo, S.; Rossman, H.; Kalkstein, N.; Marcus, K.; Barer, Y.; Keshet, A.; Shamir-Stein, N.; Shalev, V.; Zohar, A.E.; et al. Longitudinal symptom dynamics of COVID-19 infection. Nat. Commun. 2020, 11, 6208. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, G.T.; Hession, P.; Liew, C.H.; Lim, B.C.W.; Leong, T.K.; Lim, V.; Sulaiman, L.H. COVID-19 in adult patients with pre-existing chronic cardiac, respiratory and metabolic disease: A critical literature review with clinical recommendations. Trop. Dis. Travel Med. Vaccines 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liang, W.-Q.; Li, Y.-R.; He, J.-X.; Guan, W.-J. The Short- and Long-Term Clinical, Radiological and Functional Consequences of COVID-19. Arch. Bronconeumol. 2022, 58, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Shivani, F.; Kumari, N.; Bai, P.; Rakesh, F.; Haseeb, M.; Kumar, S.; Jamil, A.; Zaidi, M.; Shaukat, F.; Rizwan, A. Long-Term Symptoms of COVID-19: One-Year Follow-up Study. Cureus 2022, 14, e25937. [Google Scholar] [CrossRef] [PubMed]
- Imoto, W.; Yamada, K.; Kawai, R.; Imai, T.; Kawamoto, K.; Uji, M.; Kanda, H.; Takada, M.; Ohno, Y.; Ohtani, H.; et al. A cross-sectional, multicenter survey of the prevalence and risk factors for Long COVID. Sci. Rep. 2022, 12, 22413. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, B.; Sudry, T.; Flaks-Manov, N.; Yehezkelli, Y.; Kalkstein, N.; Akiva, P.; Ekka-Zohar, A.; Ben David, S.S.; Lerner, U.; Bivas-Benita, M.; et al. Long COVID outcomes at one year after mild SARS-CoV-2 infection: Nationwide cohort study. BMJ 2023, 380, e072529. [Google Scholar] [CrossRef]
- Wongwathanavikrom, N.; Tovichien, P.; Udomittipong, K.; Palamit, A.; Tiamduangtawan, P.; Mahoran, K.; Charoensittisup, P. Incidence and risk factors for long COVID in children with COVID-19 pneumonia. Pediatr. Pulmonol. 2024, 59, 1330–1338. [Google Scholar] [CrossRef]
- Bellan, M.; Baricich, A.; Patrucco, F.; Zeppegno, P.; Gramaglia, C.; Balbo, P.E.; Carriero, A.; Amico, C.S.; Avanzi, G.C.; Barini, M.; et al. Long-term sequelae are highly prevalent one year after hospitalization for severe COVID-19. Sci. Rep. 2021, 11, 22666. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, V.; Rafailidis, P.; Terzi, I.; Dragoumani, I.; Markatou, F.; Papanas, N.; Vradelis, S.; Gouveri, E.; Panopoulou, M.; Papazoglou, D.; et al. The prevalence of long COVID-19 syndrome in hospitalized patients with COVID-19 pneumonia. Infez. Med. 2024, 32, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Kurys-Denis, E.; Grzywa-Celińska, A.; Podgórska, K.; Kawa, M.P. What Remains up to 7 Months after Severe and Moderate Pneumonia in Non-Vaccinated Patients with Long COVID? Results of a CT Study. J. Clin. Med. 2023, 12, 5388. [Google Scholar] [CrossRef] [PubMed]
- McMichael, T.M.; Currie, D.W.; Clark, S.; Pogosjans, S.; Kay, M.; Schwartz, N.G.; Lewis, J.; Baer, A.; Kawakami, V.; Lukoff, M.D.; et al. Epidemiology of COVID-19 in a Long-Term Care Facility in King County, Washington. N. Engl. J. Med. 2020, 382, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Augustin, M.; Schommers, P.; Stecher, M.; Dewald, F.; Gieselmann, L.; Gruell, H.; Horn, C.; Vanshylla, K.; Di Cristanziano, V.; Osebold, L.; et al. Post-COVID syndrome in non-hospitalised patients with COVID-19: A longitudinal prospective cohort study. Lancet Reg. Health Eur. 2021, 6, 100122. [Google Scholar] [CrossRef] [PubMed]
- Legler, F.; Meyer-Arndt, L.; Mödl, L.; Kedor, C.; Freitag, H.; Stein, E.; Hoppmann, U.; Rust, R.; Wittke, K.; Siebert, N.; et al. Long-term symptom severity and clinical biomarkers in post-COVID-19/chronic fatigue syndrome: Results from a prospective observational cohort. eClinicalMedicine 2023, 63, 102146. [Google Scholar] [CrossRef]
- Han, Q.; Zheng, B.; Daines, L.; Sheikh, A. Long-Term Sequelae of COVID-19: A Systematic Review and Meta-Analysis of One-Year Follow-Up Studies on Post-COVID Symptoms. Pathogens 2022, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Chen, Y. Transcriptomics: Advances and approaches. Sci. China Life Sci. 2013, 56, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Dhooria, S.; Arora, S.; Chaudhary, S.; Sehgal, I.; Prabhakar, N.; Mohammad, N.; Sharma, R.; Das, P.; Kumar, Y.; Garg, M.; et al. Risk factors for clinically significant diffuse parenchymal lung abnormalities persisting after severe COVID-19 pneumonia. Indian J. Med. Res. 2023, 157, 427–437. [Google Scholar] [CrossRef]
- Menezes, S.M.; Jamoulle, M.; Carletto, M.P.; Moens, L.; Meyts, I.; Maes, P.; Van Weyenbergh, J. Blood transcriptomic analyses reveal persistent SARS-CoV-2 RNA and candidate biomarkers in post-COVID-19 condition. Lancet Microbe. 2024. [Google Scholar] [CrossRef]
- Argun Baris, S.; Toprak, O.B.; Cetinkaya, P.D.; Fakili, F.; Kokturk, N.; Kul, S.; Kayalar, O.; Tutuncu, Y.; Azak, E.; Kuluozturk, M.; et al. The predictors of long–COVID in the cohort of Turkish Thoracic Society–TURCOVID multicenter registry: One year follow–up results. Asian Pac. J. Trop. Med. 2022, 15, 400–409. [Google Scholar] [CrossRef]
- Kokturk, N.; Babayigit, C.; Kul, S.; Cetinkaya, P.D.; Nayci, S.A.; Argun Baris, S.; Karcioglu, O.; Aysert, P.; Irmak, I.; Yuksel, A.A.; et al. The predictors of COVID-19 mortality in a nationwide cohort of Turkish patients. Respir. Med. 2021, 183, 106433. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kucukural, A.; Yukselen, O.; Ozata, D.M.; Moore, M.J.; Garber, M. DEBrowser: Interactive differential expression analysis and visualization tool for count data. BMC Genom. 2019, 20, 6. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R Package Version 1.22.0 2019. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Fang, J.; Xu, Z.; Zhang, H.; Mao, M.; Chen, Y.; Zhang, L.; Pian, C. NcPath: A novel platform for visualization and enrichment analysis of human non-coding RNA and KEGG signaling pathways. Bioinformatics 2023, 39, btac812. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene set knowledge discovery with enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Herazo-Maya, J.D.; Noth, I.; Duncan, S.R.; Kim, S.; Ma, S.-F.; Tseng, G.C.; Feingold, E.; Juan-Guardela, B.M.; Richards, T.J.; Lussier, Y.; et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2013, 5, 205ra136. [Google Scholar] [CrossRef] [PubMed]
- Herazo-Maya, J.D.; Sun, J.; Molyneaux, P.L.; Li, Q.; Villalba, J.A.; Tzouvelekis, A.; Lynn, H.; Juan-Guardela, B.M.; Risquez, C.; Osorio, J.C.; et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: An international, multicentre, cohort study. Lancet Respir. Med. 2017, 5, 857–868. [Google Scholar] [CrossRef]
- Guardela, B.M.J.; Sun, J.; Zhang, T.; Xu, B.; Balnis, J.; Huang, Y.; Ma, S.-F.; Molyneaux, P.L.; Maher, T.M.; Noth, I.; et al. 50-gene risk profiles in peripheral blood predict COVID-19 outcomes: A retrospective, multicenter cohort study. EBioMedicine 2021, 69, 103439. [Google Scholar] [CrossRef]
- George, P.M.; Reed, A.; Desai, S.R.; Devaraj, A.; Faiez, T.S.; Laverty, S.; Kanwal, A.; Esneau, C.; Liu, M.K.; Kamal, F.; et al. A persistent neutrophil-associated immune signature characterizes post–COVID-19 pulmonary sequelae. Sci. Transl. Med. 2022, 14, eabo5795. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kadono, Y.; Takami, M.; Lee, J.; Lee, S.H.; Okada, F.; Kim, J.H.; Kobayashi, T.; Odgren, P.R.; Nakano, H.; et al. Osteoclast differentiation independent of the TRANCE–RANK–TRAF6 axis. J. Exp. Med. 2005, 202, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Lei, W.; Duan, R.; Li, Y.; Luo, L.; Boyce, B.F. RANKL cytokine enhances TNF-induced osteoclastogenesis independently of TNF receptor associated factor (TRAF) 6 by degrading TRAF3 in osteoclast precursors. J. Biol. Chem. 2017, 292, 10169–10179. [Google Scholar] [CrossRef] [PubMed]
- Ke, D.; Fu, X.; Xue, Y.; Wu, H.; Zhang, Y.; Chen, X.; Hou, J. IL-17A regulates the autophagic activity of osteoclast precursors through RANKL-JNK1 signaling during osteoclastogenesis in vitro. Biochem. Biophys. Res. Commun. 2018, 497, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Chung, Y.H.; Ahn, H.; Kim, H.; Rho, J.; Jeong, D. Selective regulation of MAPK signaling mediates RANKL-dependent osteoclast differentiation. Int. J. Biol. Sci. 2016, 12, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Franks, J.M.; Tanaka, Y.; Uehara, Y.; Read, D.F.; Williams, C.; Srivatsan, S.; Pitstick, L.B.; Nikolaidis, N.M.; Shaver, C.M.; et al. Pulmonary osteoclast-like cells in silica induced pulmonary fibrosis. Sci. Adv. 2024, 10, eadl4913. [Google Scholar] [CrossRef] [PubMed]
- Bondar, G.; Togashi, R.; Cadeiras, M.; Schaenman, J.; Cheng, R.K.; Masukawa, L.; Hai, J.; Bao, T.-M.; Chu, D.; Chang, E.; et al. Association between preoperative peripheral blood mononuclear cell gene expression profiles, early postoperative organ function recovery potential and long-term survival in advanced heart failure patients undergoing mechanical circulatory support. PLoS ONE 2017, 12, e0189420. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.C. Multi-dimensional COVID-19 short-and long-term outcome prediction algorithm. Expert Rev. Precis. Med. Drug Dev. 2020, 5, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yao, X.; Ma, S.; Ping, Y.; Fan, Y.; Sun, S.; He, Z.; Shi, Y.; Sun, L.; Xiao, S.; et al. A single-cell transcriptomic landscape of the lungs of patients with COVID-19. Nat. Cell Biol. 2021, 23, 1314–1328. [Google Scholar] [CrossRef]
- Sadanandam, A.; Bopp, T.; Dixit, S.; Knapp, D.J.H.F.; Emperumal, C.P.; Vergidis, P.; Rajalingam, K.; Melcher, A.; Kannan, N. A blood transcriptome-based analysis of disease progression, immune regulation, and symptoms in coronavirus-infected patients. Cell Death Discov. 2020, 6, 141. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Watanabe, S.; Verma, R.; Jablonski, R.P.; Chen, C.-I.; Cheresh, P.; Markov, N.S.; Reyfman, P.A.; McQuattie-Pimentel, A.C.; Sichizya, L.; et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir. J. 2020, 55, 1900646. [Google Scholar] [CrossRef] [PubMed]
- Dinnon, K.H., 3rd; Leist, S.R.; Okuda, K.; Dang, H.; Fritch, E.J.; Gully, K.L.; De la Cruz, G.; Evangelista, M.D.; Asakura, T.; Gilmore, R.C.; et al. A model of persistent post SARS-CoV-2 induced lung disease for target identification and testing of therapeutic strategies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Oldham, J.M.; Johnson, K.W.; Albers, G.J.; Calamita, E.; Mah, J.; Ghai, P.; Hewitt, R.J.; Maher, T.M.; Molyneaux, P.L.; Huang, M.; et al. Airway soluble CSF1R predicts progression in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2023, 9, 00690–02022. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Healthy Controls (n = 13) | No Pneumonia (n = 11) | Severe Pneumonia (n = 10) |
|---|---|---|---|
| Gender * (F/M) | 5/8 | 4/7 | 3/7 |
| Age ** | 45 (41.5–52) | 56 (41–65) | 47 (45.5–65.25) |
| Gene Symbol | Number of Genes Interacted | Names of Genes Interacted |
|---|---|---|
| ICAM1 | 6 | AREG, CSF1, IL1R1, IL1R2, PRKCD, TUBB4B |
| MARCKS | 6 | F3, FSCN1, PRKCD, RAB11FIP1, TUBB4B, ULK1 |
| TUBB4B | 6 | F3, GABARAPL1, ICAM1, MARCKS, SNAI1, ULK1 |
| HDAC5 | 5 | ATF3, DDIT3, NFKB2, NFKBIA, NFKBIE |
| NFKBIE | 5 | HDAC5, NFKB2, NFKBIA, NFKBIB, RELB |
| NFKB2 | 5 | HDAC5, NFKBIA, NFKBIB, NFKBIE, RELB |
| NFKBIA | 5 | HDAC5, NFKBIB, NFKBIB2, NFKBIE, RELB |
| ATF3 | 4 | DDIT3, FOSL1, HDAC5, JUND |
| F3 | 4 | ICAM1, MARCKS, PRKCD, TUBB4B |
| DDIT3 | 4 | ATF3, FOSL1, HDAC5, JUND |
| IL1R1 | 4 | CSF1, ICAM1, IL1R2, TOM1 |
| PRKCD | 4 | F3, FSCN1, ICAM1, MARCKS |
| AREG | 3 | ERG, HBEGF, ICAM1 |
| CSF1 | 3 | ICAM1, IL1R1, IL1R2 |
| FOSL1 | 3 | ATF3, DDIT3, JUND |
| FSCN1 | 3 | MARCKS, MCL1, PRKCD |
| IL1R2 | 3 | CSF1, ICAM1, IL1R1 |
| JUND | 3 | ATF3, DDIT3, FOSL1 |
| NFKBIB | 3 | NFKB2, NFKBIA, NFKBIE |
| RAB11FIP1 | 3 | IFITM3, MARCKS, TOM1 |
| RELB | 3 | NFKB2, NFKBIA, NFKBIE |
| TOM1 | 3 | IL1R1, IFITM3, RAB11FIP1 |
| ULK1 | 3 | GABARAPL1, MARCKS, TUBB4B |
| ERG | 2 | AREG HBEGF |
| GABARAPL1 | 2 | TUBB4B, ULK1 |
| HBEGF | 2 | AREG, ERG |
| IFITM3 | 2 | RAB11FIP1, TOM1 |
| MCL1 | 2 | FSCN1, USP2 |
| SNAI1 | 2 | TUBB4B, USP2 |
| USP2 | 2 | MCL1, SNAI1 |
| GADD45B | 2 | GADD45G, MAP2K3 |
| GADD45G | 2 | GADD45B, MAP2K3 |
| MAP2K3 | 2 | GADD45B, GADD45G |
| CSNK1D | 2 | HSPA5, NOP2 |
| HSPA5 | 2 | CSNK1D, NOP2 |
| NOP2 | 2 | CSNK1D, HSPA5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayalar, O.; Cetinkaya, P.D.; Eldem, V.; Argun Baris, S.; Kokturk, N.; Kuralay, S.C.; Rajabi, H.; Konyalilar, N.; Mortazavi, D.; Korkunc, S.K.; et al. Comparative Transcriptomic Analyses of Peripheral Blood Mononuclear Cells of COVID-19 Patients without Pneumonia and with Severe Pneumonia in the First Year of Follow-Up. Viruses 2024, 16, 1211. https://doi.org/10.3390/v16081211
Kayalar O, Cetinkaya PD, Eldem V, Argun Baris S, Kokturk N, Kuralay SC, Rajabi H, Konyalilar N, Mortazavi D, Korkunc SK, et al. Comparative Transcriptomic Analyses of Peripheral Blood Mononuclear Cells of COVID-19 Patients without Pneumonia and with Severe Pneumonia in the First Year of Follow-Up. Viruses. 2024; 16(8):1211. https://doi.org/10.3390/v16081211
Chicago/Turabian StyleKayalar, Ozgecan, Pelin Duru Cetinkaya, Vahap Eldem, Serap Argun Baris, Nurdan Kokturk, Selim Can Kuralay, Hadi Rajabi, Nur Konyalilar, Deniz Mortazavi, Seval Kubra Korkunc, and et al. 2024. "Comparative Transcriptomic Analyses of Peripheral Blood Mononuclear Cells of COVID-19 Patients without Pneumonia and with Severe Pneumonia in the First Year of Follow-Up" Viruses 16, no. 8: 1211. https://doi.org/10.3390/v16081211