
Poly(rC)-Binding Protein 2 Does Not Directly Participate in HCV Translation or Replication, but Rather Modulates Genome Packaging


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Viral RNAs
2.3. Generation of Infectious HCV Stocks
2.4. Focus-Forming Unit (FFU) Assays
2.5. MicroRNAs and siRNA Sequences
2.6. HCV and VSV Pseudoparticles (HCVpp and VSVpp)
2.7. Infections
2.8. Electroporations
2.9. Western Blot Analysis
2.10. RNA Isolation and Northern Blot Analysis
2.11. RT-qPCR Analysis
2.12. Luciferase Assays
2.13. Data Analysis
3. Results
3.1. PCBP2 Is Required for Optimal HCVcc Accumulation in Cell Culture
3.2. PCBP2 Knockdown Has No Effect on HCV Entry
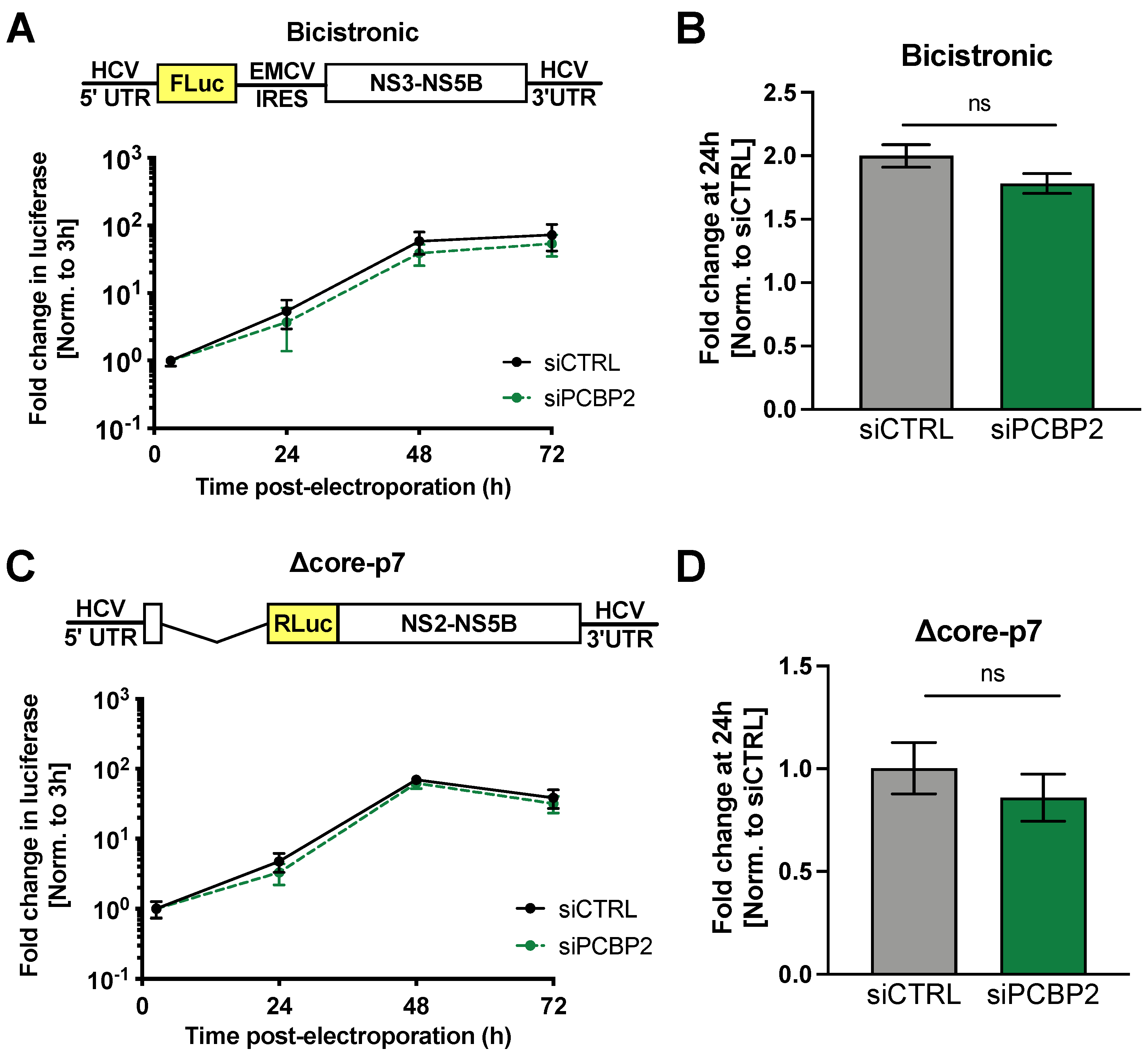
3.3. PCBP2 Knockdown Has No Effect on HCV IRES-Mediated Translation or Genome Stability
3.4. PCBP2 Knockdown Has No Effect on Viral RNA Replication
3.5. PCBP2 Sensitivity Maps to the HCV Core-Coding Region
3.6. PCBP2 Modulates Viral Genome Packaging
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Lancaster, A.M.; Jan, E.; Sarnow, P. Initiation factor-independent translation mediated by the hepatitis C virus internal ribosome entry site. RNA 2006, 12, 894–902. [Google Scholar] [CrossRef]
- Tsukiyama-Kohara, K.; Iizuka, N.; Kohara, M.; Nomoto, A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992, 66, 1476–1483. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef]
- Jones, D.M.; Atoom, A.M.; Zhang, X.; Kottilil, S.; Russell, R.S. A genetic interaction between the core and NS3 proteins of hepatitis C virus is essential for production of infectious virus. J. Virol. 2011, 85, 12351–12361. [Google Scholar] [CrossRef]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; et al. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef]
- Roder, A.E.; Vazquez, C.; Horner, S.M. The acidic domain of the hepatitis C virus NS4A protein is required for viral assembly and envelopment through interactions with the viral E1 glycoprotein. PLoS Pathog. 2019, 15, e1007163. [Google Scholar] [CrossRef]
- Makeyev, A.V.; Chkheidze, A.N.; Liebhaber, S.A. A set of highly conserved RNA-binding proteins, alphaCP-1 and alphaCP-2, implicated in mRNA stabilization, are coexpressed from an intronless gene and its intron-containing paralog. J. Biol. Chem. 1999, 274, 24849–24857. [Google Scholar] [CrossRef]
- Matunis, M.J.; Michael, W.M.; Dreyfuss, G. Characterization and primary structure of the poly(C)-binding heterogeneous nuclear ribonucleoprotein complex K protein. Mol. Cell Biol. 1992, 12, 164–171. [Google Scholar]
- Waggoner, S.A.; Liebhaber, S.A. Identification of mRNAs associated with alphaCP2-containing RNP complexes. Mol. Cell Biol. 2003, 23, 7055–7067. [Google Scholar] [CrossRef]
- Gonzalez-Moro, I.; Olazagoitia-Garmendia, A.; Colli, M.L.; Cobo-Vuilleumier, N.; Postler, T.S.; Marselli, L.; Marchetti, P.; Ghosh, S.; Gauthier, B.R.; Eizirik, D.L.; et al. The T1D-associated lncRNA Lnc13 modulates human pancreatic beta cell inflammation by allele-specific stabilization of STAT1 mRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 9022–9031. [Google Scholar] [CrossRef]
- Wang, Z.; Day, N.; Trifillis, P.; Kiledjian, M. An mRNA stability complex functions with poly(A)-binding protein to stabilize mRNA in vitro. Mol. Cell Biol. 1999, 19, 4552–4560. [Google Scholar] [CrossRef]
- Beura, L.K.; Dinh, P.X.; Osorio, F.A.; Pattnaik, A.K. Cellular poly(c) binding proteins 1 and 2 interact with porcine reproductive and respiratory syndrome virus nonstructural protein 1beta and support viral replication. J. Virol. 2011, 85, 12939–12949. [Google Scholar] [CrossRef]
- Collier, B.; Goobar-Larsson, L.; Sokolowski, M.; Schwartz, S. Translational inhibition in vitro of human papillomavirus type 16 L2 mRNA mediated through interaction with heterogenous ribonucleoprotein K and poly(rC)-binding proteins 1 and 2. J. Biol. Chem. 1998, 273, 22648–22656. [Google Scholar] [CrossRef]
- Graff, J.; Cha, J.; Blyn, L.B.; Ehrenfeld, E. Interaction of poly(rC) binding protein 2 with the 5’ noncoding region of hepatitis A virus RNA and its effects on translation. J. Virol. 1998, 72, 9668–9675. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Li, M.L.; Huang, P.N.; Chien, K.Y.; Horng, J.T.; Shih, S.R. Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5’ untranslated region and participates in virus replication. J. Gen. Virol. 2008, 89 Pt 10, 2540–2549. [Google Scholar] [CrossRef]
- Lopez-Manriquez, E.; Vashist, S.; Urena, L.; Goodfellow, I.; Chavez, P.; Mora-Heredia, J.E.; Cancio-Lonches, C.; Garrido, E.; Gutierrez-Escolano, A.L. Norovirus genome circularization and efficient replication are facilitated by binding of PCBP2 and hnRNP A1. J. Virol. 2013, 87, 11371–11387. [Google Scholar] [CrossRef] [PubMed]
- Palusa, S.; Ndaluka, C.; Bowen, R.A.; Wilusz, C.J.; Wilusz, J. The 3’ untranslated region of the rabies virus glycoprotein mRNA specifically interacts with cellular PCBP2 protein and promotes transcript stability. PLoS ONE 2012, 7, e33561. [Google Scholar] [CrossRef]
- Pingale, K.D.; Kanade, G.D.; Karpe, Y.A. Heterogeneous Nuclear Ribonucleoproteins Participate in Hepatitis E Virus Replication. J. Mol. Biol. 2020, 432, 2369–2387. [Google Scholar] [CrossRef] [PubMed]
- Woolaway, K.; Asai, K.; Emili, A.; Cochrane, A. hnRNP E1 and E2 have distinct roles in modulating HIV-1 gene expression. Retrovirology 2007, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Ihle, Y.; Seitz, S.; Gundel, U.; Wutzler, P.; Gorlach, M. Poly(rC)-binding protein 2 interacts with the oligo(rC) tract of coxsackievirus B3. Biochem. Biophys. Res. Commun. 2008, 366, 917–921. [Google Scholar] [CrossRef]
- Bedard, K.M.; Walter, B.L.; Semler, B.L. Multimerization of poly(rC) binding protein 2 is required for translation initiation mediated by a viral IRES. RNA 2004, 10, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Blyn, L.B.; Towner, J.S.; Semler, B.L.; Ehrenfeld, E. Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J. Virol. 1997, 71, 6243–6246. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.J.; Daijogo, S.; Semler, B.L. Inhibition of poliovirus-induced cleavage of cellular protein PCBP2 reduces the levels of viral RNA replication. J. Virol. 2014, 88, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Gamarnik, A.V.; Andino, R. Two functional complexes formed by KH domain containing proteins with the 5’ noncoding region of poliovirus RNA. RNA 1997, 3, 882–892. [Google Scholar] [PubMed]
- Parsley, T.B.; Towner, J.S.; Blyn, L.B.; Ehrenfeld, E.; Semler, B.L. Poly (rC) binding protein 2 forms a ternary complex with the 5’-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA 1997, 3, 1124–1134. [Google Scholar] [PubMed]
- Perera, R.; Daijogo, S.; Walter, B.L.; Nguyen, J.H.; Semler, B.L. Cellular protein modification by poliovirus: The two faces of poly(rC)-binding protein. J. Virol. 2007, 81, 8919–8932. [Google Scholar] [CrossRef]
- Randall, G.; Panis, M.; Cooper, J.D.; Tellinghuisen, T.L.; Sukhodolets, K.E.; Pfeffer, S.; Landthaler, M.; Landgraf, P.; Kan, S.; Lindenbach, B.D.; et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. USA 2007, 104, 12884–12889. [Google Scholar] [CrossRef]
- Choi, K.; Kim, J.H.; Li, X.; Paek, K.Y.; Ha, S.H.; Ryu, S.H.; Wimmer, E.; Jang, S.K. Identification of cellular proteins enhancing activities of internal ribosomal entry sites by competition with oligodeoxynucleotides. Nucleic Acids Res. 2004, 32, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Fontanes, V.; Raychaudhuri, S.; Dasgupta, A. A cell-permeable peptide inhibits hepatitis C virus replication by sequestering IRES transacting factors. Virology 2009, 394, 82–90. [Google Scholar] [CrossRef]
- Fukushi, S.; Okada, M.; Kageyama, T.; Hoshino, F.B.; Nagai, K.; Katayama, K. Interaction of poly(rC)-binding protein 2 with the 5’-terminal stem loop of the hepatitis C-virus genome. Virus Res. 2001, 73, 67–79. [Google Scholar] [CrossRef]
- Masaki, T.; Arend, K.C.; Li, Y.; Yamane, D.; McGivern, D.R.; Kato, T.; Wakita, T.; Moorman, N.J.; Lemon, S.M. miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe 2015, 17, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, A.B.; Racaniello, V.R. Hepatitis C virus internal ribosome entry site-dependent translation in Saccharomyces cerevisiae is independent of polypyrimidine tract-binding protein, poly(rC)-binding protein 2, and La protein. J. Virol. 2005, 79, 10126–10137. [Google Scholar] [CrossRef]
- Shirasaki, T.; Honda, M.; Mizuno, H.; Shimakami, T.; Okada, H.; Sakai, Y.; Murakami, S.; Wakita, T.; Kaneko, S. La protein required for internal ribosome entry site-directed translation is a potential therapeutic target for hepatitis C virus replication. J. Infect. Dis. 2010, 202, 75–85. [Google Scholar] [CrossRef]
- Tingting, P.; Caiyun, F.; Zhigang, Y.; Pengyuan, Y.; Zhenghong, Y. Subproteomic analysis of the cellular proteins associated with the 3’ untranslated region of the hepatitis C virus genome in human liver cells. Biochem. Biophys. Res. Commun. 2006, 347, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jeng, K.S.; Lai, M.M. Poly(C)-binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5’ untranslated region and directs HCV RNA replication through circularizing the viral genome. J. Virol. 2011, 85, 7954–7964. [Google Scholar] [CrossRef]
- Scott, S.; Li, Y.; Bermek, O.; Griffith, J.D.; Lemon, S.M.; Choi, K.H. Binding of microRNA-122 to the hepatitis C virus 5’ untranslated region modifies interactions with poly(C) binding protein 2 and the NS5B viral polymerase. Nucleic Acids Res. 2023, 51, 12397–12413. [Google Scholar] [CrossRef]
- Flynn, R.A.; Martin, L.; Spitale, R.C.; Do, B.T.; Sagan, S.M.; Zarnegar, B.; Qu, K.; Khavari, P.A.; Quake, S.R.; Sarnow, P.; et al. Dissecting noncoding and pathogen RNA-protein interactomes. RNA 2015, 21, 135–143. [Google Scholar] [CrossRef]
- Gontarek, R.R.; Gutshall, L.L.; Herold, K.M.; Tsai, J.; Sathe, G.M.; Mao, J.; Prescott, C.; Del Vecchio, A.M. hnRNP C and polypyrimidine tract-binding protein specifically interact with the pyrimidine-rich region within the 3’NTR of the HCV RNA genome. Nucleic Acids Res. 1999, 27, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Sp Ngberg, K.; Schwartz, S. Poly(C)-binding protein interacts with the hepatitis C virus 5’ untranslated region. J. Gen. Virol. 1999, 80 Pt 6, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Svitkin, Y.V.; Imataka, H.; Khaleghpour, K.; Kahvejian, A.; Liebig, H.D.; Sonenberg, N. Poly(A)-binding protein interaction with elF4G stimulates picornavirus IRES-dependent translation. RNA 2001, 7, 1743–1752. [Google Scholar]
- Amador-Canizares, Y.; Bernier, A.; Wilson, J.A.; Sagan, S.M. miR-122 does not impact recognition of the HCV genome by innate sensors of RNA but rather protects the 5’ end from the cellular pyrophosphatases, DOM3Z and DUSP11. Nucleic Acids Res. 2018, 46, 5139–5158. [Google Scholar] [CrossRef]
- Russell, R.S.; Meunier, J.C.; Takikawa, S.; Faulk, K.; Engle, R.E.; Bukh, J.; Purcell, R.H.; Emerson, S.U. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2008, 105, 4370–4375. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef]
- Panigrahi, M.; Thibault, P.A.; Wilson, J.A. MicroRNA 122 Affects both the Initiation and the Maintenance of Hepatitis C Virus Infections. J. Virol. 2022, 96, e0190321. [Google Scholar] [CrossRef]
- Cousineau, S.E.; Rheault, M.; Sagan, S.M. Poly(rC)-Binding Protein 1 Limits Hepatitis C Virus Virion Assembly and Secretion. Viruses 2022, 14, 291. [Google Scholar] [CrossRef]
- Machlin, E.S.; Sarnow, P.; Sagan, S.M. Masking the 5’ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. USA 2011, 108, 3193–3198. [Google Scholar] [CrossRef]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 2003, 100, 7271–7276. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.R.; Mitchell, S.A.; Spriggs, K.A.; Ostrowski, J.; Bomsztyk, K.; Ostarek, D.; Willis, A.E. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene 2003, 22, 8012–8020. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Kim, I.; Choi, K.; Choi, J.H.; Kim, E.; Lee, H.Y.; Park, J.; Kim Yoon, S. Poly(rC) binding protein 2 acts as a negative regulator of IRES-mediated translation of Hr mRNA. Exp. Mol. Med. 2018, 50, e441. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.L.; Nguyen, J.H.; Ehrenfeld, E.; Semler, B.L. Differential utilization of poly(rC) binding protein 2 in translation directed by picornavirus IRES elements. RNA 1999, 5, 1570–1585. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.; Gebert, L.F.R.; Camargo, C.; MacRae, I.J.; Sagan, S.M. miR-122-based therapies select for three distinct resistance mechanisms based on alterations in RNA structure. Proc. Natl. Acad. Sci. USA 2021, 118, e2103671118. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.; Gebert, L.F.R.; Gan, H.H.; Camacho, E.; Gunsalus, K.C.; MacRae, I.J.; Sagan, S.M. miR-122 and Ago interactions with the HCV genome alter the structure of the viral 5’ terminus. Nucleic Acids Res. 2019, 47, 5307–5324. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuttler, C.G.; Fehr, C.; Junemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Israelow, B.; Mullokandov, G.; Agudo, J.; Sourisseau, M.; Bashir, A.; Maldonado, A.Y.; Dar, A.C.; Brown, B.D.; Evans, M.J. Hepatitis C virus genetics affects miR-122 requirements and response to miR-122 inhibitors. Nat. Commun. 2014, 5, 5408. [Google Scholar] [CrossRef]
- Jangra, R.K.; Yi, M.; Lemon, S.M. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol. 2010, 84, 6615–6625. [Google Scholar] [CrossRef]
- Jopling, C.L.; Schutz, S.; Sarnow, P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 2008, 4, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Masaki, T.; Yamane, D.; McGivern, D.R.; Lemon, S.M. Competing and noncompeting activities of miR-122 and the 5’ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2013, 110, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, M.; Palmer, M.A.; Wilson, J.A. Enhanced Virus Translation Enables miR-122-Independent Hepatitis C Virus Propagation. J. Virol. 2023, 97, e0085821. [Google Scholar] [CrossRef] [PubMed]
- Rheault, M.; Cousineau, S.E.; Fox, D.R.; Abram, Q.H.; Sagan, S.M. Elucidating the Distinct Contrubutions of miR-122 in the HCV Life Cycle Reveals Insights into Virion Assembly. Nucleic Acids Res. 2023, 51, 2447–2463. [Google Scholar] [CrossRef] [PubMed]
- Schult, P.; Roth, H.; Adams, R.L.; Mas, C.; Imbert, L.; Orlik, C.; Ruggieri, A.; Pyle, A.M.; Lohmann, V. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 2018, 9, 2613. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Yamane, D.; Welsch, C.; Hensley, L.; Jangra, R.K.; Lemon, S.M. Base pairing between hepatitis C virus RNA and microRNA 122 3’ of its seed sequence is essential for genome stabilization and production of infectious virus. J. Virol. 2012, 86, 7372–7383. [Google Scholar] [CrossRef] [PubMed]
- Thibault, P.A.; Huys, A.; Amador-Canizares, Y.; Gailius, J.E.; Pinel, D.E.; Wilson, J.A. Regulation of Hepatitis C Virus Genome Replication by Xrn1 and MicroRNA-122 Binding to Individual Sites in the 5’ Untranslated Region. J. Virol. 2015, 89, 6294–6311. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; Law, M.; MacRae, I.J. A structured RNA motif locks Argonaute2:miR-122 onto the 5’ end of the HCV genome. Nat. Commun. 2021, 12, 6836. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef] [PubMed]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V., Jr.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Poenisch, M.; Metz, P.; Blankenburg, H.; Ruggieri, A.; Lee, J.Y.; Rupp, D.; Rebhan, I.; Diederich, K.; Kaderali, L.; Domingues, F.S.; et al. Identification of HNRNPK as regulator of hepatitis C virus particle production. PLoS Pathog. 2015, 11, e1004573. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Evans, M.J.; Gosert, R.; Yuan, Z.; Blum, H.E.; Goff, S.P.; Lindenbach, B.D.; Rice, C.M. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J. Virol. 2004, 78, 7400–7409. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.; Appel, N.; Koutsoudakis, G.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 2007, 81, 4591–4603. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar] [PubMed]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Ando, T.; Suzuki, R.; Matsuda, M.; Nakashima, K.; Ito, M.; Omatsu, T.; Oba, M.; Ochiai, H.; Kato, T.; et al. Involvement of the 3’ Untranslated Region in Encapsidation of the Hepatitis C Virus. PLoS Pathog. 2016, 12, e1005441. [Google Scholar] [CrossRef] [PubMed]
- Shimoike, T.; Mimori, S.; Tani, H.; Matsuura, Y.; Miyamura, T. Interaction of hepatitis C virus core protein with viral sense RNA and suppression of its translation. J. Virol. 1999, 73, 9718–9725. [Google Scholar] [CrossRef]
- Stewart, H.; Bingham, R.J.; White, S.J.; Dykeman, E.C.; Zothner, C.; Tuplin, A.K.; Stockley, P.G.; Twarock, R.; Harris, M. Identification of novel RNA secondary structures within the hepatitis C virus genome reveals a cooperative involvement in genome packaging. Sci. Rep. 2016, 6, 22952. [Google Scholar] [CrossRef]
- Germain, M.A.; Chatel-Chaix, L.; Gagne, B.; Bonneil, E.; Thibault, P.; Pradezynski, F.; de Chassey, B.; Meyniel-Schicklin, L.; Lotteau, V.; Baril, M.; et al. Elucidating novel hepatitis C virus-host interactions using combined mass spectrometry and functional genomics approaches. Mol. Cell Proteomics 2014, 13, 184–203. [Google Scholar] [CrossRef] [PubMed]
- Zayas, M.; Long, G.; Madan, V.; Bartenschlager, R. Coordination of Hepatitis C Virus Assembly by Distinct Regulatory Regions in Nonstructural Protein 5A. PLoS Pathog. 2016, 12, e1005376. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Belyaeva, T.; Stonehouse, N.J.; Pearson, A.R.; Harris, M. All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J. Virol. 2010, 84, 9267–9277. [Google Scholar] [CrossRef]
- Hsieh, T.Y.; Matsumoto, M.; Chou, H.C.; Schneider, R.; Hwang, S.B.; Lee, A.S.; Lai, M.M. Hepatitis C virus core protein interacts with heterogeneous nuclear ribonucleoprotein K. J. Biol. Chem. 1998, 273, 17651–17659. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cousineau, S.E.; Camargo, C.; Sagan, S.M. Poly(rC)-Binding Protein 2 Does Not Directly Participate in HCV Translation or Replication, but Rather Modulates Genome Packaging. Viruses 2024, 16, 1220. https://doi.org/10.3390/v16081220
Cousineau SE, Camargo C, Sagan SM. Poly(rC)-Binding Protein 2 Does Not Directly Participate in HCV Translation or Replication, but Rather Modulates Genome Packaging. Viruses. 2024; 16(8):1220. https://doi.org/10.3390/v16081220
Chicago/Turabian StyleCousineau, Sophie E., Carolina Camargo, and Selena M. Sagan. 2024. "Poly(rC)-Binding Protein 2 Does Not Directly Participate in HCV Translation or Replication, but Rather Modulates Genome Packaging" Viruses 16, no. 8: 1220. https://doi.org/10.3390/v16081220