

Novel Pyrazino[1,2-a]indole-1,3(2H,4H)-dione Derivatives Targeting the Replication of Flaviviridae Viruses: Structural and Mechanistic Insights

 , , , and
, , , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cells and Viral Constructs
2.2. In Vitro Transcription
2.3. Transfection with In Vitro Transcribed RNA
2.4. Cell-Based Antiviral and Cytotoxicity Assays
2.5. Chemicals
2.6. Luciferase and Bradford Assays
2.7. Measurement of Intracellular ATP Levels
2.8. Indirect Immunofluorescence
2.9. Gel Electrophoresis and Western Blot Analysis
2.10. Total RNA Extraction and Quantification of Viral Replicons
2.11. Selection of Drug-Resistance Mutations
2.12. Next Generation Sequencing
2.13. Eradication of HCV RNA
2.14. Construction of Viral Plasmids Containing NS5B Mutant T181I
2.15. Statistical Analysis
2.16. In Silico Studies
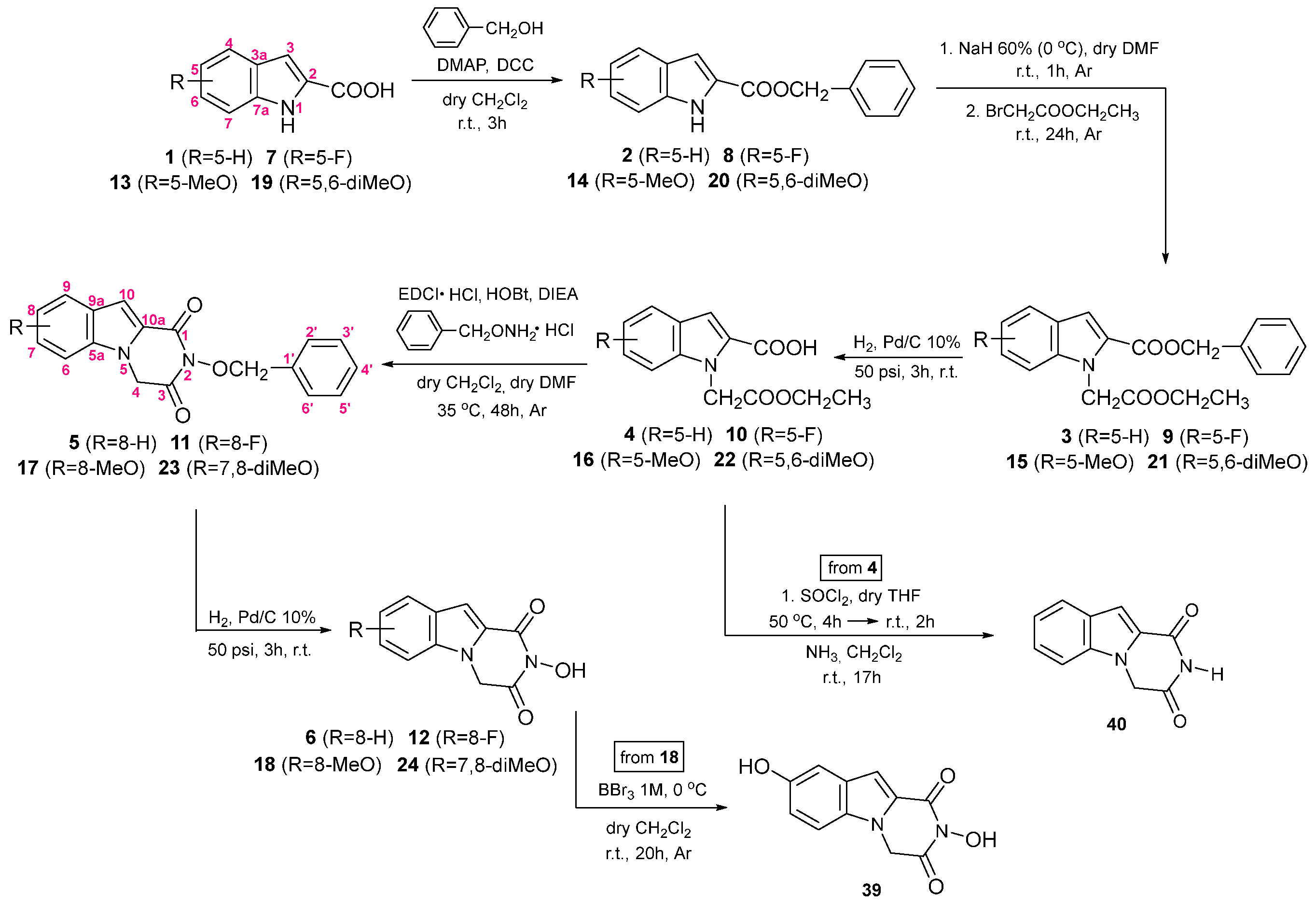
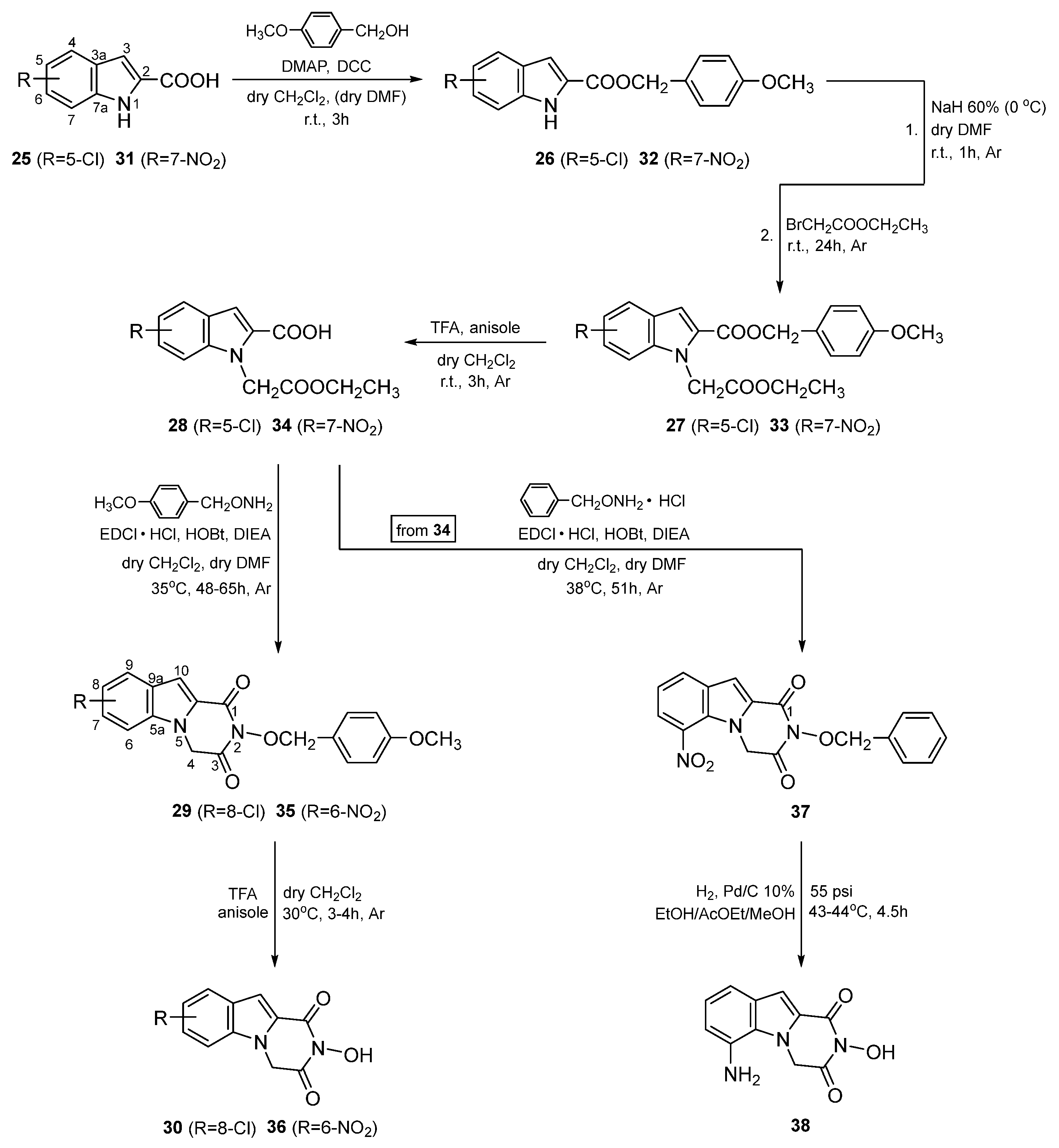
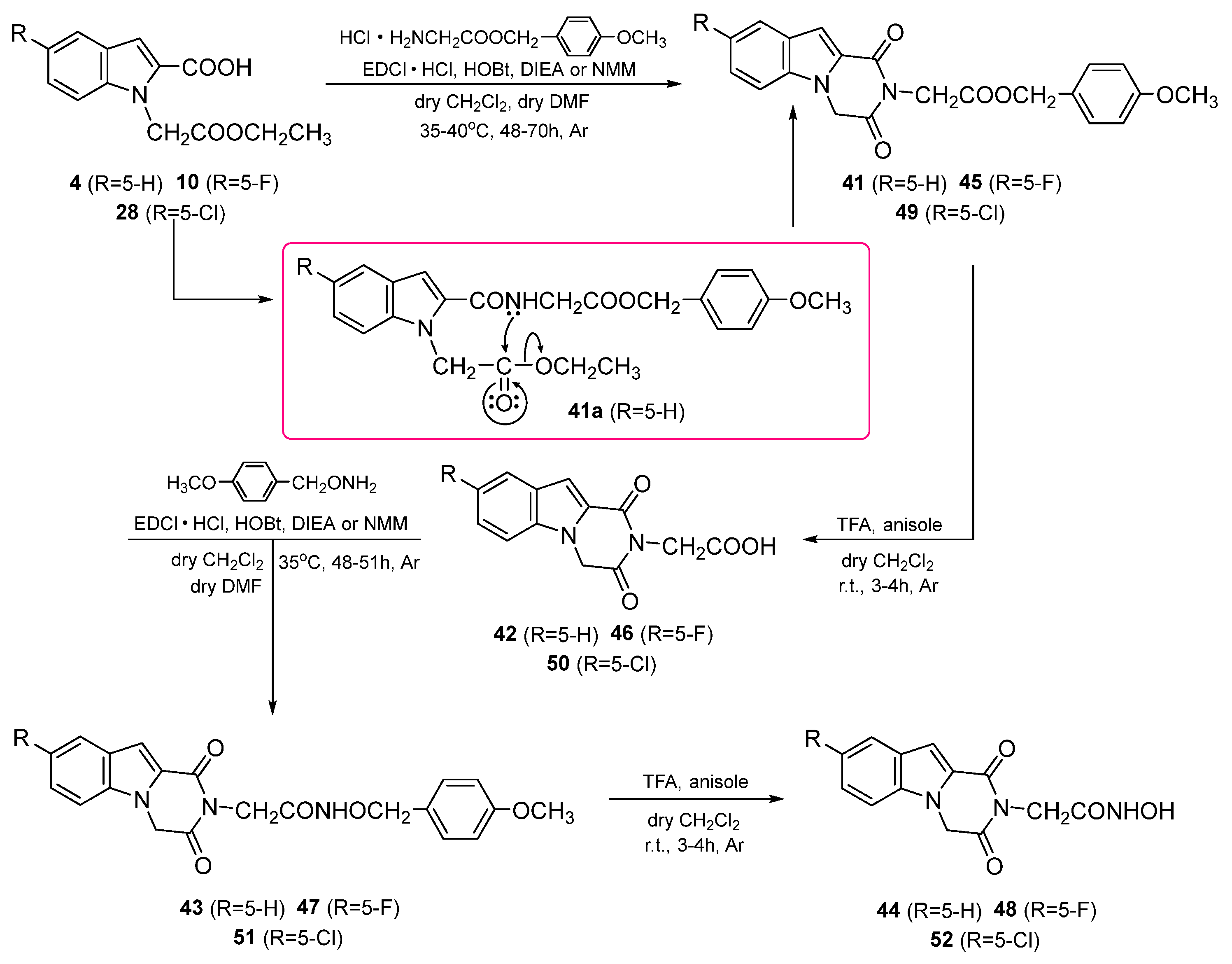
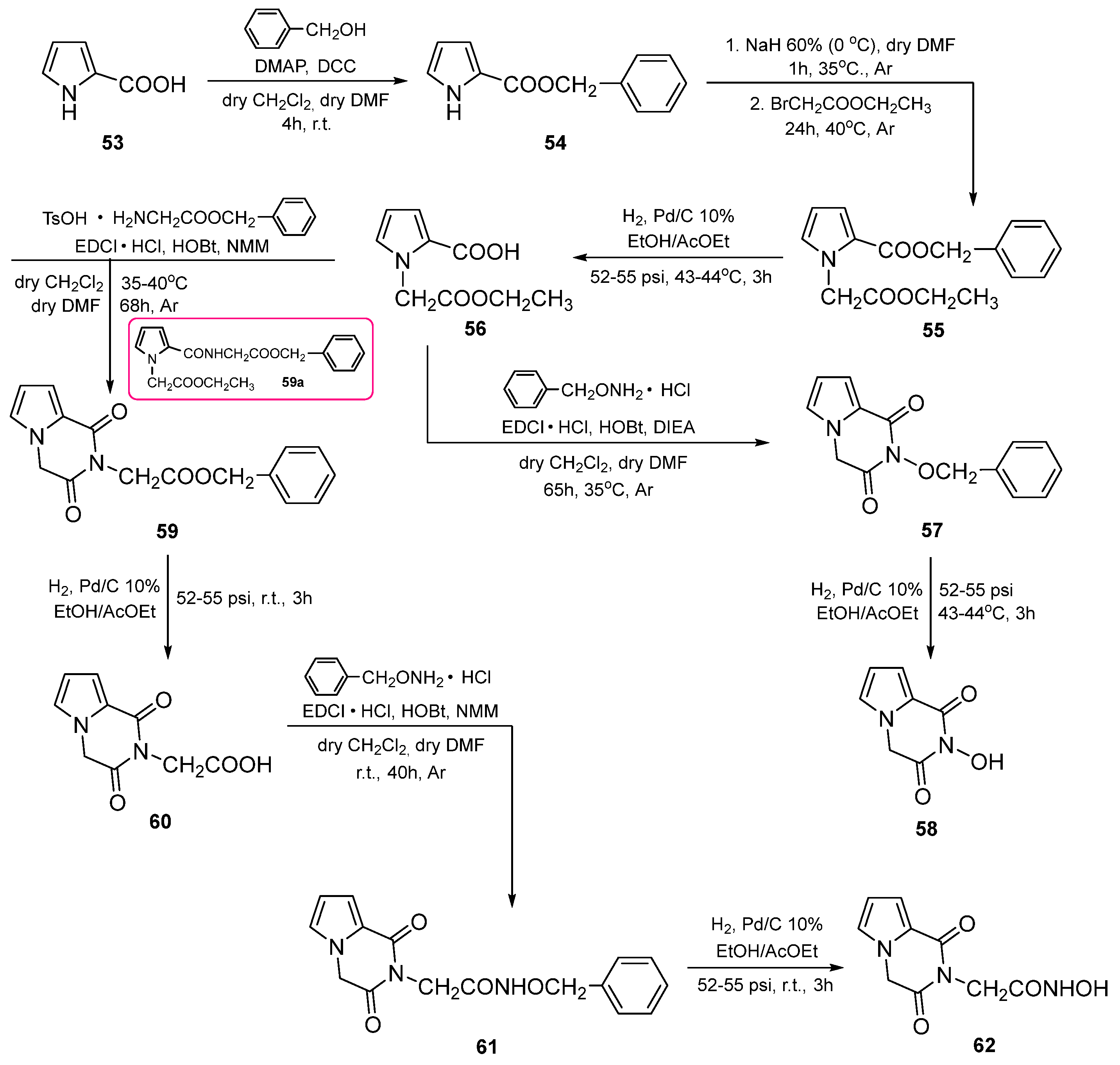
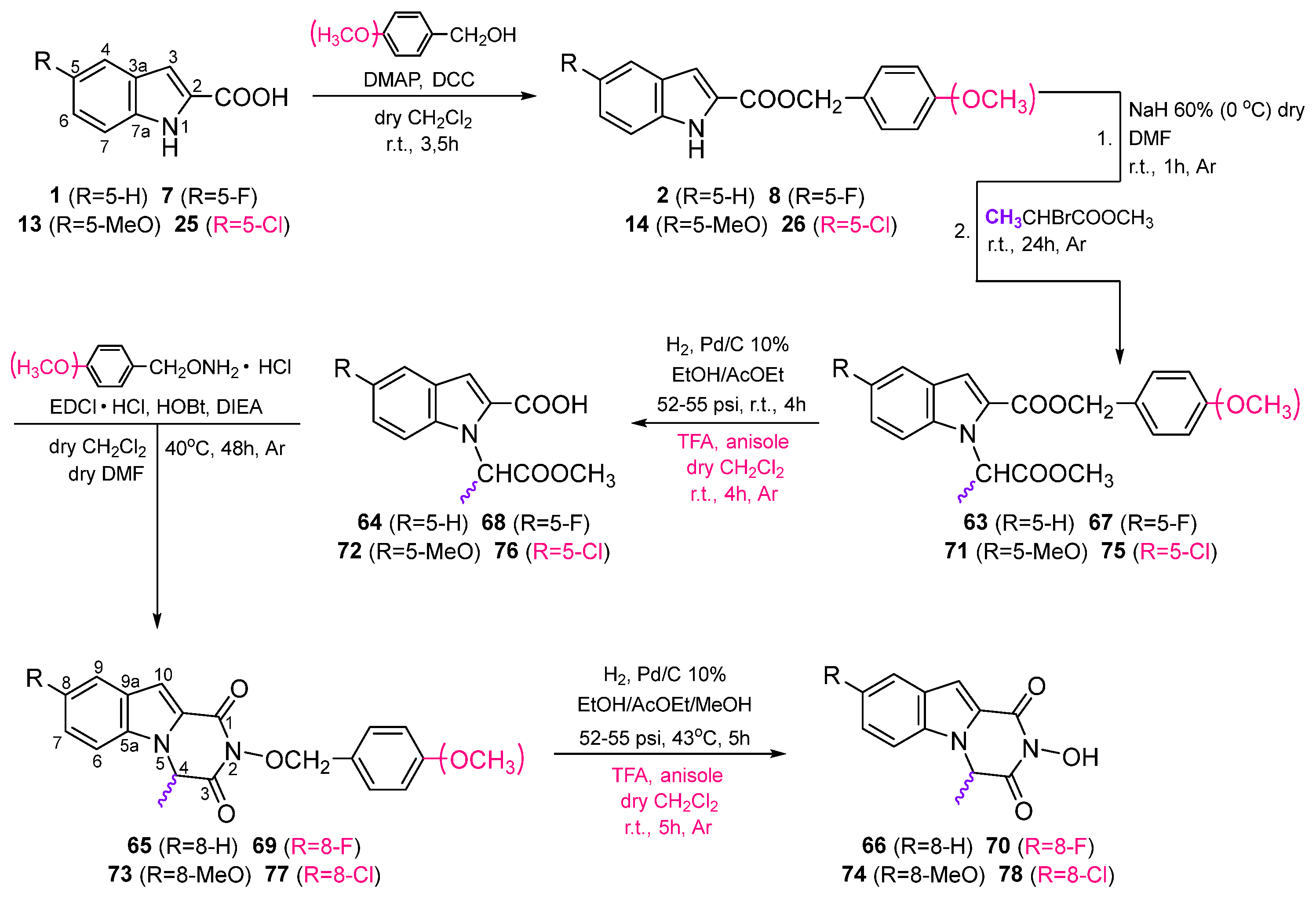
3. Chemistry
4. Results
4.1. Biological Evaluation
4.1.1. Screening of Compounds in HCV Genotype 1b Replicon System
4.1.2. Comparing the Anti-HCV Intergenotypic Activity of Compounds 36 to 30
4.1.3. Screening of Compounds against DENV and YFV Replicon Systems
4.2. Further Characterization and Mechanism of Action Studies
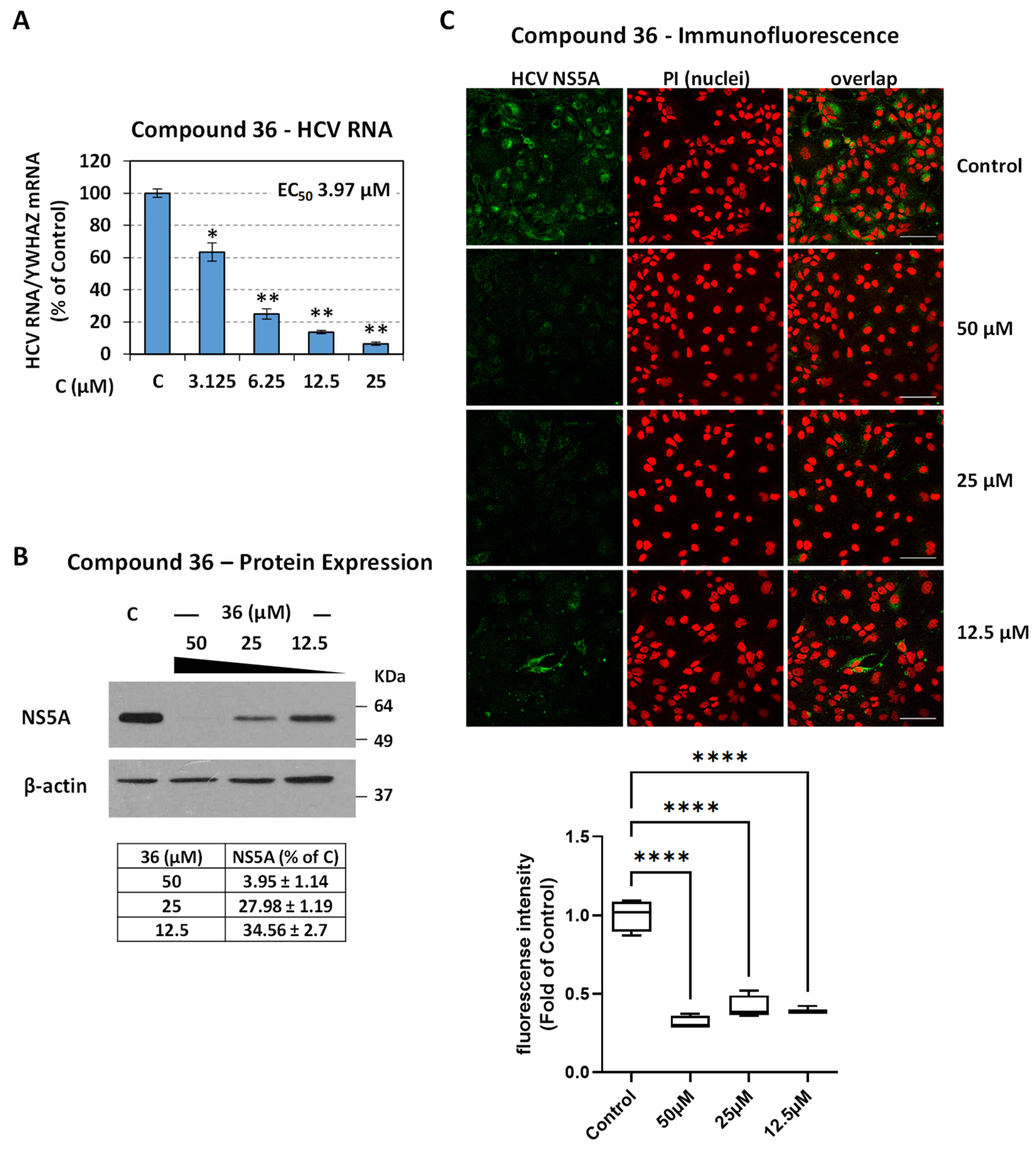
4.2.1. Validation of Compound Activity with Additional Assays
4.2.2. Drug–Drug Interaction Studies of Pyrazino[1,2-a]indole-1,3(2H,4H)-dione Derivatives with Approved HCV Inhibitors
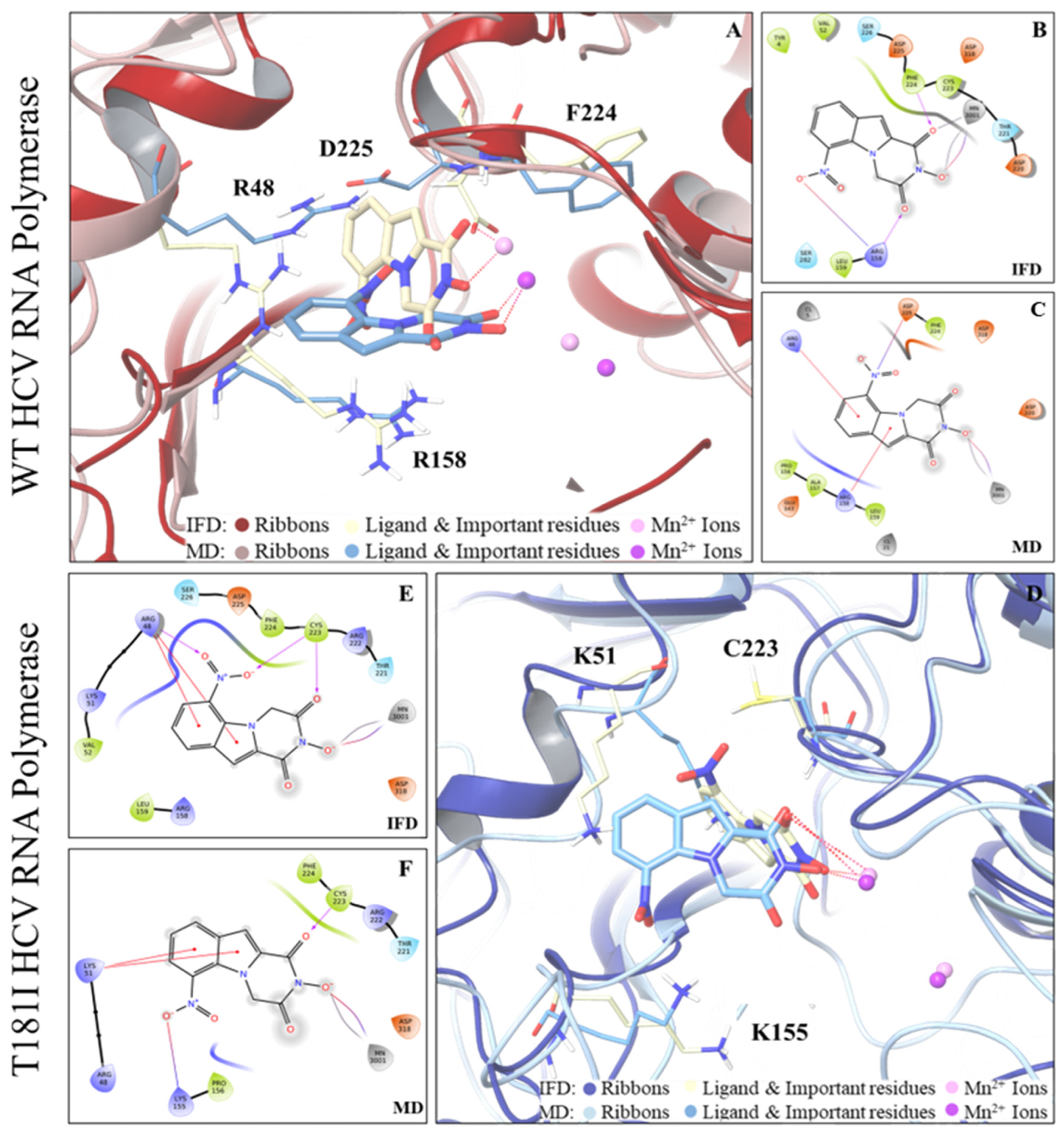
4.2.3. Compound 36 Shows a High Barrier to Resistance and Targets RdRp Polymerase
4.3. In Silico Studies
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hepatitis C. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 20 February 2024).
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Dengue and Severe Dengue. Available online: https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 20 February 2024).
- Yellow Fever. Available online: https://www.who.int/news-room/fact-sheets/detail/yellow-fever (accessed on 20 February 2024).
- Stanaway, J.D.; Shepard, D.S.; Undurraga, E.A.; Halasa, Y.A.; Coffeng, L.E.; Brady, O.J.; Hay, S.I.; Bedi, N.; Bensenor, I.M.; Castañeda-Orjuela, C.A.; et al. The Global Burden of Dengue: An Analysis from the Global Burden of Disease Study 2013. Lancet Infect. Dis. 2016, 16, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Vasconcelos, P.F.C. Yellow Fever. J. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The Continued Threat of Emerging Flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Heinz, F.X.; Stiasny, K. Flaviviruses and Flavivirus Vaccines. Vaccine 2012, 30, 4301–4306. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The Molecular and Structural Basis of Advanced Antiviral Therapy for Hepatitis C Virus Infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Miller, S. Molecular Aspects of Dengue Virus Replication. Future Microbiol. 2008, 3, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Pierson, T.C. Molecular Insight into Dengue Virus Pathogenesis and Its Implications for Disease Control. Cell 2015, 162, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Ploss, A. Yellow Fever Virus: Knowledge Gaps Impeding the Fight against an Old Foe. Trends Microbiol. 2018, 26, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of Hepatitis C Virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue Virus- and Hepatitis C Virus-Induced Replication and Assembly Compartments: The Enemy Inside—Caught in the Web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Peters, M.G.; Stefan, Z. New Therapies for Hepatitis C Virus Infection. Clin. Infect. Dis. 2009, 48, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; De Clercq, E. Current Therapy for Chronic Hepatitis C: The Role of Direct-Acting Antivirals. Antiviral Res. 2017, 142, 83–122. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, V.; Banzi, R.; Cariani, E.; Chester, J.; Villa, E.; D’Amico, R.; Bertele, V.; Trenti, T. New Direct-Acting Antivirals for the Treatment of Patients with Hepatitis C Virus Infection: A Systematic Review of Randomized Controlled Trials. J. Clin. Exp. Hepatol. 2019, 9, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Li, J.; Zhang, Y.; Ji, S.; Li, Y.; Zhao, Y.; Li, B.; Li, W.; Quan, M.; Duan, Y.; et al. Real-World Effectiveness of Direct-Acting Antiviral Regimens against Hepatitis C Virus (HCV) Genotype 3 Infection: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2021, 23, 100268. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Marcellin, P.; Schinazi, R.F. Treatment of Hepatitis C Virus Infection with Direct-Acting Antiviral Agents: 100% Cure? Liver Int. 2018, 38, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, L.; Ceccherini-Silberstein, F.; Van Laethem, K.; Li, G.; Vandamme, A.-M.; Rockstroh, J.K. Impact of HCV Genotype on Treatment Regimens and Drug Resistance: A Snapshot in Time. Rev. Med. Virol. 2016, 26, 408–434. [Google Scholar] [CrossRef] [PubMed]
- Barth, H. Hepatitis C Virus: Is It Time to Say Goodbye yet? Perspectives and Challenges for the next Decade. World J. Hepatol. 2015, 7, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Julander, J.G. Experimental Therapies for Yellow Fever. Antiviral Res. 2013, 97, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Low, J.G.H.; Ooi, E.E.; Vasudevan, S.G. Current Status of Dengue Therapeutics Research and Development. J. Infect. Dis. 2017, 215, S96–S102. [Google Scholar] [CrossRef] [PubMed]
- Caillet-Saguy, C.; Lim, S.P.; Shi, P.-Y.; Lescar, J.; Bressanelli, S. Polymerases of Hepatitis C Viruses and Flaviviruses: Structural and Mechanistic Insights and Drug Development. Antiviral Res. 2014, 105, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Sesmero, E.; Thorpe, I.F. Using the Hepatitis C Virus RNA-Dependent RNA Polymerase as a Model to Understand Viral Polymerase Structure, Function and Dynamics. Viruses 2015, 7, 3974–3994. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, W.; Gong, P. A Structural Overview of RNA-Dependent RNA Polymerases from the Flaviviridae Family. Int. J. Mol. Sci. 2015, 16, 12943–12957. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulou, E.; Pardali, V.; Zoidis, G. Metal-Chelating Agents against Viruses and Parasites. Future Med. Chem. 2018, 10, 1283–1285. [Google Scholar] [CrossRef] [PubMed]
- Moianos, D.; Prifti, G.-M.; Makri, M.; Zoidis, G. Targeting Metalloenzymes: The “Achilles’ Heel” of Viruses and Parasites. Pharmaceuticals 2023, 16, 901. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.C.; Lomonosova, E.; Patel, J.A.; Li, Q.; Villa, J.A.; Gupta, A.K.; Morrison, L.A.; Bailly, F.; Cotelle, P.; Giannakopoulou, E.; et al. Inhibition of Hepatitis B Virus Replication by N-Hydroxyisoquinolinediones and Related Polyoxygenated Heterocycles. Antiviral Res. 2017, 143, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulou, E.; Pardali, V.; Edwards, T.C.; Woodson, M.; Tajwar, R.; Tavis, J.E.; Zoidis, G. Identification and Assessment of the 1,6-Dihydroxy-Pyridin-2-One Moiety as Privileged Scaffold for HBV Ribonuclease H Inhibition. Antiviral Res. 2024, 223, 105833. [Google Scholar] [CrossRef] [PubMed]
- Prifti, G.-M.; Moianos, D.; Giannakopoulou, E.; Pardali, V.; Tavis, J.E.; Zoidis, G. Recent Advances in Hepatitis B Treatment. Pharmaceuticals 2021, 14, 417. [Google Scholar] [CrossRef] [PubMed]
- Tavis, J.E.; Zoidis, G.; Meyers, M.J.; Murelli, R.P. Chemical Approaches to Inhibiting the Hepatitis B Virus Ribonuclease H. ACS Infect. Dis. 2019, 5, 655–658. [Google Scholar] [CrossRef]
- Kowalinski, E.; Zubieta, C.; Wolkerstorfer, A.; Szolar, O.H.J.; Ruigrok, R.W.H.; Cusack, S. Structural Analysis of Specific Metal Chelating Inhibitor Binding to the Endonuclease Domain of Influenza pH1N1 (2009) Polymerase. PLoS Pathog. 2012, 8, e1002831. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.Q.; Rajendran, S.; Yang, Y.; Li, Y.; Wong Kai In, P.; Overton, H.; Parkes, K.E.B.; Cammack, N.; Martin, J.A.; Klumpp, K. Activity of the Isolated HIV RNase H Domain and Specific Inhibition by N-Hydroxyimides. Biochem. Biophys. Res. Commun. 2004, 317, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Cianci, C.; Chung, T.D.Y.; Meanwell, N.; Putz, H.; Hagen, M.; Colonno, R.J.; Krystal, M. Identification of N-Hydroxamic Acid and N-Hydroxyimide Compounds That Inhibit the Influenza Virus Polymerase. Antiviral Chem. Chemother. 1996, 7, 353–360. [Google Scholar] [CrossRef]
- Geerling, E.; Murphy, V.; Mai, M.C.; Stone, E.T.; Casals, A.G.; Hassert, M.; O’Dea, A.T.; Cao, F.; Donlin, M.J.; Elagawany, M.; et al. Metal Coordinating Inhibitors of Rift Valley Fever Virus Replication. PLoS ONE 2022, 17, e0274266. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, G.; Giannakopoulou, E.; Stevaert, A.; Frakolaki, E.; Myrianthopoulos, V.; Fytas, G.; Mavromara, P.; Mikros, E.; Bartenschlager, R.; Vassilaki, N.; et al. Novel Indole—Flutimide Heterocycles with Activity against Influenza PA Endonuclease and Hepatitis C Virus. Med. Chem. Commun. 2016, 7, 447–456. [Google Scholar] [CrossRef]
- Vrolijk, J.M.; Kaul, A.; Hansen, B.E.; Lohmann, V.; Haagmans, B.L.; Schalm, S.W.; Bartenschlager, R. A Replicon-Based Bioassay for the Measurement of Interferons in Patients with Chronic Hepatitis C. J. Virol. Methods 2003, 110, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Aichele, U.; Kersting, N.; Klein, R.; Aichele, P.; Bisse, E.; Sewell, A.K.; Blum, H.E.; Bartenschlager, R.; Lohmann, V.; et al. Analysis of CD8+ T-Cell—Mediated Inhibition of Hepatitis C Virus Replication Using a Novel Immunological Model. Gastroenterology 2009, 136, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Scheel, T.K.H.; Gottwein, J.M.; Marukian, S.; Dustin, L.B.; Bukh, J.; Rice, C.M. Efficient Replication of Genotype 3a and 4a Hepatitis C Virus Replicons in Human Hepatoma Cells. Antimicrob. Agents Chemother. 2012, 56, 5365–5373. [Google Scholar] [CrossRef] [PubMed]
- Dufner-Beattie, J.; O’Guin, A.; O’Guin, S.; Briley, A.; Wang, B.; Balsarotti, J.; Roth, R.; Starkey, G.; Slomczynska, U.; Noueiry, A.; et al. Identification of AP80978, a Novel Small-Molecule Inhibitor of Hepatitis C Virus Replication That Targets NS4B. Antimicrob. Agents Chemother. 2014, 58, 3399–3410. [Google Scholar] [CrossRef] [PubMed]
- Noueiry, A.O.; Olivo, P.D.; Slomczynska, U.; Zhou, Y.; Buscher, B.; Geiss, B.; Engle, M.; Roth, R.M.; Chung, K.M.; Samuel, M.; et al. Identification of Novel Small-Molecule Inhibitors of West Nile Virus Infection. J. Virol. 2007, 81, 11992–12004. [Google Scholar] [CrossRef] [PubMed]
- Lougiakis, N.; Frakolaki, E.; Karmou, P.; Pouli, N.; Marakos, P.; Madan, V.; Bartenschlager, R.; Vassilaki, N. Novel Nucleoside Analogues Targeting HCV Replication through an NS5A-Dependent Inhibition Mechanism. Chem. Biol. Drug Des. 2017, 90, 352–367. [Google Scholar] [CrossRef]
- Giannakopoulou, E.; Pardali, V.; Frakolaki, E.; Siozos, V.; Myrianthopoulos, V.; Mikros, E.; Taylor, M.C.; Kelly, J.M.; Vassilaki, N.; Zoidis, G. Scaffold Hybridization Strategy towards Potent Hydroxamate-Based Inhibitors of Flaviviridae Viruses and Trypanosoma Species. Med. Chem. Commun. 2019, 10, 991–1006. [Google Scholar] [CrossRef]
- Shimakami, T.; Welsch, C.; Yamane, D.; McGivern, D.R.; Yi, M.; Zeuzem, S.; Lemon, S.M. Protease Inhibitor–Resistant Hepatitis C Virus Mutants with Reduced Fitness from Impaired Production of Infectious Virus. Gastroenterology 2011, 140, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; McGivern, D.R.; Wauthier, E.; Yi, M.; Madden, V.J.; Welsch, C.; Antes, I.; Wen, Y.; Chugh, P.E.; McGee, C.E.; et al. Regulation of the Hepatitis C Virus RNA Replicase by Endogenous Lipid Peroxidation. Nat. Med. 2014, 20, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Friebe, P.; Boudet, J.; Simorre, J.-P.; Bartenschlager, R. Kissing-Loop Interaction in the 3’ End of the Hepatitis C Virus Genome Essential for RNA Replication. J. Virol. 2005, 79, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Woerz, I.; Meuleman, P.; Leroux-Roels, G.; Bartenschlager, R. Cell Culture Adaptation of Hepatitis C Virus and In Vivo Viability of an Adapted Variant. J. Virol. 2007, 81, 13168–13179. [Google Scholar] [CrossRef]
- Vassilaki, N.; Friebe, P.; Meuleman, P.; Kallis, S.; Kaul, A.; Paranhos-Baccalà, G.; Leroux-Roels, G.; Mavromara, P.; Bartenschlager, R. Role of the Hepatitis C Virus Core+1 Open Reading Frame and Core Cis-Acting RNA Elements in Viral RNA Translation and Replication. J. Virol. 2008, 82, 11503–11515. [Google Scholar] [CrossRef] [PubMed]
- van den Hoff, M.J.B.; Christoffels, V.M.; Labruyère, W.T.; Moorman, A.F.M.; Lamers, W.H. Electrotransfection with “Intracellular” Buffer. In Animal Cell Electroporation and Electrofusion Protocols; Nickoloff, J.A., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 1995; pp. 185–197. ISBN 978-1-59259-535-8. [Google Scholar]
- Vassilaki, N.; Boleti, H.; Mavromara, P. Expression Studies of the HCV-1a Core+1 Open Reading Frame in Mammalian Cells. Virus Res. 2008, 133, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.; Hamed, M.M.; Frakolaki, E.; Katsamakas, S.; Vassilaki, N.; Bartenschlager, R.; Zoidis, G.; Hirsch, A.K.; Abdel-Halim, M.; Abadi, A.H. Redesigning of the Cap Conformation and Symmetry of the Diphenylethyne Core to Yield Highly Potent Pan-Genotypic NS5A Inhibitors with High Potency and High Resistance Barrier. Eur. J. Med. Chem. 2022, 229, 114034. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Mulakala, C.; Viswanadhan, V.N. Could MM-GBSA Be Accurate Enough for Calculation of Absolute Protein/Ligand Binding Free Energies? J. Mol. Graph. Model. 2013, 46, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: New York, NY, USA, 2006; p. 84. [Google Scholar]
- Kempf, D.J.; Condon, S.L. Synthesis of Rigid, Heterocyclic Dipeptide Analogs. J. Org. Chem. 1990, 55, 1390–1394. [Google Scholar] [CrossRef]
- Fytas, G.; Zoidis, G.; Taylor, M.C.; Kelly, J.M.; Tsatsaroni, A.; Tsotinis, A. Novel 2,6-Diketopiperazine-Derived Acetohydroxamic Acids as Promising Anti—Trypanosoma Brucei Agents. Future Med. Chem. 2019, 11, 1259–1266. [Google Scholar] [CrossRef]
- Pawlotsky, J.-M. What Are the Pros and Cons of the Use of Host-Targeted Agents against Hepatitis C? Antiviral Res. 2014, 105, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C Virus Drug Resistance Associated Substitutions and Their Clinical Relevance: Update 2018. Drug Resist. Updates 2018, 37, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Bressanelli, S.; Tomei, L.; Rey, F.A.; De Francesco, R. Structural Analysis of the Hepatitis C Virus RNA Polymerase in Complex with Ribonucleotides. J. Virol. 2002, 76, 3482–3492. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | EC50 a (μΜ) | CC50 b (μΜ) | SI c |
|---|---|---|---|---|
| 6 | 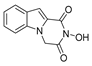 | 126.70 ± 8.98 | >200 | >1.6 |
| 12 |  | 80.98 ± 8.24 | >200 | >2.5 |
| 18 |  | >200 | >200 | - |
| 24 |  | >200 | >200 | - |
| 25 | 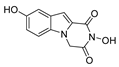 | >200 | >200 | - |
| 26 |  | >200 | >200 | - |
| 30 | 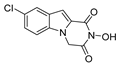 | 9.9 ± 1.75 | >200 | >20.2 |
| 36 | 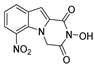 | 1.61 ± 0.32 | 175.4 ± 13.45 | 108.9 |
| 38 |  | >200 | >200 | - |
| 44 | 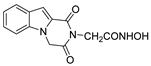 | >200 | >200 | - |
| 46 | 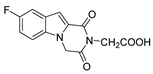 | >200 | >200 | - |
| 48 | 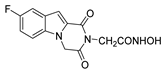 | >200 | >200 | - |
| 52 | 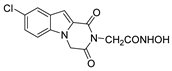 | >200 | >200 | - |
| 58 | 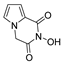 | >200 | >200 | - |
| 62 | 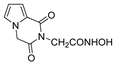 | >200 | >200 | - |
| 66 |  | 99.68 ± 5.52 | 177.3 ± 5.71 | 1.8 |
| 70 | 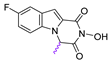 | 32.87 ± 1.93 | 86.45 ± 3.36 | 2.6 |
| 74 | 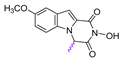 | 61.15 ± 1.72 | 100.6 ± 4.05 | 1.6 |
| 78 | 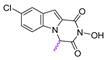 | 12.86 ± 0.39 | 99.91 ± 3.54 | 7.8 |
| Compound | Structure | EC50 a (μM) | |||
|---|---|---|---|---|---|
| GT 1a | GT 2a | GT 3a | GT 4a | ||
| 30 | 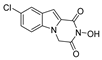 | 2.79 ± 0.48 | >100 | >100 | 84.73 ± 5.28 |
| 36 |  | 2.57 ± 0.34 | 78.31 ± 3.44 | 27.86 ± 1.64 | >100 |
| Compound | Structure | CC50 a (μΜ) | YFV | DENV | ||
|---|---|---|---|---|---|---|
| EC50 b (μΜ) | SI c | EC50 (μΜ) | SI | |||
| 6 |  | >200 | 57.55 ± 2.64 | >3.48 | >200 | - |
| 12 | 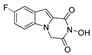 | >200 | >200 | - | >200 | - |
| 18 | 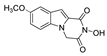 | >200 | >200 | - | >200 | - |
| 24 | 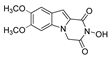 | >200 | >200 | - | >200 | - |
| 39 |  | >200 | >200 | - | >200 | - |
| 40 | 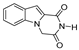 | >200 | >200 | - | >200 | - |
| 30 | 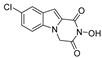 | >200 | >200 | - | >200 | - |
| 36 | 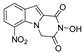 | 175.4 ± 13.45 | 143.9 ± 4.90 | 1.22 | 124.3 ± 6.49 | 1.41 |
| 38 |  | >200 | >200 | - | 75.24 ± 6.58 | >2.66 |
| 44 | 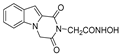 | >200 | >200 | - | >200 | - |
| 46 | 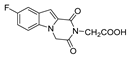 | >200 | >200 | - | >200 | - |
| 48 | 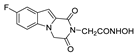 | >200 | >200 | - | >200 | - |
| 52 | 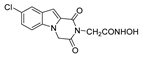 | >200 | >200 | - | 61.3 ± 3.10 | >3.26 |
| 58 | 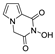 | >200 | >200 | - | 131.2 ± 10.09 | >1.52 |
| 62 |  | >200 | >200 | - | >200 | - |
| 66 | 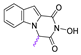 | 177.3 ± 5.71 | 30.33 ± 2.10 | 5.85 | >100 | - |
| 70 | 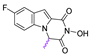 | 86.45 ± 3.36 | 11.67 ± 0.66 | 7.41 | >50 | - |
| 74 | 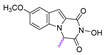 | 100.6 ± 4.05 | 20.87 ± 1.28 | 4.82 | >50 | - |
| 78 | 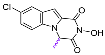 | 99.91 ± 3.54 | 6.57 ± 0.33 | 15.22 | >50 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannakopoulou, E.; Akrani, I.; Mpekoulis, G.; Frakolaki, E.; Dimitriou, M.; Myrianthopoulos, V.; Vassilaki, N.; Zoidis, G. Novel Pyrazino[1,2-a]indole-1,3(2H,4H)-dione Derivatives Targeting the Replication of Flaviviridae Viruses: Structural and Mechanistic Insights. Viruses 2024, 16, 1238. https://doi.org/10.3390/v16081238
Giannakopoulou E, Akrani I, Mpekoulis G, Frakolaki E, Dimitriou M, Myrianthopoulos V, Vassilaki N, Zoidis G. Novel Pyrazino[1,2-a]indole-1,3(2H,4H)-dione Derivatives Targeting the Replication of Flaviviridae Viruses: Structural and Mechanistic Insights. Viruses. 2024; 16(8):1238. https://doi.org/10.3390/v16081238
Chicago/Turabian StyleGiannakopoulou, Erofili, Ifigeneia Akrani, George Mpekoulis, Efseveia Frakolaki, Marios Dimitriou, Vassilios Myrianthopoulos, Niki Vassilaki, and Grigoris Zoidis. 2024. "Novel Pyrazino[1,2-a]indole-1,3(2H,4H)-dione Derivatives Targeting the Replication of Flaviviridae Viruses: Structural and Mechanistic Insights" Viruses 16, no. 8: 1238. https://doi.org/10.3390/v16081238