Abstract
The COVID-19 pandemic has revealed a bidirectional relationship between SARS-CoV-2 infection and diabetes mellitus. Existing evidence strongly suggests hyperglycemia as an independent risk factor for severe COVID-19, resulting in increased morbidity and mortality. Conversely, recent studies have reported new-onset diabetes following SARS-CoV-2 infection, hinting at a potential direct viral attack on pancreatic beta cells. In this review, we explore how hyperglycemia, a hallmark of diabetes, might influence SARS-CoV-2 entry and accessory proteins in pancreatic β-cells. We examine how the virus may enter and manipulate such cells, focusing on the role of the spike protein and its interaction with host receptors. Additionally, we analyze potential effects on endosomal processing and accessory proteins involved in viral infection. Our analysis suggests a complex interplay between hyperglycemia and SARS-CoV-2 in pancreatic β-cells. Understanding these mechanisms may help unlock urgent therapeutic strategies to mitigate the detrimental effects of COVID-19 in diabetic patients and unveil if the virus itself can trigger diabetes onset.
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic is driven by the novel coronavirus, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). Since January 2020, the World Health Organization has recorded approximately 775.6 million cases, with 7 million deaths due to COVID-19 [1]. These statistics illustrate that despite global efforts, COVID-19 remains a pressing health concern. Moreover, the constant emergence of novel SARS-CoV-2 variants hinders progress to eliminate threats posed by this virus.
Individuals infected with SARS-CoV-2 exhibit a wide range of symptoms, from asymptomatic to mild to severe illness [2]. Moreover, COVID-19 patients, either of advanced ages or with comorbidities such as diabetes mellitus (DM), were found to be predisposed to severe disease onset, often leading to ICU admission and/or mortality [3,4,5,6].
Coincident DM may be of particular concern in COVID-19 patients. For example, one study showed an associated increased risk of severity and mortality with DM [7], while others reported higher ICU admissions and higher mortality rates among diabetic COVID-19 patients compared to their non-diabetic counterparts [5]. Several other studies further reported significantly increased in-hospital deaths and severe disease outcomes associated with diabetes in COVID-19 patients [3,8,9,10].
Hyperglycemia, which is a defining characteristic of DM, has since been determined as an independent risk factor for COVID-19 severity and/or mortality [11,12]. Furthermore, several studies found that COVID-19 is associated with hyperglycemia in patients with and without known diabetes [13,14]. Patients who maintained euglycemia also showed improved outcomes of COVID-19 compared to those with hyperglycemia [15]. Another study further confirmed that a relatively high level of blood glucose upon hospital admission was an independent risk factor for the mortality of COVID-19 patients with diabetes [16]. Furthermore, improved glycemic control correlated with better outcomes in patients with COVID-19 and pre-existing type 2 DM (T2DM) [17]. These findings highlight the importance of monitoring and treating hyperglycemia, irrespective of diabetic history, to lessen the severity of COVID-19 progression.
In addition to the impact of hyperglycemia on COVID-19 severity, the emergence of new-onset diabetes (NOD) is of concern. NOD, which is also referred to as new-onset hyperglycemia, was reported in COVID-19 patients without any history of diabetes [10,18]. Several researchers have investigated cases of NOD, and Table 1 summarizes the key results from a selection of recent studies. It is important to note that this table is not exhaustive, and further research is warranted on this topic.

Table 1.
Studies reporting on NOD cases.
One explanation for the emergence of NOD cases may be the possibility that SARS-CoV-2 directly infects pancreatic β-cells which may result in dysregulated pancreas functioning and altered glucose metabolism in healthy individuals [25,26]. Recent evidence also showed that COVID-19 may cause direct damage to the pancreas, in addition to insulin resistance, which could exacerbate hyperglycemia in diabetic patients or result in NOD in previously non-diabetic individuals [27,28]. More specifically, SARS-CoV-2 could infect insulin-producing pancreatic β-cells and elicit its impairment [29,30].
The loss and/or dysfunction of β-cells is an underlying feature of DM, and the possibility that SARS-CoV-2 may induce similar effects alerts to an emerging health crisis. Pancreatic β-cells reportedly express most of the known SARS-CoV-2 entry receptors, such as angiotensin-converting enzyme-2 (ACE2) and Neuropilin-1 (NRP1) [29,31,32], hence rendering them vulnerable to SARS-CoV-2 infection [30].
These findings emphasize the bidirectional nature of diabetes and COVID-19. Not only does diabetes increase the risk of developing severe COVID-19, but SARS-CoV-2 is also capable of inducing diabetogenic effects. However, much remains to be elucidated about the exact alterations occurring in this bidirectional relationship. This review investigates the potential interplay between diabetes, more specifically hyperglycemia, and putative SARS-CoV-2 entry mechanisms into pancreatic β-cells.
2. The Bidirectional Relationship between Diabetes and COVID-19
Several studies demonstrated that elevated glucose levels upon hospital admission were an independent risk factor for the progression of severe COVID-19 cases and mortality [11,33]. For example, some researchers found that 40% of their subjects displayed uncontrolled hyperglycemia upon admission and that in-patient mortality was greater for patients with diabetes [34]. They also noted that mortality was higher for patients without pre-existing diabetes who developed hyperglycemia in the hospital. These findings were further strengthened by Lazarus et al. [35], who showed a relationship between admission fasting glucose levels and COVID-19 severity. Overall, this highlights the important role of hyperglycemia in SARS-CoV-2 infection and, subsequently, the severity of COVID-19 onset.
COVID-19 can also affect glucose homeostasis in individuals without a history of diabetes [36]. The accompanying increase in cytokine production and dysregulated metabolic responses observed during severe COVID-19 may contribute to the onset of insulin resistance and subsequent hyperglycemia [37]. Recent findings also suggest a direct effect of SARS-CoV-2 on glucose metabolism as NOD, diabetic ketoacidosis, and unusually high insulin requirements to achieve glycemic control were reported for COVID-19 patients [14,17,22,26,38,39,40,41].
A plausible explanation for metabolic dysregulation during SARS-CoV-2 infections may be due to the direct damage of pancreatic β-cells to thereby impair insulin secretion [42]. Pancreatic endocrine cells, specifically β-cells, are responsible for insulin synthesis, storage, and secretion in response to changes in blood glucose levels [43]. To be able to respond to high glucose levels, glucose transporter 2 (GLUT2) is utilized for glucose uptake into β-cells. Glucose is then metabolized via glycolysis and, subsequently, oxidative phosphorylation to generate ATP. This increases the ATP/ADP ratio, leading to the closure of the ATP-sensitive K+ channels and the depolarization of the membrane potential. Ca2+ channels open because of this depolarization, allowing for extracellular Ca2+ ions to enter the β-cells and induce the exocytosis of secretory vesicles to secrete insulin [44].
An immune response via the release of cytokines and chemokines mediated by SARS-CoV-2 may induce damage in β-cells and attenuate their functionality through a loss of the ability to sense glycemic levels and insulin release [14,26,45]. Multiple studies reported cases of acute pancreatitis or pancreatic injury in patients who developed post-SARS-CoV-2 infections [46,47,48,49,50,51,52,53]. This emphasizes the ability of SARS-CoV-2 to directly affect the endocrine system, which of course includes the pancreas [54,55,56].
SARS-CoV-2 enters host cells via its primary receptor, ACE2 [57,58,59], with its expression increased upon inflammatory stress. This suggests an enhancement of β-cell sensitivity to SARS-CoV-2 infection during inflammatory conditions [60] such as DM. In agreement, there was increased ACE2 expression together with a pro-inflammatory profile in patients with diabetes and COVID-19 [61]. An upregulation of ACE2 receptors was also postulated to favor viral entry into host cells, which results in a relatively higher viral load and poor prognosis. Following viral infection, the loss of ACE2′s physiological function may enhance systemic adverse effects of the renin–angiotensin–aldosterone system (RAAS) [62].
Various studies revealed that ACE2 participates in the regulation of metabolism and, specifically, glucose homeostasis. For example, ACE2 deletion worsened glucose tolerance in their non-obese diabetic mice models, eventually resulting in NOD [63]. Moreover, infusion of angiotensin-2 (Ang2) in a C57bl/6J mouse model, which is cleaved by ACE2 to form angiotensin-1-7 (Ang1-7), results in decreased ACE2 pancreatic activity and expression accompanied by increased angiotensin type-1 receptor (AT1R) expression and oxidative stress [64]. Furthermore, ACE2 deletion increases oxidative stress and ACE and AT1R expression in pancreatic islets [63].
Overall, these findings confirm the role of ACE2 (SARS-CoV-2 receptor) in causing metabolic dysregulations, especially regarding glucose control and insulin secretion. The controversy surrounding ACE2 expression in β-cells leads to the hypothesis that other receptors, such as NRP1, Dipeptidyl peptidase-4 (DPP4), aminopeptidase N (APN), and Basigin, may be recruited in terms of SARS-CoV-2 entry mechanisms. Other proteases, such as members of the A disintegrin and metalloproteinase (ADAM) family, may also be recruited together with or in the absence of the primary SARS-CoV-2 protease, transmembrane protease serine-2 (TMPRSS2). Nevertheless, it is evident that there is a potential bidirectional relationship between COVID-19 and diabetes.
3. Unveiling the Viral Structure and Function of SARS-CoV-2
Coronaviruses (CoVs) are a family of enveloped, positive-strand RNA viruses that can cause respiratory and enteric disease in animals and humans [65]. These viruses are becoming an increasing health concern following three recent zoonotic outbreaks of highly pathogenic human CoVs (HCoVs), namely the severe acute respiratory syndrome coronavirus (SARS-CoV) from 2002 to 2003, the Middle East respiratory syndrome CoV (MERS-CoV) in 2012, and the current SARS-CoV-2 pandemic that began in 2020 [66,67,68,69].
The general structure of CoVs includes four structural proteins: the spike (S), envelope (E), matrix (M), and nucleocapsid (N). The E- and M-proteins play central roles in viral assembly, while the N-protein is required for the virus’s RNA synthesis [70]. The S-protein mediates receptor binding and the fusion of viral particles with target host cells. Figure 1 illustrates the general structure of a CoV, with a specific focus on the S-protein.
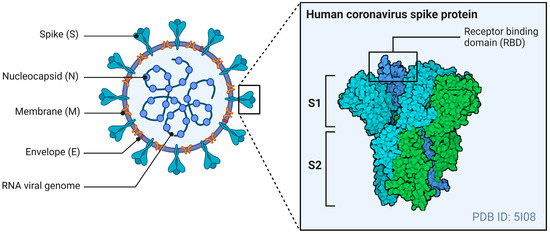
Figure 1.
The general structure of a CoV. The S-protein of an HCoV, containing the RBD, mediates membrane fusion by binding to various cellular receptors. Adapted from “Coronavirus Structure and Protein Visualization”, by Biorender.com (2024). Retrieved from https://app.biorender.com/biorender-templates (accessed on 24 May 2024).
The S-protein is a type 1 fusion protein that functions as a trimer. Each monomer is divided into two subunits: S1, which mediates receptor binding, and S2, which contains the transmembrane domain and mediates fusion with the host cell membrane [71]. The receptor binding domain (RBD) is found within the S1 subunit and is responsible for binding to receptors of the CoVs [72]. The S-protein also undergoes post-translational cleavages by host cell proteases to expose this RBD to host cell receptors and allow viral entry.
Proteases involved in CoV S-protein activation can act at different stages of the viral life cycle. These include proprotein convertases (Furin) during viral packaging, cell surface proteases (transmembrane serine proteases) after receptor binding, and lysosomal proteases (Cathepsins B/L) after virion endocytosis [73,74]. The role of S-protein priming by host cell proteases is particularly important in SARS-CoV-2 viral entry and tropism. The S1/S2 junction is cleavable by either trypsin-like proteases (i.e., TMPRSS2) that are present on the host cell membrane and endosomal cathepsins that are activated by a drop in pH (i.e., Cathepsins B/L) [59,75].
The different types of S-protein priming may be crucial factors in explaining SARS-CoV-2 cell tropism and the features of COVID-19 symptoms [76]. The type of protease priming may also determine whether the membrane fusion process occurs directly at the plasma membrane or endosomal levels [77]. Several studies showed that both TMPRSS2 and Cathepsins B/L are important for SARS-CoV-2 entry [59,78]. Figure 2 summarizes the key events in each main entry pathway, namely the membrane fusion and the receptor-mediated endosomal entry pathways.
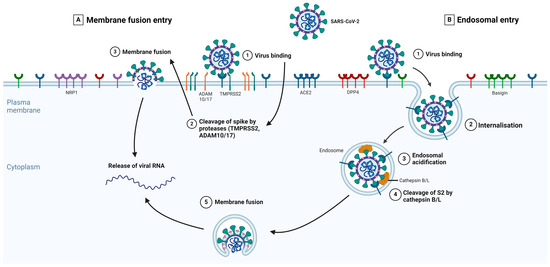
Figure 2.
The expanded entry pathways of SARS-CoV-2. The S-protein of SARS-CoV-2 binds to its primary receptor, ACE2, or potentially to Basigin (step 1), which induces conformational changes in the S1 subunit and exposes the S2 site for cleavage. The S-protein can bind to NRP1 and DPP4, enabling membrane fusion. Depending on the entry pathway taken by SARS-CoV-2, the S2 site is cleaved by either TMPRSS2 or ADAM 10/17 (membrane fusion entry, part A) or Cathepsins B/L (receptor-mediated endosomal entry, part B). In the presence of proteases on the plasma membrane, S-protein cleavage occurs at the cell surface (part A, step 2). With endosomal entry, the S-protein-receptor complex is internalized via clathrin-mediated endocytosis (part B, step 2) into endolysosomes, where the S2 site is cleaved by Cathepsins B/L (part B, step 4). Before this, the cleavage via cathepsins requires an acidic environment for its activity; therefore, endosomal acidification occurs (part B, step 3). Fusion between the virus and host cellular membranes forms a fusion pore where viral RNA is released into the host cytoplasm for viral uncoating and replication. Adapted from Jackson et al. [79]. Created in BioRender.com.
4. Membrane Fusion Entry Pathway
The SARS-CoV-2 infection of host cells is completed in four steps: (1) prebinding, (2) receptor binding, (3) proteolytic cleavage, and (4) membrane fusion. Before SARS-CoV-2 can enter cells, S-proteins exhibit a prebinding conformation with an exposed RBD to enable its attachment to host membrane receptors [80], such as ACE2, in the second step. Upon binding to the receptor, S-proteins are cleaved by proteases (TMPRSS2) present in the host cell membrane [81] to reveal the S2 subunit through the shedding of the S1 subunit [82] and activating membrane fusion. After the final step, when the cell membranes of the virus and host are fused, SARS-CoV-2 releases its viral material into the host cell cytosol and initiates other infectious processes [83,84].
To understand how SARS-CoV-2 gains entry into pancreatic β-cells, we previously discussed the mechanisms of viral membrane fusion. Now, we will explore how hyperglycemia can influence proteins involved in membrane fusion, potentially enhancing the susceptibility of β-cells to infection. We also highlight other potential receptors and proteases that may be part of the SARS-CoV-2 membrane fusion entry into β-cells.
4.1. Angiotensin-Converting Enzyme-2
The primary receptor for SARS-CoV-2 is ACE2, the plasma membrane-bound carboxypeptidase [57,59,85]. SARS-CoV-2 S1 subunits induce ACE2 downregulation upon binding. Cells combat this viral infectivity toward adjacent cells by decreasing the amount of ACE2 on their surfaces [86]. This results in an overall lowering of ACE2 activity, leading to marked elevations in Ang2 with a reduction in Ang1-7 that induces pancreatic dysfunction, insulin secretion inhibition, and resultant hyperglycemia [87].
While a homolog of the angiotensin-converting enzyme (ACE), ACE2 possesses recognized roles in the RAAS [88]. The RAAS is an endocrine system that regulates blood pressure, fluid, electrolyte balance, and volume homeostasis through active metabolites [89]. ACE2 is a potent negative RAAS regulator that is crucial for maintaining the system’s homeostasis [90,91]. The main function of ACE2 is to cleave Ang2, which has potent vasoconstriction, pro-inflammatory, and pro-fibrotic effects, into Ang1-7 to induce vasodilatory, antiproliferative, and apoptotic effects [88,90,92,93]. Ang2 binds to AT1R and angiotensin type-2 receptor (AT2R), and Ang1-7 binds to the Mas receptor [94] to exert its antagonistic effects.
Tissues such as the lungs, kidney, small and large intestine, and vasculature express ACE2 [95,96,97]. The expression of ACE2 in the pancreas, however, has been contradictory. ACE2 can be expressed in β-cells, acinar cells, ductal cells, and in the islet microvasculature and pericytes [32,52]. Variation in ACE2 localization within islets was demonstrated in deceased COVID-19 patients [98], indicating differences in severity and outcome reported as well as contradictions between studies. Several studies reported that ACE2 is expressed in both endocrine and exocrine pancreatic tissues, specifically in the islets of Langerhans [29,99,100,101,102,103]. In the human pancreas, ACE2 is highly expressed in β-cells compared to other islets, in addition to pericytes and endothelial cells surrounding the islets [60].
Contrary to these findings, some studies found that β-cells do not express ACE2 [52,104,105], leading to the hypothesis that ACE2-dependent entry of SARS-CoV-2 is questionable in β-cells. This also highlights the potential coordination of ACE2 with other surface receptors as entry factors for SARS-CoV-2 invasion into the pancreas.
The discrepancies between research work that identified ACE2 in β-cells and those that did not could be explained by differences in experimental approaches and contexts for such studies. For instance, Fignani et al. [60] reported ACE2 in some islet endocrine cells from seven non-diabetic donors and one T1DM donor. However, ACE2 was mainly observed in the cytoplasm with a small fraction on the plasma membrane. These same authors utilized three ACE2 antibodies that recognize different epitopes of the full-length ACE2 in immunofluorescence staining. Coate et al. [104] employed two of these antibodies and did not detect either form of ACE2 in β-cells. These two studies, therefore, reveal how differing protocols may be a source of variability in terms of protein expression. Later sections of this review will highlight controversies surrounding the expression of other proteins in β-cells and why this may be the case.
Regarding the role of ACE2 in the pancreas, local RAAS in the islets regulates glucose homeostasis through ACE2 and Ang1-7 [106]. ACE2 overexpression in the pancreas of T2DM mice improved glucose tolerance and increased insulin secretion and β-cell proliferation [107]. ACE2 deficiency can also impact islet development and β-cell function [108,109]. Moreover, Ang2 can impair insulin signaling and islet function [110,111,112,113]. The ACE2/Ang1-7/Mas axis within β-cells is involved in the proliferation, differentiation, functioning, protection, and insulin production [114,115,116,117]. These findings emphasize the crucial role of ACE2 in islet functioning.
Uncontrolled hyperglycemia observed in patients with DM could also regulate ACE2-S-protein binding [118,119], possibly through the glycosylation and glycation of ACE2. Glycosylation is a post-translational process whereby glycans are enzymatically attached to macromolecules [120,121,122], whereas glycation is the non-enzymatic attachment of glucose to macromolecules [123]. ACE2 is a metalloprotease with a relatively long half-life and can hence be easily glycated by enzymatic and non-enzymatic mechanisms in patients with chronic hyperglycemia. This would favor the formation of advanced glycation end-products (AGEs) [118,124] that were linked to an increased risk for COVID-19 [125].
In accordance, relatively high total and glycosylated ACE2 expression levels were detected in COVID-19 heart samples from diabetic patients compared to non-diabetic samples [118]. Interestingly, one in silico study reported that the glycation of residues in both the S-protein and ACE2 resulted in a loss of interaction between them [119]. Considering this, alternative viral entry proteins may be involved [126] as viruses can develop new effective ways to enter host cells when their main entry mechanism is impaired [127,128].
4.2. Dipeptidyl Peptidase-4
Another potential entry factor is DPP4, or Cluster of Differentiation (CD) 26, which functions as a receptor for SARS-CoV-2 as it is the primary receptor for MERS-CoV [129,130,131]. Some researchers predicted that the S-protein of SARS-CoV-2 directly interacts with DPP4 in host cells [132], while another demonstrated how the RBD of the S-protein directly binds to DPP4 [133]. In contrast, some studies report that DPP4 may not be used for cellular entry by SARS-CoV-2 [57,59].
A molecular docking study by Cameron et al. [134] showed that the SARS-CoV-2 S-protein RBD interaction with DPP4 may be significantly weaker compared to that of the MERS-CoV-DPP4 complex, attributed to the differences in amino acid sequences between the two viral S-protein RBD regions. However, a later docking study showed that mutations in the RBD of the delta variant may enhance the SARS-CoV-2-DPP4 interaction to a similar degree compared to that of MERS-CoV [135]. These authors concluded that the more virulent variants may be better suited for DPP4 binding.
There is increased DPP4 expression in airway samples from COVID-19 patients [136,137], while its inhibition was associated with lower COVID-19 mortality [138]. Considering that DPP4 is a highly proteolytic protein, it was postulated as a protease for the cleavage of SARS-CoV-2 S-proteins [132]. Although the role of DPP4 as a receptor and protease is not yet fully elucidated, these findings highlight the potential function of DPP4 in SARS-CoV-2 entry mechanisms.
It is also a proteolytic enzyme that is involved in the degradation and inactivation of incretins, a group of gut hormones that stimulate insulin secretion [139,140]. These include glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) that function to augment glucose-induced insulin release from pancreatic β-cells, suppress glucagon secretion, and slow gastric emptying [141,142]. Inhibition of DPP4 can improve glycemic control and exert the beneficial effects of GLP-1 and GIP [143]; for example, the former positively stimulates the proliferation of β-cells to regulate their cell mass [144].
According to the available research, the expression of DPP4 in islets varies, similar to that of ACE2. Some studies support its expression [98,145], while others dispute it [146,147]. Interestingly, Shah et al. [148] found that DPP4 was detected in conditioned medium of human islets, suggesting that DPP4 is released from islets and also present in them.
Fadzeyeva et al. [149] found that a mouse model systemically lacking DPP4 displayed improved islet health and glucoregulation compared to their wild-type counterparts. The direct inhibition of DPP4 in human islets improved β-cell function, survival, and insulin secretion [150,151,152]. Therefore, an increase in DPP4 could translate to a loss of β-cell mass and attenuated insulin secretion.
Emerging evidence suggests that there is a crosstalk between DPP4 and the RAAS. For example, Ang2-infused mice kidneys displayed increased DPP4 activity [153]. These authors also found that supraphysiological Ang2 concentrations stimulated DPP4 activity in cultured proximal tubule cells. Similar findings were reported by Lee et al. [154] in podocytes that may apply to other cell types, such as β-cells. Given that the RAAS in the islets regulates glucose homeostasis [106], it is plausible that an increase in DPP4 results in impaired insulin secretion through the increase in Ang2 [155].
Relatively high levels of DPP4 are observed in patients with T2DM compared to controls [156]. Following this, hyperglycemia could stimulate DPP4 mRNA expression and enzyme activity in human glomerular endothelial cells [157]. Mannucci et al. [158] presented data showing that the degree of hyperglycemia in both T1DM and T2DM directly correlates with circulating DPP4 activity. Additionally, they showed that alterations in DPP4 were correlated with variations in HbA1c. These findings suggest that chronic hyperglycemia could stimulate DPP4 activity. This could result in a reduction in active GLP-1 levels, possibly contributing to hyperglycemia.
Evidence suggests a positive correlation between DPP4 elevations in diabetic and SARS-CoV-2-induced effects in host cells and its potential role in facilitating viral entry. In addition, the increase in DPP4 results in enhanced degradation of GLP-1, which can stimulate β-cell proliferation. A lack of GLP-1 could decrease β-cell mass and attenuate insulin secretion, with hyperglycemia as the overall outcome. This interplay between dysregulated DPP4 and hyperglycemia may contribute to developing NOD in susceptible individuals.
4.3. Neuropilin-1
Neuropilin-1 (NRP1) is a cell surface receptor involved in angiogenesis, organ development, and immune function [159,160]. It possesses a large extracellular domain that binds ligands in several signaling pathways associated with cell migration, growth, and development [161].
NRP1 was recently identified as a potential receptor for SARS-CoV-2 entry [31,162]. Some researchers found that SARS-CoV-2 utilizes NRP1 as a receptor in β-cells in the absence of ACE2 [29], which would lead to β-cell apoptosis and a reduction in insulin secretion. These authors also reported upregulated NRP1 levels in β-cells from individuals with COVID-19. Islet NRP1 is highly expressed, mostly confined to β-cells and rarely in pancreatic α-cells [29,30]. In contrast, Ji et al. [163] found that NRP1 was weakly expressed in islets. While the presence of NRP1 in pancreatic islets cannot be entirely ruled out, the lower levels observed by Ji et al. [163] suggest a potentially limited role in this specific tissue. More research is warranted to definitively determine NRP1’s involvement in SARS-CoV-2 infection and its expression patterns across various human tissues.
SARS-CoV-2 S-protein cleavage, mediated by Furin, results in a C-terminal motif on the S1 subunit which binds to NRP1 [31,162,164]. NRP1 stabilizes this C-terminus to allow for more efficient S1/S2 cleavage and to enable the S2 subunit to mediate membrane fusion more rapidly [164]. Inhibiting the binding between this C-terminal motif and NRP1 results in reduced viral infection in various cell types, including human islets [31,162,164].
Of note, research indicates that NRP1 may be a co-receptor for ACE2. For example, NRP1-knockout (KO) ACE2-expressing HeLa cells displayed less viral entry than those expressing ACE2 and NRP1 [162]. Furthermore, human islets infected with SARS-CoV-2 ex vivo showed that ACE2- and NRP1-expressing cells had more SARS-CoV-2 N-protein transcripts than those expressing either ACE2 or NRP1 [30]. Furthermore, these authors found that ACE2-expressing NRP1-KO cells contained fewer RNA transcripts than the ACE2-KO NRP1-expressing cells, while the double-KO cells exhibited little to no infection. Together with Cantuti-Castelvetri et al. [31] who reported on the independent facilitation of SARS-CoV-2 S-protein pseudoviral entry by ACE2 but not TMPRSS2 or NRP1, it can be postulated that NRP1 is a co-receptor that potentiates ACE2-mediated SARS-CoV-2 infection [165].
The role that NRP1 plays in the context of diabetes is controversial. Minor alleles of NRP1 have been associated with T1DM in children [166], which suggests that it could influence the development of some T1DM cases. Moreover, an analysis of a (cryopreserved) human diabetic kidney RNA sequencing dataset revealed that NRP1 was significantly upregulated [167]. However, others demonstrated decreased NRP1 protein expression in cultured differentiated podocytes, podocytes from diabetic db/db mice, and diabetic patients diagnosed with diabetic nephropathy [168,169]. Further studies are therefore needed to help navigate such contradictory findings and to demonstrate whether the upregulation and participation of NRP-1 in COVID-19 could result in long-term complications.
4.4. Glucose-Regulated Protein-78
Glucose-regulated protein-78 (GRP78), also referred to as heat shock protein A5, is an essential endoplasmic reticulum (ER)-localized chaperone that regulates ER signaling molecules to ensure proper protein folding or direct unfolded proteins to cellular degradation systems [170,171,172].
Viral infection may be promoted by GRP78 by its functioning as an alternative receptor and/or stabilizing S-protein binding [173]. For example, research work predicted that GRP78 can dock at the RBD of the SARS-CoV-2 S-protein [171], which has the same GRP78 recognition site as other human and bat CoVs [174]. Han et al. [175] provide evidence that cell surface GRP78 is utilized by SARS-CoV-2 to enter cells that express low or no ACE2, such as monocytes and macrophages. Considering the variable expression of ACE2 in β-cells (discussed in Section 4.1), SARS-CoV-2 may use GRP78 to enter these cells in an ACE2-independent manner. GRP78 also binds to the MERS-CoV S-protein in vitro, and some researchers showed that GRP78 knockdown attenuated MERS-CoV infection [176]. Based on such evidence, it can be postulated that GRP78 is involved in the entry mechanisms of SARS-CoV-2.
SARS-CoV-2 infection induces ER stress in host cells [177,178], possibly due to the inability to accommodate the significant increase in protein production that occurs during viral replication [179]. CoV infections can upregulate GRP78 levels [176,180]. For example, research studies found increased GRP78 serum levels in COVID-19 patients compared to COVID-19-negative pneumonia patients and healthy individuals [177,181].
Other potential SARS-CoV-2 host entry factors can also modulate GRP78 levels under conditions of ER stress. For instance, Basigin mediates ER stress-induced GRP78 upregulation [182]. This indicates that Basigin upregulation following ER stress may exacerbate GRP78’s mediation of viral entry. GRP78 can also influence cellular ACE2 levels; for example, Carlos et al. [183] observed decreased cell surface ACE2 levels, but not total ACE2, due to GRP78 knockdown (independent of ER stress). This suggests a possible role for GRP78 in ACE2 trafficking to the cell surface.
GRP78 is expressed in all pancreatic cell types, particularly β-cells [147,184]. It has also been reported that GRP78 increased in diabetic mouse models [185,186] and islets from donors with T2DM compared to non-diabetic donors [185,187]. Others also noted significantly increased serum concentrations of GRP78 in T2DM patients, with positive correlations between GRP78 levels and HbA1c and AGEs [188]. In contrast, some studies have found no significant increases in islet GRP78 levels in T2DM donors [189,190].
Chronic hyperglycemia disrupts ER homeostasis to cause ER stress [191]. This could increase the levels of GRP78 within β-cells and provide an additional entry for SARS-CoV-2. The same outcome of increased GRP78 levels could be achieved with SARS-CoV-2 inducing ER stress. As reviewed by Cao et al. [192] and Lee et al. [193], ER stress plays an important role in β-cell dysfunction and loss, resulting in augmented insulin responses to hyperglycemia as observed during diabetes. Therefore, the shared mechanisms between hyperglycemia, SARS-CoV-2, and GRP78 may offer valuable insights for developing therapeutic strategies to target ER stress to improve patient outcomes.
4.5. Transmembrane Protease Serine-2
TMPRSS2 is a type 2 transmembrane serine protease located on the surface of host cells and whose expression is influenced by androgen [194]. Besides its role in cancer, particularly prostate cancer [195,196], its function in other pathologies was relatively unknown until SARS-CoV emerged. TMPRSS2 activates the S-protein of SARS-CoV for membrane fusion of the viral and host membranes [197,198,199]. Research has established that this protease activates S-proteins of SARS-CoV-2 to allow entry via membrane fusion into host cells [59]. For example, SARS-CoV-2 S-protein-mediated cellular entry was attenuated through in vitro androgen deprivation or antagonism [200].
According to RNA and protein expression data retrieved from the Human Protein Atlas database [201], TMPRSS2 is mainly expressed in the lungs, salivary glands, thyroid, gastrointestinal tract, kidneys, liver, and pancreas. As with ACE2 and DPP4 expression, the expression of TMPRSS2 in endocrine β-cells is controversial. Here, some studies support the notion that TMPRSS2 is indeed expressed in β-cells [17,29,32,98,101], whereas others concluded that its expression is either extremely low or undetectable [104,105]. Of note, Piva et al. [202] observed that TMPRSS2 expression is higher in pancreatic β-cells of T2DM patients compared to controls.
Some researchers found relatively high expression levels of TMPRSS2 in diabetic COVID-19 heart sample autopsies compared to non-diabetic ones [118]. It has also been highlighted that SARS-CoV-2 infection-related damage to pancreatic tissues is mediated by TMPRSS2 [203], suggesting a fundamental role for the virus in disease outcomes with diabetes. This is a possible explanation for why DM is an important risk factor for hospitalization and death in COVID-19 patients. However, Zhang et al. [204] reported that elevated glucose levels did not affect TMPRSS2 levels.
While some studies suggest that relatively high glucose conditions may elevate TMPRSS2 expression, the results remain inconclusive. Despite its potential glucose dependence, TMPRSS2 is a crucial protease for SARS-CoV-2 entry by facilitating S-protein cleavage at the cell surface. However, it is not the sole role player, and other proteases, such as ADAM10 and ADAM17 [205], can also cleave the S-protein. This can be potentially competitive with TMPRSS2, as observed for SARS-CoV [206]. This redundancy in the cleavage process highlights the adaptability of SARS-CoV-2 and the potential for therapeutic strategies that target not just TMPRSS2, but a broader range of host cell proteases involved in viral entry.
4.6. A Disintegrin and Metalloproteinase Family
The ADAM family includes a group of multidomain transmembrane proteolytic enzymes that are involved in regulating many physiological processes such as cell adhesion and migration, intracellular signaling, the regulation of growth factors and cytokines, and proteolysis of the intracellular matrix. These processes maintain homeostasis within the body but also play an important role in pathophysiologic complications such as diabetes [207,208]. ADAM9, ADAM10, and ADAM17 are prominent proteases involved in various physiological processes such as development, regeneration, immunity, and the regulation of cell differentiation and proliferation through cleaving ligands and receptors [209,210,211,212,213,214].
ADAM9 was identified as a potential host factor for SARS-CoV-2 S-protein-mediated infection [215] as ADAM9 knockdown resulted in significantly diminished SARS-CoV-2 infection. The authors also highlighted how the use of ADAM9 in SARS-CoV-2 entry in low-expressing ACE2 cells primarily depends on the latter. Although ADAM9 is associated with SARS-CoV-2 entry, it does not cleave the S-protein. Some showed that ADAM9 directly interacts with the RBD of the SARS-CoV-2 S-protein [216] which shifts its role from a potential S-protein protease to a potential receptor.
Of note, ADAM9 was identified as a key driver of SARS-CoV-2 severity in a multi-omics analysis of a young, comorbidity-free COVID-19 patient cohort [217]. This analysis found that increased ADAM9 RNA expression levels were detected in the serum of critically ill COVID-19 patients, further emphasizing the role that ADAM9 plays in SARS-CoV-2 infection.
Jocher et al. [205] established the role of both ADAM10 and ADAM17 as host cell factors for SARS-CoV-2 entry, as it cleaves the S-protein in a TMPRSS2-independent manner. Yamamoto et al. [218] found that SARS-CoV-2 pseudoviral entry was significantly inhibited using an ADAM10 knockdown model. Furthermore, circulating levels of ADAM17 were increased in patients with COVID-19 [219]. Inhibition of ADAM10 significantly blunts SARS-CoV-2 infection, whereas ADAM17 did not [218]. This demonstrates the potential role of ADAM10 and ADAM17 in SARS-CoV-2 viral entry, where SARS-CoV-2 entry is more dependent on ADAM10 than ADAM17.
Both ADAM10 and ADAM17 can shed ACE2 [206,220,221,222,223] and promote SARS-CoV-2 pathogenesis [224]. In contrast, ADAM9 does not possess the ability to cleave ACE2 [220]. The proteolysis or shedding of ACE2 releases a soluble, enzymatically active form corresponding to the ACE2 ectodomain [222,225,226]. While the function of soluble ACE2 (sACE2) is still obscure, the current literature shows that the shedding mechanism is under strict molecular control [227]. Patel et al. [228] identified that Ang2 activates ADAM17, which increases the release of sACE2. Other research work further confirmed ADAM17’s role in regulating ACE2, where its overexpression increased ACE2 shedding and subsequently decreased cellularly bound ACE2 in mouse pancreatic islets [229].
Multiple studies reported ADAM9’s expression in Langerhans’s islets [230,231], while others found that ADAM17 is expressed in islets, including β-cells [104,147,184]. ADAM10 is present in the pancreas [232] and is localized in the cell membranes of endocrine and exocrine cells [231].
Regarding the effects of hyperglycemia on ADAM9, Puddu et al. [233] found increased expression under high glucose conditions in a retinal pigment epithelial cell model. Similarly, Moin et al. [234] found that ADAM9 levels were elevated in T2DM, while Suresh Babu et al. [235] reported increased ADAM9 expression in macrophages exposed to high glucose and in human diabetic hearts.
C-X-C motif chemokine ligand-16 (CXCL16) is a chemokine that exists either in its soluble form to bind to immune cells that express its receptor or in its native form to drive such immune cells to the site of inflammation [236]. Of note, ADAM10 and/or ADAM17 can cleave CXCL16 to its soluble form [212]. Considering that CXCL16 levels are elevated in T2DM patients compared to healthy individuals [237,238] and there is a marked upregulation of serum CXCL16 levels in β-cells in a T1DM model [232], we postulate that there will be increased ADAM10 and/or ADAM17 levels under such conditions. In support, Herman-Edelstein et al. [239] reported elevated ADAM10 and ADAM17 mRNA levels in diabetic hearts.
While the impact of diabetes on ADAM9, ADAM10, and ADAM17 expression remains under investigation, these family members have emerged as important contributors to SARS-CoV-2 infection. Their ability to cleave the viral S-protein (like TMPRSS2) can facilitate cellular entry. Despite the potential influence of diabetes on ADAM expression, their presence offers a level of redundancy in the viral entry process. This highlights the vulnerability of the virus, as targeting a single protease as a therapeutic intervention may be less effective. Future strategies should therefore focus on broader inhibition of multiple host cell proteases like ADAMs and TMPRSS2 to block SARS-CoV-2 entry into host cells more effectively.
4.7. Furin
As a transmembrane endoprotease, Furin cleaves various proproteins prior to secretion and as part of their maturation process [240]. As a proprotein convertase, Furin cleaves and activates many viral proteins, such as SARS-CoV-2 S-proteins, to facilitate its entry into host cells [241]. This cleavage occurs either during viral production in an infected cell or on virus entry into host cells [75,78,242]. The S-protein of SARS-CoV-2 contains a polybasic Furin cleavage site at the S1/S2 junction, which is absent in other SARS-CoVs [243]. The insertion of similar polybasic Furin cleavage sites increases virulence [244]. Based on the structure of this Furin cleavage site, it can be suggested that SARS-CoV-2 mimics host cell machinery for viral entry [245].
Here, an in vitro study showed that Furin KO decreased viral production by 100-fold [242], while others demonstrated that loss of the Furin cleavage site resulted in lowered viral pathogenicity [246]. Furin-mediated cleavage of this S1/S2 junction may only be valid in cells where the membrane fusion pathway prevails over the endocytosis entry pathway. This observation is based on results from Hoffmann et al. [75], where these authors observed how variants of SARS-CoV-2 S-proteins that were resistant to S-protein cleavage displayed significantly diminished entry into TMPRSS2-positive cells but not TMPRSS2-negative cells.
The expression of Furin in human pancreatic endocrine cells (including in β-cells) is moderate to high [30,147,247]. Furin controls proliferation and differentiation in islet cells [248] while playing a role in secretory granule acidification [249], with this being crucial for β-cell granule maturation and proinsulin-to-insulin conversion.
Increased Furin levels are associated with T2DM [250,251,252]. Moreover, elevated serum Furin levels can serve as a marker for diabetes progression and are correlated with metabolic dysregulations and an increased risk of diabetes-associated death [253]. This is mainly attributed to increased levels of osteopontin (pro-inflammatory glycoprotein) in diabetic individuals, and that can then upregulate Furin expression [253]. As a result of such a correlation between Furin and diabetes, it is plausible that this can aid SARS-CoV-2 entry by increasing viral entry and load, resulting in a poor prognosis for diabetic COVID-19 patients.
The intricate process of SARS-CoV-2 cell entry via the membrane fusion pathway is contingent on a complex interplay of viral and host proteins. The S-protein is pivotal for viral attachment and membrane fusion. Host cell receptors like ACE2 and other potential host factors (DPP4, NRP1, and GRP78) serve as the initial attachment points for the virus. Subsequent proteolytic processing of the viral S-protein by host proteases (TMPRSS2, ADAMs proteins, and Furin) is essential for membrane fusion. While these key proteins are currently being extensively characterized, the impact of comorbidities, particularly hyperglycemia, on their expression and function remains an area of active investigation. The potential influence of hyperglycemia on these proteins is summarized in Table 2. A comprehensive understanding of these molecular interactions is essential for developing effective therapeutic interventions.
Table 2.
A summary of the potential influence of hyperglycemia on the proteins that mediate the membrane fusion entry of SARS-CoV-2.
5. Receptor-Mediated Endosomal Entry Pathway
Like other viruses, SARS-CoV-2 also uses host endosomal routes for viral entry [254]. Although the membrane fusion entry pathway is significantly more efficient than the endocytosis pathway, the viral entry mode amongst different cells depends on protease expression [255,256]. In the absence of TMPRSS2, SARS-CoV-2 is transported into the host endosome via endocytosis, either through clathrin-dependent or clathrin- and caveolae-independent entry pathways, for uptake and transport before they fuse with the lysosomes [257]. This is the case for SARS-CoV viral entry routes [257,258]. SARS-CoV-2, in particular, makes use of clathrin-mediated endocytosis to infect cells [259]. Endolysosomal fusion also activates lysosomal proteases (i.e., cathepsins), which cleave the S-protein to facilitate the fusion of the viral and host membranes to allow for the entry of viral RNA [260,261].
5.1. Cathepsins B/L
Cathepsins are non-specific proteases with endopeptidase and exopeptidase activities that participate in protein degradation in late endosomes and lysosomes. Cysteine proteases (Cathepsin B, L, and S) can contribute the most to viral entry by mediating the conversion of the virion into an infectious subvirion via partial proteolysis [262,263,264,265].
Cathepsin B is a lysosomal cysteine primarily involved in degrading or processing lysosomal proteins. In certain conditions, it is also involved in cell invasion, vesicle trafficking, inflammasome formation, and cell death [266]. However, Cathepsin L is integral in degrading extracellular, cytoplasmic, and nuclear proteins. It is also involved in many processes, including autophagy, apoptosis, cell cycle regulation, bone resorption, antigen processing, and tumor invasion/metastasis [267].
The role of cathepsins in viral entry is derived mostly from studies involving reoviruses, Ebola virus, and SARS-CoV [263,268,269] with limited studies on SARS-CoV-2 [270]. Cathepsin B plays an essential role in Ebola viral entry, whereas Cathepsin L plays a larger role in SARS-CoV [269,271,272] and SARS-CoV-2 [59,270] entry. This does not, however, exclude the possible role that Cathepsin B may play in terms of SARS-CoV-2 entry.
For example, Jaimes et al. [76] found that Cathepsin B can cleave SARS-CoV-2 S-proteins by using a biochemical peptide cleavage assay, while others found that Cathepsin B gene expression levels were upregulated in patients with severe COVID-19 [273]. Moreover, selective Cathepsin B inhibitors impaired SARS-CoV-2 infection in vitro, strengthening the notion that it plays an important role in SARS-CoV-2 entry [274]. However, conflicting data question the role of Cathepsin B in SARS-CoV-2 viral entry. Here, the authors showed that Cathepsin L was critical for S-protein activation by employing an S-protein pseudovirus system [275,276]. This indicates that SARS-CoV-2 may have a preference for Cathepsin L rather than Cathepsin B in terms of its entry mechanisms. However, further research work is required to better elucidate this vexing question.
Cathepsin L levels were increased following SARS-CoV-2 pseudovirus entry into a human hepatoma cell line (Huh7) [276]. The authors also found that Cathepsin L overexpression increased pseudoviral entry, while its knockdown and ACE2 inhibition in humanized ACE2 mice decreased SARS-CoV-2 entry. Moreover, Cathepsin L inactivation attenuated SARS-CoV-2 entry, although its inhibition was weaker in TMPRSS2-positive versus TMPRSS2-negative cells [59]. This indicates a compensatory relationship between TMPRSS2 and Cathepsin L, where the inactivity of one compensates for the other.
Cathepsin L cleaves the S-protein S1/S2 site (which differs from the TMPRSS2 cleavage site [241]) to enhance the entry of SARS-CoV-2. Cathepsin L is also secreted under systemic or local inflammatory conditions [277]; for example, Zhao et al. [276] reported increased circulating Cathepsin L levels in COVID-19 patients that positively correlated with disease severity. This indicates that circulating Cathepsin L may exacerbate viral entry during the inflammation that accompanies COVID-19.
Several studies revealed that TMPRSS2 (a fusion of the viral and host membrane) and Cathepsins B/L (a fusion of the viral and endosomal membrane) are dissimilar and work independently. Multiple studies reported inhibition of viral entry through the combined usage of TMPRSS2 and cathepsin inhibitors [59,256,274,278,279]. This emphasizes the importance of not only TMPRSS2 but also cathepsins in terms of SARS-CoV-2 entry into host cells.
Previous studies could detect Cathepsin B from isolated islets of Langerhans [280] and islet endocrine cells from the rat pancreas [281]. Others also identified Cathepsin B in insulin-secreting cell lines, such as HIT T15 [282] and INS-1 [283]. Furthermore, significant quantities of Cathepsin B mRNA were detected in total RNA from isolated islets of Langerhans but not in equivalent amounts of RNA from the whole pancreas [284]. In contrast, Cathepsin L is present in all pancreatic cell types [30,147] and is moderately expressed in pancreatic endocrine cells (i.e., α- and β-cells) [105].
Vidotti et al. [285] reported that high glucose levels stimulated the mRNA levels of Cathepsin B, with Liu et al. [286] reporting similar findings of increased Cathepsin B expression under high glucose stimulation. Contrastingly, Peres et al. [287] found that exposure to high glucose had no effect on its activity, while others found decreased Cathepsin B levels in the pancreatic β-cells of T2DM patients [288].
Cathepsin L expression in T2DM donors was also higher than in non-diabetic donors [105], while it was required to develop T1DM in mouse models [289] and regulate human and mouse islet cell proliferation [290]. These findings confirm a relationship between the presence of diabetes and greater Cathepsin L expression levels. There is also a relationship between Cathepsins B/L and hyperglycemia in the pancreas, with Jung et al. [283] reporting that Cathepsin B/L inhibition induced lysosomal dysfunction which enhanced pancreatic β-cell apoptosis under high glucose.
Research studies completed thus far have been unable to resolve conflicting findings regarding the effect of diabetes on Cathepsin B/L expression, and this requires further investigation. Regardless, Cathepsins B/L contribute to a complementary route for viral entry into endosomes. Such an alternative pathway highlights the adaptability of SARS-CoV-2 and the potential importance of this mechanism, particularly in instances where surface proteases are relatively limited. Thus, further research is needed to understand how diabetes may influence such cathepsins in terms of expression and function and also to ascertain their role in terms of SARS-CoV-2 infection.
5.2. Basigin
Basigin is a transmembrane glycoprotein that is a member of the immunoglobulin superfamily [291] and is also known as extracellular matrix metalloproteinase inducer (EMMPRIN) or CD147 [292]. It plays an important role in pathologies such as stroke, heart disease, and Alzheimer’s disease, and in the development, progression, metastasis, and prognosis of some human cancers [293,294]. Its role in infections by pathogens such as human immunodeficiency virus, hepatitis B and C viruses, and Kaposi’s sarcoma-associated herpesvirus is well studied, revealing associated mechanisms in terms of viral pathogenesis [295]. These findings allude to its possible role in SARS-CoV-2 pathogenicity.
Basigin was first highlighted as a SARS-CoV-2 S-protein co-receptor by Wang et al. [296], where the authors demonstrated that S-protein binding to Basigin elicited important functional implications in terms of viral entry via endocytosis. Since then, numerous studies revealed Basigin as a possible SARS-CoV-2 co-receptor [136,297,298,299,300,301,302,303,304,305,306,307,308]. Moreover, some also highlighted its potential role in the viral entry of SARS-CoV-2 into host cells with a relatively low abundance of ACE2 [309].
Basigin expression is also upregulated in various cell types upon ER and oxidative stress [292,310], both of which could occur in COVID-19 patients [178] and could propagate viral infection. Additionally, others detected Basigin in the pancreas of patients with COVID-19 [163]. Basigin is also a receptor for cyclophilin A (CyPA) [311], a member of the immunophilin family that is crucial in promoting viral infections. Here, CyPA is involved in viral invasion by human immunodeficiency virus-1 [312] and SARS-CoV [313], with their ability to infect host cells depending on CyPA and Basigin interactions. Such interactions occur when the viral N-protein binds to CyPA which in turn recognizes Basigin on the host cell membrane [313] to allow for the internalization of the virus.
Basigin is also expressed in the pancreas, with some studies reporting that it is highly expressed in α-, β-, and γ-cells [147,184,314], while others did not detect it in the islets [315]. CyPA was detected in pancreatic β-cells and its expression increased in high glucose-stimulated β-cells [316]. Moreover, these authors demonstrated how CyPA knockdown also enhanced insulin secretion and decreased β-cell apoptosis.
Basigin becomes active when glycosylated and participates in several physiological processes [317]. Due to its inherent glycosylation properties, Basigin is a candidate for modifications caused by hyperglycemia or by excess AGEs accompanying diabetes and metabolic diseases. In agreement, Basigin expression was upregulated by relatively high glucose levels and AGEs [302,318,319] and hence may play a crucial role in priming virus binding to host cells during diabetes. Mahmoud et al. [320] further confirmed this by reporting on the increased expression of Basigin in response to relatively high glucose levels in primary human preadipocytes. The increased expression of Basigin in response to high glucose levels may promote SARS-CoV-2 entry into the host cells, which may help explain the relatively high mortality rate in diabetic COVID-19 patients. The role of CyPA in SARS-CoV-2 infection may also be of importance in the diabetic context.
However, studies question the role of Basigin as a SARS-CoV-2 co-receptor. For example, Ragotte et al. [321] found that it did not directly interact with SARS-CoV-2 S-proteins. Shilts et al. [322] further supported this finding, showing no direct interaction between Basigin and the full-length S-protein or its S1 subunit. Thus, the role of Basigin in SARS-CoV-2 pathogenesis remains unclear and requires further investigation.
5.3. Aminopeptidase N
Aminopeptidase N (APN) (or CD13) is also referred to as a ‘moonlighting enzyme’ due to its multiple functions [323]. Here, several studies found that APNs execute crucial regulatory functions during normal and pathologic immune responses [324,325,326]. For example, it plays a role in antigen processing [327,328], cell trafficking [329,330,331,332], and the processing of inflammatory mediators, all of which are crucial features of immune responses. It is also involved in peptide cleavage, viral infection, endocytosis, and cell signal transduction [333].
APN has been identified as a viral receptor for other CoVs such as the porcine respiratory CoV, porcine transmissible gastroenteritis virus, feline infectious peritonitis virus, feline enteric CoV, and canine CoV [334,335]. These data indicate that APNs may also be involved in SARS-CoV-2 host entry [336,337]. Although some dispute this [57,59], we postulate that APN is indeed a receptor. In support of our argument, another CoV (HCoV-229E) uses APN as a receptor and utilizes the endosomal pathway for cellular entry [338].
It is also involved in an alternative RAAS axis that includes aminopeptidase A (APA) and insulin-regulated aminopeptidase (IRAP). Ang2 is cleaved by APA into Ang3, which is then cleaved into Ang4 by APN and bound to IRAP [339]. IRAP serves multiple functions in different organs (e.g., adipocytes and muscle cells) where it can co-localize with GLUTs. For example, in response to insulin, it can be redistributed from the endosome to the cell surface of GLUT4 specialized vesicles [340]. Of note, this translocation is impaired in T2DM patients [341]. Others also link the APN/Ang4/IRAP axis to glucose homeostasis, where there was decreased basal and insulin-stimulated glucose uptake into muscle cells and adipocytes in IRAP-deficient mice [342].
As APN is expressed in the pancreas [343,344] and IRAP is expressed in several cell types (liver, kidney, lung, pancreas, and neurons) [345], a postulate can be made that the APN/Ang4/IRAP axis plays a similar role in pancreatic glucose homeostasis. In agreement, some researchers detected GLUT4 expression in human pancreatic islets [346], while Härdtner et al. [115] found an activation of the APN/Ang4/IRAP axis in pancreatic β-cells under hyperglycemic conditions. In support, others also reported that Ang4 stimulates insulin signaling and secretion [347,348].
Thus, it is our opinion that a greater understanding of the potential interaction between APN and SARS-CoV-2, and particularly the role of the APN/Ang4/IRAP axis in the context of hyperglycemia, may lead to valuable insights regarding viral entry mechanisms and susceptibility. Further research is, therefore, crucial to determine whether APN plays a meaningful role in terms of SARS-CoV-2 infection and if its regulation under hyperglycemic conditions offers a potential target for therapeutic interventions.
5.4. Early Endosomal Antigen-1 and Rab Proteins
Early endosomal antigen-1 (EEA1) is a crucial component of the endosomal fusion process [349,350]. Rab proteins regulate endocytic pathways and belong to the Ras superfamily of small GTPases [351] that regulate intracellular material transport and innate immunity [352,353]. Of interest, Rab5 is considered the master regulator of endocytic trafficking and regulates the transport of newly endocytosed vesicles from the plasma membrane to early endosomes [354] and the membrane association of EEA1 [355,356,357]. It participates in the formation of clathrin-coated vesicles, the fusion of clathrin-coated vesicles with early endosomes, and the fusion between early endosomes [358,359]. Rab7 regulates early endosome maturation and trafficking from late endosomes to lysosomes [360]. Rab9 can promote late endosome entry into the trans-Golgi network [361], while Rab11 is involved in vesicle cycling [362].
Various viruses often use Rab proteins, and hence EEA1, to regulate the viral endosome transport process. These include the human immunodeficiency virus [361], herpes simplex virus [363,364], classical swine fever virus [365], CoVs (including porcine delta-CoV [366]), and porcine epidemic diarrhea virus [367]. In addition, beta-CoVs, including SARS-CoV-2, utilize lysosomes for egress [368].
Although the role of Rab proteins in CoV infection remains unclear, the knockdown of early endosome-related genes (EEA1 and Rab5), late endosome-related genes (Rab7A and Rab7B), and late endosome to lysosomal maturation-related genes significantly inhibited the entry of the mouse hepatitis virus into host cells [260]. Furthermore, Atik et al. [369] showed that Rab5 expression was significantly increased in COVID-19 patients, indicating that SARS-CoV-2 entry into host cells may be utilizing host early endosomes and that EEA1 may be a marker of this process. Others also showed that SARS-CoV-2 can enter host cells through clathrin-mediated endocytosis and that the genome is trafficked to early endosomes in a Rab5-dependent manner [370]. In agreement, COVID-19 patients displayed increased Rab5 expression and decreased Rab7 and Rab11B expression [369]. Others also found that the EEA1 gene residing in an associated locus linked to COVID-19 disease severity suggests an overactive cellular response that may facilitate SARS-CoV-2 entry and processing, thereby contributing to COVID-19 severity [371]. These findings indicate that SARS-CoV-2 can utilize the host cell’s early endosomes to enter. However, more research is required to fully elucidate the full picture in this instance.
EEA1 is expressed in the cytosol, endosome, and plasma membrane in various organs, including the pancreas [372]. For example, Jung et al. [373] detected Rab5, EEA1, and clathrin expression in a rat insulinoma cell line (INS-1E). Moreover, others found Rab7A in β-cells, Rab5 and Rab7 in human pancreatic cell lines, and Rab11 in a MIN6 insulin-secreting cell line [374,375,376].
Of note, Rab5/7/11 are all involved in insulin secretion; for example, Rab5 overexpression upregulated both glucose-stimulated insulin secretion and the GLP-1 receptor-mediated potentiation of insulin secretion in cultured β-cells [377]. GLP-1 can improve the glucose sensing, proinsulin biosynthesis, survival, and proliferation of β-cells [378,379].
Rab7 interacting lysosomal protein (RILP) acts as a downstream effector for Rab7 and is responsible for stabilizing Rab7 on the endosomal membrane [380,381]. RILP regulates insulin secretion via the mediation of lysosomal degradation of proinsulin in a Rab7-dependent manner [382]. These authors also demonstrated that RILP was upregulated in diabetic rat and mouse models, implying a potential pathogenic role in metabolic diseases such as DM. In agreement, some found that the inhibition of Rab7A may promote β-cell survival in the context of metabolic stress and prevent the onset of T2DM [375].
Rab11 participates in insulin granule exocytosis in β-cells [374] and is also involved in the endosomal recycling, sorting, and exocytotic GLUT4 movement in cardiac muscle [383]. It will be interesting to determine whether it plays a similar role in pancreatic β-cells.
Thus, EEA1 and Rab proteins play a crucial role in the intracellular trafficking and fusion of endosomes, a potential entry pathway for SARS-CoV-2. A greater understanding of how diabetes may influence the expression/activity of such proteins should provide valuable insights into alternative entry mechanisms, particularly in scenarios where the expression of surface proteases is limited. Further research is therefore necessary to determine whether hyperglycemia directly disrupts the regulation of EEA1 and Rab proteins, potentially affecting SARS-CoV-2 endosomal entry and susceptibility, especially in diabetic patients.
5.5. Valosin-Containing Protein
Valosin-containing protein (VCP) is a stress response protein involved in cell death and survival and is also referred to as ATPase p97 [384]. VCP can function as an apoptotic regulator and is an essential target in Akt signaling cascades [385]. It also functions in several other cellular functions, such as ER-associated degradation (ERAD), mitochondrial-associated protein degradation [386,387], the ubiquitin–proteasome system (UPS) [388], DNA replication and break repair [389,390,391], nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation [392,393], and endomembrane fusion [394].
VCP also plays a role in endosomal trafficking [395,396], where it mediates endosome fusion through binding to clathrin, syntaxin, and EEA1 [394,397]. It can also control the EEA1 oligomeric state to regulate endosome fusion and trafficking [394]. This indicates a potential relationship between VCP and EEA1.
VCP also participates in multiple stages of virus production, including entry and uncoating, intracellular trafficking, nucleic acid replication, and egress [388,389,398,399,400,401,402,403]. Regarding its role in CoVs, VCP depletion decreased HCoV-229E replication in a Huh7 hepatoma cell line [404]. These authors also demonstrated how VCP silencing inhibited early-stage degradation of viral N-proteins and the consequent accumulation of virion-associated N-proteins. Since VCP is linked to the UPS, the accumulation of N-proteins could indicate that VCP is directly required for N-protein degradation. VCP inhibition also triggers antiviral effects, especially during the early stages of the viral life cycle, during virus uncoating and the RNA replication of HCoVs (HCoV-229E and HCoV-OC43) [405]. This suggests that VCP plays a role in maturing endosomes loaded with viral material for CoV replication. Research by Ramanathan et al. [394] and Meyer et al. [388] further confirms this.
Although it is evolutionarily conserved and ubiquitously expressed [397], VCP is predominantly localized in the cytosol while also found on ER membranes, the Golgi apparatus, the nucleus, mitochondria, and endosomes. Moreover, Nicolls et al. [406] found that it is expressed in normal adult mouse islets, while others detected VCP in the rat insulin-producing (RINm5F) cell line and human pancreatic islets [384]. Of note, the Human Protein Atlas [407] indicates that VCP protein levels in pancreatic endocrine cells are relatively low as it is mostly expressed in pancreatic α-cells.
Focusing on insulin synthesis, proinsulin molecules initially translocate into the ER lumen and then the Golgi apparatus, where they are subsequently processed into insulin and C-peptides [408]. Proinsulin may also enter the ERAD and undergo degradation in pancreatic β-cells [409]. As pancreatic β-cells from T1DM patients display relatively higher degrees of ER stress [410], ERAD proteins such as VCP are upregulated [411]. We therefore postulate that such accumulated substrates will be targeted for degradation and therefore not undergo processing to insulin. In support, VCP is associated with a model of T2DM [412] and is an ERAD protein that plays a crucial role in proinsulin degradation.
High glucose can trigger GLUT2 degradation in lysosomes in insulin-secreting Min6 cells [413]. GLUT2 mediates the entry and exit of glucose in organs such as the pancreas [414] and is integral for insulin secretion from β-cells. In agreement, Guillam et al. [415] found that GLUT2-KO mice exhibited diminished glucose clearance and plasma insulin levels due to impaired glucose-stimulated insulin secretion. GLUT2 is expressed on the apical membrane of β-cells [416,417], but lower levels are found in the diabetic pancreas [414]. Considering that EEA1 is an early endosome marker [418] and VCP facilitates protein degradation via lysosomal degradation [419], we propose that both proteins are involved in GLUT2 degradation and would be expected to be upregulated under high glucose conditions in pancreatic β-cells.
While VCP is known to be involved in various cellular processes, its specific contribution to viral entry or replication for SARS-CoV-2 needs further exploration. Here, an understanding of how hyperglycemia may influence VCP function could provide insights into potential mechanisms affecting viral pathogenesis in diabetic patients. Further research is, therefore, crucial to elucidate the specific role of VCP in SARS-CoV-2 infection and to determine if its regulation under hyperglycemic conditions presents a target for therapeutic interventions.
SARS-CoV-2 can also enter host cells via endocytosis. This pathway necessitates a complex interplay between viral and host factors. Upon internalization, the virus is trafficked through the endosomal compartment, where acidic conditions facilitate viral entry. Endosomal proteases such as Cathepsins B/L are crucial for viral S-protein processing. Additionally, host proteins (Basigin, APN, EEA1 together with Rab5/7/11, and VCP) possibly play pivotal roles in endosomal trafficking and viral uncoating. The precise mechanisms underlying the impact of hyperglycemia on these proteins and subsequent viral entry remain to be fully elucidated, but emerging evidence suggests potential alterations in protein expression and possibly activity. A comprehensive understanding of these factors is essential for developing targeted therapeutic interventions. The following Table 3 summarizes the key proteins involved in the endosomal entry of SARS-CoV-2 and explores the potential impact of hyperglycemia on them.
Table 3.
A summary of the potential influence of hyperglycemia on the proteins that mediate the endosomal entry of SARS-CoV-2.
6. Conclusions
As the specific mechanisms of SARS-CoV-2 host cell entry are becoming increasingly understood, it remains unclear how pre-existing conditions like DM and hyperglycemia can worsen COVID-19 outcomes. This review highlighted the potential interplay between hyperglycemia, SARS-CoV-2 entry, and accessory proteins in pancreatic β-cells. Current research reveals how hyperglycemia is potentiating the viral entry of SARS-CoV-2 into pancreatic β-cells, resulting in their dysfunction and inability to produce insulin. Further research is warranted to elucidate the specific mechanisms involved and explore potential therapeutic targets to mitigate the detrimental effects of hyperglycemia on COVID-19 pathogenesis in diabetic individuals.
Author Contributions
Conceptualization, T.M.M. and D.E.J.; writing—original draft presentation, T.M.M.; writing—review and editing, M.F.E. and D.E.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the South African Medical Research Council (SAMRC) and Stellenbosch University to D.E.J.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable.
Acknowledgments
Figure 1 and Figure 2 were created with BioRender.com, a scientific image and illustration software. The authors wish to acknowledge the financial support provided by the South African Medical Research Council and Stellenbosch University (to D.E.J.) and the National Research Foundation (NRF; to T.M.M.).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- World Health Organization. COVID-19 Cases|WHO COVID-19 Dashboard. Available online: https://data.who.int/dashboards/covid19/cases (accessed on 7 July 2024).
- Center for Disease Control and Prevention. Coronavirus Disease 2019 (COVID-19)—Symptoms. Available online: https://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.html (accessed on 10 June 2024).
- Barron, E.; Bakhai, C.; Kar, P.; Weaver, A.; Bradley, D.; Ismail, H.; Knighton, P.; Holman, N.; Khunti, K.; Sattar, N.; et al. Associations of Type 1 and Type 2 Diabetes with COVID-19-Related Mortality in England: A Whole-Population Study. Lancet Diabetes Endocrinol. 2020, 8, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and Impact of Cardiovascular Metabolic Diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhang, X.; Jiang, F.; Zhang, X.; Hu, N.; Bimu, C.; Feng, J.; Yan, S.; Guan, Y.; Xu, D.; et al. Clinical Characteristics and Risk Factors for Mortality of COVID-19 Patients with Diabetes in Wuhan, China: A Two-Center, Retrospective Study. Diabetes Care 2020, 43, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, H.; Szarpak, L.; Kosior, D.; Wieczorek, W.; Szarpak, A.; Al-Jeabory, M.; Gawel, W.; Gasecka, A.; Jaguszewski, M.J.; Jarosz-Chobot, P. Impact of Diabetes Mellitus on In-Hospital Mortality in Adult Patients with COVID-19: A Systematic Review and Meta-Analysis. Acta Diabetol. 2021, 58, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, J.; Sun, X.; Wang, L.; Xu, Y.; Zhang, Y.; Liu, X.; Dong, C. Influence of Diabetes Mellitus on the Severity and Fatality of SARS-CoV-2 (COVID-19) Infection. Diabetes Obes. Metab. 2020, 22, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Maddaloni, E.; D’Onofrio, L.; Alessandri, F.; Mignogna, C.; Leto, G.; Coraggio, L.; Sterpetti, S.; Pascarella, G.; Mezzaroma, I.; Lichtner, M.; et al. Clinical Features of Patients with Type 2 Diabetes with and without COVID-19: A Case Control Study (CoViDiab I). Diabetes Res. Clin. Pract. 2020, 169, 108454. [Google Scholar] [CrossRef] [PubMed]
- Dey, R.K.; Hilmy, A.I.; Imad, H.A.; Yoosuf, A.A.; Latheef, A.A. COVID-19 and Emergencies in Patients with Diabetes: Two Case Reports. J. Med. Case Rep. 2021, 15, 57. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, D.B.; Budhathoki, P.; Raut, S.; Adhikari, S.; Ghimire, P.; Thapaliya, S.; Rabaan, A.A.; Karki, B.J. New-Onset Diabetes in COVID-19 and Clinical Outcomes: A Systematic Review and Meta-Analysis. World J. Virol. 2021, 10, 275–287. [Google Scholar] [CrossRef]
- Wang, S.; Ma, P.; Zhang, S.; Song, S.; Wang, Z.; Ma, Y.; Xu, J.; Wu, F.; Duan, L.; Yin, Z.; et al. Fasting Blood Glucose at Admission Is an Independent Predictor for 28-Day Mortality in Patients with COVID-19 without Previous Diagnosis of Diabetes: A Multi-Centre Retrospective Study. Diabetologia 2020, 63, 2102–2111. [Google Scholar] [CrossRef]
- Carrasco-Sánchez, F.J.; López-Carmona, M.D.; Martínez-Marcos, F.J.; Pérez-Belmonte, L.M.; Hidalgo-Jiménez, A.; Buonaiuto, V.; Suárez Fernández, C.; Freire Castro, S.J.; Luordo, D.; Pesqueira Fontan, P.M.; et al. Admission Hyperglycaemia as a Predictor of Mortality in Patients Hospitalized with COVID-19 Regardless of Diabetes Status: Data from the Spanish SEMI-COVID-19 Registry. Ann. Med. 2021, 53, 103–116. [Google Scholar] [CrossRef]
- Jivanji, C.J.; Asrani, V.M.; Windsor, J.A.; Petrov, M.S. New-Onset Diabetes After Acute and Critical Illness: A Systematic Review. Mayo Clin. Proc. 2017, 92, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tian, S.; Chen, T.; Cui, Z.; Shi, N.; Zhong, X.; Qiu, K.; Zhang, J.; Zeng, T.; Chen, L.; et al. Newly Diagnosed Diabetes Is Associated with a Higher Risk of Mortality than Known Diabetes in Hospitalized Patients with COVID-19. Diabetes Obes. Metab. 2020, 22, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Hoong, C.W.S.; Yeo, X.E.; Lin, Y.; Ooi, S.T.; Yeoh, E. High Glycaemic Variability Is Associated with Progression of COVID-19. Acta Diabetol. 2021, 58, 1701–1704. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, S.R.; Majumder, A.; Sanyal, D.; Biswas, A.; Bhattacharjee, K. 1098-P: Admission Blood Glucose but Not HbA1c Predicts Mortality in People with Diabetes Hospitalized with COVID-19 Infection. Diabetes 2021, 70, 1098-P. [Google Scholar] [CrossRef]
- Zhu, L.; She, Z.-G.; Cheng, X.; Qin, J.-J.; Zhang, X.-J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-Existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077.e3. [Google Scholar] [CrossRef]
- Chee, Y.J.; Tan, S.K.; Yeoh, E. Dissecting the Interaction between COVID-19 and Diabetes Mellitus. J. Diabetes Investig. 2020, 11, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, R.; Wang, J.; Cheng, Q.; Zhang, R.; Zhang, S.; Le, Y.; Wang, H.; Xiao, W.; Gao, H.; et al. Diabetes, Even Newly Defined by HbA1c Testing, Is Associated with an Increased Risk of in-Hospital Death in Adults with COVID-19. BMC Endocr. Disord. 2021, 21, 56. [Google Scholar] [CrossRef] [PubMed]
- Unsworth, R.; Wallace, S.; Oliver, N.S.; Yeung, S.; Kshirsagar, A.; Naidu, H.; Kwong, R.M.W.; Kumar, P.; Logan, K.M. New-Onset Type 1 Diabetes in Children During COVID-19: Multicenter Regional Findings in the U.K. Diabetes Care 2020, 43, e170–e171. [Google Scholar] [CrossRef]
- Ebekozien, O.A.; Noor, N.; Gallagher, M.P.; Alonso, G.T. Type 1 Diabetes and COVID-19: Preliminary Findings From a Multicenter Surveillance Study in the U.S. Diabetes Care 2020, 43, e83–e85. [Google Scholar] [CrossRef]
- Sathish, T.; Kapoor, N.; Cao, Y.; Tapp, R.J.; Zimmet, P. Proportion of Newly Diagnosed Diabetes in COVID-19 Patients: A Systematic Review and Meta-Analysis. Diabetes Obes. Metab. 2021, 23, 870–874. [Google Scholar] [CrossRef]
- Fadini, G.P.; Morieri, M.L.; Boscari, F.; Fioretto, P.; Maran, A.; Busetto, L.; Bonora, B.M.; Selmin, E.; Arcidiacono, G.; Pinelli, S.; et al. Newly-Diagnosed Diabetes and Admission Hyperglycemia Predict COVID-19 Severity by Aggravating Respiratory Deterioration. Diabetes Res. Clin. Pract. 2020, 168, 108374. [Google Scholar] [CrossRef] [PubMed]
- Armeni, E.; Aziz, U.; Qamar, S.; Nasir, S.; Nethaji, C.; Negus, R.; Murch, N.; Beynon, H.C.; Bouloux, P.; Rosenthal, M.; et al. Protracted Ketonaemia in Hyperglycaemic Emergencies in COVID-19: A Retrospective Case Series. Lancet Diabetes Endocrinol. 2020, 8, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Chee, Y.J.; Ng, S.J.H.; Yeoh, E. Diabetic Ketoacidosis Precipitated by COVID-19 in a Patient with Newly Diagnosed Diabetes Mellitus. Diabetes Res. Clin. Pract. 2020, 164, 108166. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.; Amiel, S.A.; Zimmet, P.; Alberti, G.; Bornstein, S.; Eckel, R.H.; Mingrone, G.; Boehm, B.; Cooper, M.E.; Chai, Z.; et al. New-Onset Diabetes in COVID-19. N. Engl. J. Med. 2020, 383, 789–790. [Google Scholar] [CrossRef]
- Azar, W.S.; Njeim, R.; Fares, A.H.; Azar, N.S.; Azar, S.T.; El Sayed, M.; Eid, A.A. COVID-19 and Diabetes Mellitus: How One Pandemic Worsens the Other. Rev. Endocr. Metab. Disord. 2020, 21, 451–463. [Google Scholar] [CrossRef]
- Lima-Martínez, M.M.; Carrera Boada, C.; Madera-Silva, M.D.; Marín, W.; Contreras, M. COVID-19 and Diabetes: A Bidirectional Relationship. Clin. Investig. Arter. 2021, 33, 151–157. [Google Scholar] [CrossRef]
- Wu, C.-T.; Lidsky, P.V.; Xiao, Y.; Lee, I.T.; Cheng, R.; Nakayama, T.; Jiang, S.; Demeter, J.; Bevacqua, R.J.; Chang, C.A.; et al. SARS-CoV-2 Infects Human Pancreatic β Cells and Elicits β Cell Impairment. Cell Metab. 2021, 33, 1565–1576.e5. [Google Scholar] [CrossRef]
- Tang, X.; Uhl, S.; Zhang, T.; Xue, D.; Li, B.; Vandana, J.J.; Acklin, J.A.; Bonnycastle, L.L.; Narisu, N.; Erdos, M.R.; et al. SARS-CoV-2 Infection Induces Beta Cell Transdifferentiation. Cell Metab. 2021, 33, 1577–1591.e7. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Müller, J.A.; Groß, R.; Conzelmann, C.; Krüger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 Infects and Replicates in Cells of the Human Endocrine and Exocrine Pancreas. Nat. Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef]
- Wu, J.; Huang, J.; Zhu, G.; Wang, Q.; Lv, Q.; Huang, Y.; Yu, Y.; Si, X.; Yi, H.; Wang, C.; et al. Elevation of Blood Glucose Level Predicts Worse Outcomes in Hospitalized Patients with COVID-19: A Retrospective Cohort Study. BMJ Open Diabetes Res. Care 2020, 8, e001476. [Google Scholar] [CrossRef] [PubMed]
- Bode, B.; Garrett, V.; Messler, J.; McFarland, R.; Crowe, J.; Booth, R.; Klonoff, D.C. Glycemic Characteristics and Clinical Outcomes of COVID-19 Patients Hospitalized in the United States. J. Diabetes Sci. Technol. 2020, 14, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, G.; Audrey, J.; Wangsaputra, V.K.; Tamara, A.; Tahapary, D.L. High Admission Blood Glucose Independently Predicts Poor Prognosis in COVID-19 Patients: A Systematic Review and Dose-Response Meta-Analysis. Diabetes Res. Clin. Pract. 2021, 171, 108561. [Google Scholar] [CrossRef] [PubMed]
- Kazakou, P.; Paschou, S.A.; Psaltopoulou, T.; Gavriatopoulou, M.; Korompoki, E.; Stefanaki, K.; Kanouta, F.; Kassi, G.N.; Dimopoulos, M.-A.; Mitrakou, A. Early and Late Endocrine Complications of COVID-19. Endocr. Connect. 2021, 10, R229–R239. [Google Scholar] [CrossRef] [PubMed]
- Finucane, F.M.; Davenport, C. Coronavirus and Obesity: Could Insulin Resistance Mediate the Severity of COVID-19 Infection? Front. Public. Health 2020, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Alsadhan, I.; Alruwashid, S.; Alhamad, M.; Alajmi, S.; Alshehri, S.; Alfadhli, E.; Ekhzaimy, A. Diabetic Ketoacidosis Precipitated by Coronavirus Disease 2019 Infection: Case Series. Curr. Ther. Res. Clin. Exp. 2020, 93, 100609. [Google Scholar] [CrossRef]
- Kim, N.Y.; Ha, E.; Moon, J.S.; Lee, Y.H.; Choi, E.Y. Acute Hyperglycemic Crises with Coronavirus Disease-19: Case Reports. Diabetes Metab. J. 2020, 44, 349–353. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Chen, J.; Zuo, X.; Zhang, H.; Deng, A. COVID-19 Infection May Cause Ketosis and Ketoacidosis. Diabetes Obes. Metab. 2020, 22, 1935–1941. [Google Scholar] [CrossRef]
- Rayman, G.; Lumb, A.; Kennon, B.; Cottrell, C.; Nagi, D.; Page, E.; Voigt, D.; Courtney, H.; Atkins, H.; Platts, J.; et al. Guidance on the Management of Diabetic Ketoacidosis in the Exceptional Circumstances of the COVID-19 Pandemic. Diabet. Med. 2020, 37, 1214–1216. [Google Scholar] [CrossRef]
- Maddaloni, E.; Buzzetti, R. COVID-19 and Diabetes Mellitus: Unveiling the Interaction of Two Pandemics. Diabetes Metab. Res. Rev. 2020, 36, e33213321. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Rorsman, P. Diabetes Mellitus and the β Cell: The Last Ten Years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Newgard, C.B. Mechanisms Controlling Pancreatic Islet Cell Function in Insulin Secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Accili, D. Can COVID-19 Cause Diabetes? Nat. Metab. 2021, 3, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Akkus, C.; Yilmaz, H.; Mizrak, S.; Adibelli, Z.; Akdas, O.; Duran, C. Development of Pancreatic Injuries in the Course of COVID-19. Acta Gastroenterol. Belg. 2020, 83, 585–592. [Google Scholar] [PubMed]
- Aloysius, M.M.; Thatti, A.; Gupta, A.; Sharma, N.; Bansal, P.; Goyal, H. COVID-19 Presenting as Acute Pancreatitis. Pancreatology 2020, 20, 1026–1027. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Gupta, V.; Misra, A. COVID19 Induced Acute Pancreatitis and Pancreatic Necrosis in a Patient with Type 2 Diabetes. Diabetes Metab. Syndr. 2020, 14, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Hadi, A.; Werge, M.; Kristiansen, K.T.; Pedersen, U.G.; Karstensen, J.G.; Novovic, S.; Gluud, L.L. Coronavirus Disease-19 (COVID-19) Associated with Severe Acute Pancreatitis: Case Report on Three Family Members. Pancreatology 2020, 20, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Anand, E.R.; Major, C.; Pickering, O.; Nelson, M. Acute Pancreatitis in a COVID-19 Patient. Br. J. Surg. 2020, 107, e182. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, N.K.; Karmakar, B.K.; Taylor, O.M. Coronavirus Disease-19 (COVID-19) Associated with Acute Necrotising Pancreatitis (ANP). BMJ Case Rep. 2020, 13, e237903. [Google Scholar] [CrossRef]
- Liu, F.; Long, X.; Zhang, B.; Zhang, W.; Chen, X.; Zhang, Z. ACE2 Expression in Pancreas May Cause Pancreatic Damage After SARS-CoV-2 Infection. Clin. Gastroenterol. Hepatol. 2020, 18, 2128–2130.e2. [Google Scholar] [CrossRef]
- Szatmary, P.; Arora, A.; Thomas Raraty, M.G.; Joseph Dunne, D.F.; Baron, R.D.; Halloran, C.M. Emerging Phenotype of Severe Acute Respiratory Syndrome-Coronavirus 2-Associated Pancreatitis. Gastroenterology 2020, 159, 1551–1554. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Dalan, R.; Hopkins, D.; Mingrone, G.; Boehm, B.O. Endocrine and Metabolic Link to Coronavirus Infection. Nat. Rev. Endocrinol. 2020, 16, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, N.P.; Ranathunga, I.; Ratnasamy, V.; Wijewickrama, P.S.A.; Dissanayake, H.A.; Yogendranathan, N.; Gamage, K.K.K.; de Silva, N.L.; Sumanatilleke, M.; Katulanda, P.; et al. The Impact of SARS-CoV-2 Virus Infection on the Endocrine System. J. Endocr. Soc. 2020, 4, bvaa082. [Google Scholar] [CrossRef] [PubMed]
- Steenblock, C.; Todorov, V.; Kanczkowski, W.; Eisenhofer, G.; Schedl, A.; Wong, M.-L.; Licinio, J.; Bauer, M.; Young, A.H.; Gainetdinov, R.R.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and the Neuroendocrine Stress Axis. Mol. Psychiatry 2020, 25, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Fignani, D.; Licata, G.; Brusco, N.; Nigi, L.; Grieco, G.E.; Marselli, L.; Overbergh, L.; Gysemans, C.; Colli, M.L.; Marchetti, P.; et al. SARS-CoV-2 Receptor Angiotensin I-Converting Enzyme Type 2 (ACE2) Is Expressed in Human Pancreatic β-Cells and in the Human Pancreas Microvasculature. Front. Endocrinol. 2020, 11, 596898. [Google Scholar] [CrossRef] [PubMed]
- Bonyek-Silva, I.; Machado, A.F.A.; Cerqueira-Silva, T.; Nunes, S.; Silva Cruz, M.R.; Silva, J.; Santos, R.L.; Barral, A.; Oliveira, P.R.S.; Khouri, R.; et al. LTB4-Driven Inflammation and Increased Expression of ALOX5/ACE2 During Severe COVID-19 in Individuals With Diabetes. Diabetes 2021, 70, 2120–2130. [Google Scholar] [CrossRef]
- Samavati, L.; Uhal, B.D. ACE2, Much More Than Just a Receptor for SARS-CoV-2. Front. Cell Infect. Microbiol. 2020, 10, 317. [Google Scholar] [CrossRef]
- Roca-Ho, H.; Palau, V.; Gimeno, J.; Pascual, J.; Soler, M.J.; Riera, M. Angiotensin-Converting Enzyme 2 Influences Pancreatic and Renal Function in Diabetic Mice. Lab. Investig. 2020, 100, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, K.H.; Xia, H.; Pedersen, K.B.; Speth, R.C.; Lazartigues, E. Pancreatic Angiotensin-Converting Enzyme 2 Improves Glycemia in Angiotensin II-Infused Mice. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E874–E884. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Liu, Z.; Chen, D. Human Coronaviruses: Origin, Host and Receptor. J. Clin. Virol. 2022, 155, 105246. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wunderink, R.G. MERS, SARS and Other Coronaviruses as Causes of Pneumonia. Respirology 2018, 23, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-W.; Lin, C.-W. Human Coronaviruses: Clinical Features and Phylogenetic Analysis. BioMedicine 2013, 3, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic Origins of Human Coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and Bat RaTG13 Spike Glycoprotein Structures Inform on Virus Evolution and Furin-Cleavage Effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host Cell Proteases: Critical Determinants of Coronavirus Tropism and Pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus Membrane Fusion Mechanism Offers a Potential Target for Antiviral Development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andréo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e0012822. [Google Scholar] [CrossRef]
- Almehdi, A.M.; Khoder, G.; Alchakee, A.S.; Alsayyid, A.T.; Sarg, N.H.; Soliman, S.S.M. SARS-CoV-2 Spike Protein: Pathogenesis, Vaccines, and Potential Therapies. Infection 2021, 49, 855–876. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, R.; Huo, S.; Zhou, Y.; Jiang, S.; Wang, Q.; Yu, F. Molecular Mechanism of Interaction between SARS-CoV-2 and Host Cells and Interventional Therapy. Signal Transduct. Target. Ther. 2021, 6, 233. [Google Scholar] [CrossRef] [PubMed]
- Pizzato, M.; Baraldi, C.; Boscato Sopetto, G.; Finozzi, D.; Gentile, C.; Gentile, M.D.; Marconi, R.; Paladino, D.; Raoss, A.; Riedmiller, I.; et al. SARS-CoV-2 and the Host Cell: A Tale of Interactions. Front. Virol. 2022, 1, 815388. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-Converting Enzyme 2 (ACE2) as a SARS-CoV-2 Receptor: Molecular Mechanisms and Potential Therapeutic Target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The Pivotal Link between ACE2 Deficiency and SARS-CoV-2 Infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, X.; Wang, X.; Peng, L.; Song, G.; He, J. Angiotensin(1-7) Improves Islet Function in Diabetes Through Reducing JNK/Caspase-3 Signaling. Horm. Metab. Res. 2022, 54, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Bindom, S.M.; Lazartigues, E. The Sweeter Side of ACE2: Physiological Evidence for a Role in Diabetes. Mol. Cell Endocrinol. 2009, 302, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-Angiotensin-Aldosterone (RAAS): The Ubiquitous System for Homeostasis and Pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.M.E.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The Emerging Role of ACE2 in Physiology and Disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A Peptidase in the Renin-Angiotensin System, a SARS Receptor, and a Partner for Amino Acid Transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef]
- Memon, B.; Abdelalim, E.M. ACE2 Function in the Pancreatic Islet: Implications for Relationship between SARS-CoV-2 and Diabetes. Acta Physiol. 2021, 233, e13733. [Google Scholar] [CrossRef]
- Batlle, D.; Jose Soler, M.; Ye, M. ACE2 and Diabetes: ACE of ACEs? Diabetes 2010, 59, 2994–2996. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Ferreira, A.J.; Simões E Silva, A.C. Recent Advances in the Angiotensin-Converting Enzyme 2-Angiotensin(1-7)-Mas Axis. Exp. Physiol. 2008, 93, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The Protein Expression Profile of ACE2 in Human Tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.E.; Thurman, A.; Pezzulo, A.A.; Leidinger, M.R.; Klesney-Tait, J.A.; Karp, P.H.; Tan, P.; Wohlford-Lenane, C.; McCray, P.B.; Meyerholz, D.K. Heterogeneous Expression of the SARS-Coronavirus-2 Receptor ACE2 in the Human Respiratory Tract. EBioMedicine 2020, 60, 102976. [Google Scholar] [CrossRef] [PubMed]
- Steenblock, C.; Richter, S.; Berger, I.; Barovic, M.; Schmid, J.; Schubert, U.; Jarzebska, N.; von Mässenhausen, A.; Linkermann, A.; Schürmann, A.; et al. Viral Infiltration of Pancreatic Islets in Patients with COVID-19. Nat. Commun. 2021, 12, 3534. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.-J.; Yang, J.-K.; Lin, S.-S.; Ji, X.-J.; Guo, L.-M. Loss of Angiotensin-Converting Enzyme 2 Leads to Impaired Glucose Homeostasis in Mice. Endocrine 2008, 34, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.J.; Yang, J.K. Tissue-Specific Pattern of Angiotensin-Converting Enzyme 2 Expression in Rat Pancreas. J. Int. Med. Res. 2010, 38, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Taneera, J.; El-Huneidi, W.; Hamad, M.; Mohammed, A.K.; Elaraby, E.; Hachim, M.Y. Expression Profile of SARS-CoV-2 Host Receptors in Human Pancreatic Islets Revealed Upregulation of ACE2 in Diabetic Donors. Biology 2020, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffré, F.; et al. A Human Pluripotent Stem Cell-Based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136.e7. [Google Scholar] [CrossRef]
- Shaharuddin, S.H.; Wang, V.; Santos, R.S.; Gross, A.; Wang, Y.; Jawanda, H.; Zhang, Y.; Hasan, W.; Garcia, G.; Arumugaswami, V.; et al. Deleterious Effects of SARS-CoV-2 Infection on Human Pancreatic Cells. Front. Cell Infect. Microbiol. 2021, 11, 678482. [Google Scholar] [CrossRef] [PubMed]
- Coate, K.C.; Cha, J.; Shrestha, S.; Wang, W.; Gonçalves, L.M.; Almaça, J.; Kapp, M.E.; Fasolino, M.; Morgan, A.; Dai, C.; et al. SARS-CoV-2 Cell Entry Factors ACE2 and TMPRSS2 Are Expressed in the Microvasculature and Ducts of Human Pancreas but Are Not Enriched in β Cells. Cell Metab. 2020, 32, 1028–1040.e4. [Google Scholar] [CrossRef] [PubMed]
- Kusmartseva, I.; Wu, W.; Syed, F.; Van Der Heide, V.; Jorgensen, M.; Joseph, P.; Tang, X.; Candelario-Jalil, E.; Yang, C.; Nick, H.; et al. Expression of SARS-CoV-2 Entry Factors in the Pancreas of Normal Organ Donors and Individuals with COVID-19. Cell Metab. 2020, 32, 1041–1051.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Sun, W.; Jiang, P. Role of the ACE2/Ang-(1-7)/Mas Axis in Glucose Metabolism. Rev. Cardiovasc. Med. 2021, 22, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Bindom, S.M.; Hans, C.P.; Xia, H.; Boulares, A.H.; Lazartigues, E. Angiotensin I–Converting Enzyme Type 2 (ACE2) Gene Therapy Improves Glycemic Control in Diabetic Mice. Diabetes 2010, 59, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.; Yiannikouris, F.; Thatcher, S.; Cassis, L. ACE2 Deficiency Reduces β-Cell Mass and Impairs β-Cell Proliferation in Obese C57BL/6 Mice. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E621–E631. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liang, J.; Leung, P.S. The ACE2/Ang-(1-7)/Mas Axis Regulates the Development of Pancreatic Endocrine Cells in Mouse Embryos. PLoS ONE 2015, 10, e0128216. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Cooper, M.E.; Thomas, M.C. Role of the Renin-Angiotensin System in the Endocrine Pancreas: Implications for the Development of Diabetes. Int. J. Biochem. Cell Biol. 2006, 38, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Lastra, G.; Manrique, C. The Expanding Role of Oxidative Stress, Renin Angiotensin System, and Beta-Cell Dysfunction in the Cardiometabolic Syndrome and Type 2 Diabetes Mellitus. Antioxid. Redox Signal 2007, 9, 943–954. [Google Scholar] [CrossRef]
- Yuan, L.; Li, X.; Xu, G.-L.; Qi, C.-J. Effects of Renin-Angiotensin System Blockade on Islet Function in Diabetic Rats. J. Endocrinol. Investig. 2010, 33, 13–19. [Google Scholar] [CrossRef]
- Prasannarong, M.; Santos, F.R.; Henriksen, E.J. ANG-(1-7) Reduces ANG II-Induced Insulin Resistance by Enhancing Akt Phosphorylation via a Mas Receptor-Dependent Mechanism in Rat Skeletal Muscle. Biochem. Biophys. Res. Commun. 2012, 426, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leung, P.S. The Role of Renin-Angiotensin System in Cellular Differentiation: Implications in Pancreatic Islet Cell Development and Islet Transplantation. Mol. Cell Endocrinol. 2013, 381, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Härdtner, C.; Mörke, C.; Walther, R.; Wolke, C.; Lendeckel, U. High Glucose Activates the Alternative ACE2/Ang-(1-7)/Mas and APN/Ang IV/IRAP RAS Axes in Pancreatic β-Cells. Int. J. Mol. Med. 2013, 32, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-L.; Wang, Y.; Yuan, L.; Li, Y.; Li, X.-Y. The Angiotensin-Converting Enzyme 2/Angiotensin (1–7)/Mas Axis Protects the Function of Pancreatic β Cells by Improving the Function of Islet Microvascular Endothelial Cells. Int. J. Mol. Med. 2014, 34, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, C.; Wang, L.; Cao, X.; Wang, Y.Y.; Yang, J.K. Antioxidant Effect of Angiotensin (1–7) in the Protection of Pancreatic β Cell Function. Mol. Med. Rep. 2016, 14, 1963–1969. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Scisciola, L.; Sardu, C.; Trotta, M.C.; De Feo, M.; Maiello, C.; Mascolo, P.; De Micco, F.; Turriziani, F.; Municinò, E.; et al. Glycated ACE2 Receptor in Diabetes: Open Door for SARS-CoV-2 Entry in Cardiomyocyte. Cardiovasc. Diabetol. 2021, 20, 99. [Google Scholar] [CrossRef] [PubMed]
- Sartore, G.; Bassani, D.; Ragazzi, E.; Traldi, P.; Lapolla, A.; Moro, S. In Silico Evaluation of the Interaction between ACE2 and SARS-CoV-2 Spike Protein in a Hyperglycemic Environment. Sci. Rep. 2021, 11, 22860. [Google Scholar] [CrossRef] [PubMed]
- Spiro, R.G. Glycoproteins. Adv. Protein Chem. 1973, 27, 349–467. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W. Glycosylation. Curr. Opin. Cell Biol. 1992, 4, 1017–1023. [Google Scholar] [CrossRef]
- Spiro, R.G. Protein Glycosylation: Nature, Distribution, Enzymatic Formation, and Disease Implications of Glycopeptide Bonds. Glycobiology 2002, 12, 43R–56R. [Google Scholar] [CrossRef]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced Glycation End-Products: A Review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Mishra, M. Do Advanced Glycation End Products and Its Receptor Play a Role in Pathophysiology of Hypertension? Int. J. Angiol. 2017, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sellegounder, D.; Zafari, P.; Rajabinejad, M.; Taghadosi, M.; Kapahi, P. Advanced Glycation End Products (AGEs) and Its Receptor, RAGE, Modulate Age-Dependent COVID-19 Morbidity and Mortality. A Review and Hypothesis. Int. Immunopharmacol. 2021, 98, 107806. [Google Scholar] [CrossRef] [PubMed]
- Masre, S.F.; Jufri, N.F.; Ibrahim, F.W.; Abdul Raub, S.H. Classical and Alternative Receptors for SARS-CoV-2 Therapeutic Strategy. Rev. Med. Virol. 2021, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S. Virus–Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Jaimes, J.A.; Whittaker, G.R. Molecular Diversity of Coronavirus Host Cell Entry Receptors. FEMS Microbiol. Rev. 2021, 45, fuaa057. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular Basis of Binding between Novel Human Coronavirus MERS-CoV and Its Receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef] [PubMed]
- van Doremalen, N.; Miazgowicz, K.L.; Milne-Price, S.; Bushmaker, T.; Robertson, S.; Scott, D.; Kinne, J.; McLellan, J.S.; Zhu, J.; Munster, V.J. Host Species Restriction of Middle East Respiratory Syndrome Coronavirus through Its Receptor, Dipeptidyl Peptidase 4. J. Virol. 2014, 88, 9220–9232. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, Y.L.; Jeong, Y.; Jang, Y.-S. Middle East Respiratory Syndrome-Coronavirus Infection into Established hDPP4-Transgenic Mice Accelerates Lung Damage Via Activation of the Pro-Inflammatory Response and Pulmonary Fibrosis. J. Microbiol. Biotechnol. 2020, 30, 427–438. [Google Scholar] [CrossRef]
- Vankadari, N.; Wilce, J.A. Emerging WuHan (COVID-19) Coronavirus: Glycan Shield and Structure Prediction of Spike Glycoprotein and Its Interaction with Human CD26. Emerg. Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef]
- Mani, S.; Kaur, A.; Jakhar, K.; Kumari, G.; Sonar, S.; Kumar, A.; Das, S.; Kumar, S.; Kumar, V.; Kundu, R.; et al. Targeting DPP4-RBD Interactions by Sitagliptin and Linagliptin Delivers a Potential Host-Directed Therapy against Pan-SARS-CoV-2 Infections. Int. J. Biol. Macromol. 2023, 245, 125444. [Google Scholar] [CrossRef] [PubMed]
- Cameron, K.; Rozano, L.; Falasca, M.; Mancera, R.L. Does the SARS-CoV-2 Spike Protein Receptor Binding Domain Interact Effectively with the DPP4 (CD26) Receptor? A Molecular Docking Study. Int. J. Mol. Sci. 2021, 22, 7001. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.N.; Gupta, A.M.; Banerjee, D.; Chakrabarti, J.; Raghavendra, P.B. Unraveling DPP4 Receptor Interactions with SARS-CoV-2 Variants and MERS-CoV: Insights into Pulmonary Disorders via Immunoinformatics and Molecular Dynamics. Viruses 2023, 15, 2056. [Google Scholar] [CrossRef] [PubMed]
- Amati, F.; Vancheri, C.; Latini, A.; Colona, V.L.; Grelli, S.; D’Apice, M.R.; Balestrieri, E.; Passarelli, C.; Minutolo, A.; Loddo, S.; et al. Expression Profiles of the SARS-CoV-2 Host Invasion Genes in Nasopharyngeal and Oropharyngeal Swabs of COVID-19 Patients. Heliyon 2020, 6, e05143. [Google Scholar] [CrossRef] [PubMed]
- Maremanda, K.P.; Sundar, I.K.; Li, D.; Rahman, I. Age-Dependent Assessment of Genes Involved in Cellular Senescence, Telomere, and Mitochondrial Pathways in Human Lung Tissue of Smokers, COPD, and IPF: Associations With SARS-CoV-2 COVID-19 ACE2-TMPRSS2-Furin-DPP4 Axis. Front. Pharmacol. 2020, 11, 584637. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cai, Z.; Zhang, J. DPP-4 Inhibitors May Improve the Mortality of Coronavirus Disease 2019: A Meta-Analysis. PLoS ONE 2021, 16, e0251916. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B.; Hughes, T.E. Inhibition of Dipeptidyl Peptidase-4 Augments Insulin Secretion in Response to Exogenously Administered Glucagon-like Peptide-1, Glucose-Dependent Insulinotropic Polypeptide, Pituitary Adenylate Cyclase-Activating Polypeptide, and Gastrin-Releasing Peptide in Mice. Endocrinology 2005, 146, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Yazbeck, R.; Howarth, G.S.; Abbott, C.A. Dipeptidyl Peptidase Inhibitors, an Emerging Drug Class for Inflammatory Disease? Trends Pharmacol. Sci. 2009, 30, 600–607. [Google Scholar] [CrossRef]
- Kim, W.; Egan, J.M. The Role of Incretins in Glucose Homeostasis and Diabetes Treatment. Pharmacol. Rev. 2008, 60, 470–512. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Pleiotropic Effects of Glucagon-like Peptide-1 (GLP-1)-Based Therapies on Vascular Complications in Diabetes. Curr. Pharm. Des. 2011, 17, 4379–4385. [Google Scholar] [CrossRef]
- Davidson, M.H. Potential Impact of Dipeptidyl Peptidase-4 Inhibitors on Cardiovascular Pathophysiology in Type 2 Diabetes Mellitus. Postgrad. Med. 2014, 126, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Buteau, J. GLP-1 Receptor Signaling: Effects on Pancreatic Beta-Cell Proliferation and Survival. Diabetes Metab. 2008, 34 (Suppl. S2), S73–S77. [Google Scholar] [CrossRef] [PubMed]
- Bugliani, M.; Syed, F.; Paula, F.M.M.; Omar, B.A.; Suleiman, M.; Mossuto, S.; Grano, F.; Cardarelli, F.; Boggi, U.; Vistoli, F.; et al. DPP-4 Is Expressed in Human Pancreatic Beta Cells and Its Direct Inhibition Improves Beta Cell Function and Survival in Type 2 Diabetes. Mol. Cell. Endocrinol. 2018, 473, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Augstein, P.; Naselli, G.; Loudovaris, T.; Hawthorne, W.J.; Campbell, P.; Bandala-Sanchez, E.; Rogers, K.; Heinke, P.; Thomas, H.E.; Kay, T.W.; et al. Localization of Dipeptidyl Peptidase-4 (CD26) to Human Pancreatic Ducts and Islet Alpha Cells. Diabetes Res. Clin. Pract. 2015, 110, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Ardestani, A.; Dharmadhikari, G.; Laue, S.; Schumann, D.M.; Kerr-Conte, J.; Pattou, F.; Klein, T.; Maedler, K. The DPP-4 Inhibitor Linagliptin Restores β-Cell Function and Survival in Human Isolated Islets through GLP-1 Stabilization. J. Clin. Endocrinol. Metab. 2013, 98, E1163–E1172. [Google Scholar] [CrossRef] [PubMed]
- Fadzeyeva, E.; Locatelli, C.A.A.; Trzaskalski, N.A.; Nguyen, M.-A.; Capozzi, M.E.; Vulesevic, B.; Morrow, N.M.; Ghorbani, P.; Hanson, A.A.; Lorenzen-Schmidt, I.; et al. Pancreas-Derived DPP4 Is Not Essential for Glucose Homeostasis under Metabolic Stress. iScience 2023, 26, 106748. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.P.; Sorrell, J.E.; Haller, A.; Roelofs, K.; Hutch, C.R.; Kim, K.-S.; Gutierrez-Aguilar, R.; Li, B.; Drucker, D.J.; D’Alessio, D.A.; et al. The Role of Pancreatic Preproglucagon in Glucose Homeostasis in Mice. Cell Metab. 2017, 25, 927–934.e3. [Google Scholar] [CrossRef]
- Traub, S.; Meier, D.T.; Schulze, F.; Dror, E.; Nordmann, T.M.; Goetz, N.; Koch, N.; Dalmas, E.; Stawiski, M.; Makshana, V.; et al. Pancreatic α Cell-Derived Glucagon-Related Peptides Are Required for β Cell Adaptation and Glucose Homeostasis. Cell Rep. 2017, 18, 3192–3203. [Google Scholar] [CrossRef]
- Campbell, S.A.; Hubert, M.; Johnson, J.; Salamon, N.; Light, P.E. The DPP4 Inhibitor Sitagliptin Increases Active Glp-1 Levels from Human Islets and May Increase Islet Cell Survival Prior to Transplantation. OBM Transplant. 2019, 3, 069. [Google Scholar] [CrossRef]
- Aroor, A.; Zuberek, M.; Duta, C.; Meuth, A.; Sowers, J.R.; Whaley-Connell, A.; Nistala, R. Angiotensin II Stimulation of DPP4 Activity Regulates Megalin in the Proximal Tubules. Int. J. Mol. Sci. 2016, 17, 780. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kim, J.E.; Lee, M.H.; Song, H.K.; Ghee, J.Y.; Kang, Y.S.; Min, H.S.; Kim, H.W.; Cha, J.J.; Han, J.Y.; et al. DA-1229, a Dipeptidyl Peptidase IV Inhibitor, Protects against Renal Injury by Preventing Podocyte Damage in an Animal Model of Progressive Renal Injury. Lab. Investig. 2016, 96, 547–560. [Google Scholar] [CrossRef]
- Leung, P.S. Pancreatic RAS. Adv. Exp. Med. Biol. 2010, 690, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, J.; Nargis, T.; Tantia, O.; Ghosh, S.; Chakrabarti, P. Increased Plasma Dipeptidyl Peptidase-4 (DPP4) Activity Is an Obesity-Independent Parameter for Glycemic Deregulation in Type 2 Diabetes Patients. Front. Endocrinol. 2019, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Pala, L.; Mannucci, E.; Pezzatini, A.; Ciani, S.; Sardi, J.; Raimondi, L.; Ognibene, A.; Cappadona, A.; Vannelli, B.G.; Rotella, C.M. Dipeptidyl Peptidase-IV Expression and Activity in Human Glomerular Endothelial Cells. Biochem. Biophys. Res. Commun. 2003, 310, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, E.; Pala, L.; Ciani, S.; Bardini, G.; Pezzatini, A.; Sposato, I.; Cremasco, F.; Ognibene, A.; Rotella, C.M. Hyperglycaemia Increases Dipeptidyl Peptidase IV Activity in Diabetes Mellitus. Diabetologia 2005, 48, 1168–1172. [Google Scholar] [CrossRef]
- Gagnon, M.L.; Bielenberg, D.R.; Gechtman, Z.; Miao, H.-Q.; Takashima, S.; Soker, S.; Klagsbrun, M. Identification of a Natural Soluble Neuropilin-1 That Binds Vascular Endothelial Growth Factor: In Vivo Expression and Antitumor Activity. Proc. Natl. Acad. Sci. USA 2000, 97, 2573–2578. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Fujita, Y.; Yamashita, T. Neuropilin-1-Mediated Pruning of Corticospinal Tract Fibers Is Required for Motor Recovery after Spinal Cord Injury. Cell Death Dis. 2019, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Pellet-Many, C.; Frankel, P.; Jia, H.; Zachary, I. Neuropilins: Structure, Function and Role in Disease. Biochem. J. 2008, 411, 211–226. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 Is a Host Factor for SARS-CoV-2 Infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Ji, N.; Zhang, M.; Ren, L.; Wang, Y.; Hu, B.; Xiang, J.; Gong, Y.; Wu, C.; Qu, G.; Ding, W.; et al. SARS-CoV-2 in the Pancreas and the Impaired Islet Function in COVID-19 Patients. Emerg. Microbes Infect. 2022, 11, 1115–1125. [Google Scholar] [CrossRef]
- Li, Z.; Buck, M. Neuropilin-1 Assists SARS-CoV-2 Infection by Stimulating the Separation of Spike Protein S1 and S2. Biophys. J. 2021, 120, 2828–2837. [Google Scholar] [CrossRef]
- Kyrou, I.; Randeva, H.S.; Spandidos, D.A.; Karteris, E. Not Only ACE2—The Quest for Additional Host Cell Mediators of SARS-CoV-2 Infection: Neuropilin-1 (NRP1) as a Novel SARS-CoV-2 Host Cell Entry Mediator Implicated in COVID-19. Sig Transduct. Target. Ther. 2021, 6, 21. [Google Scholar] [CrossRef]
- Hasan, N.M.; Kendrick, M.A.; Druckenbrod, N.R.; Huelsmeyer, M.K.; Warner, T.F.; MacDonald, M.J. Genetic Association of the Neuropilin-1 Gene with Type 1 Diabetes in Children: Neuropilin-1 Expression in Pancreatic Islets. Diabetes Res. Clin. Pract. 2010, 87, e29–e32. [Google Scholar] [CrossRef]
- Wilson, P.C.; Wu, H.; Kirita, Y.; Uchimura, K.; Ledru, N.; Rennke, H.G.; Welling, P.A.; Waikar, S.S.; Humphreys, B.D. The Single-Cell Transcriptomic Landscape of Early Human Diabetic Nephropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 19619–19625. [Google Scholar] [CrossRef]
- Loeffler, I.; Rüster, C.; Franke, S.; Liebisch, M.; Wolf, G. Erythropoietin Ameliorates Podocyte Injury in Advanced Diabetic Nephropathy in the Db/Db Mouse. Am. J. Physiol. Ren. Physiol. 2013, 305, F911–F918. [Google Scholar] [CrossRef]
- Bondeva, T.; Rüster, C.; Franke, S.; Hammerschmid, E.; Klagsbrun, M.; Cohen, C.D.; Wolf, G. Advanced Glycation End-Products Suppress Neuropilin-1 Expression in Podocytes. Kidney Int. 2009, 75, 605–616. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A Cell’s Response to Stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 Spike-Host Cell Receptor GRP78 Binding Site Prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Ha, D.P.; Van Krieken, R.; Carlos, A.J.; Lee, A.S. The Stress-Inducible Molecular Chaperone GRP78 as Potential Therapeutic Target for Coronavirus Infection. J. Infect. 2020, 81, 452–482. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. SARS-CoV-2 Spike-Heat Shock Protein A5 (GRP78) Recognition May Be Related to the Immersed Human Coronaviruses. Front. Pharmacol. 2020, 11, 577467. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Lv, Y.; Moser, D.; Zhou, X.; Woehrle, T.; Han, L.; Osterman, A.; Rudelius, M.; Choukér, A.; Lei, P. ACE2-Independent SARS-CoV-2 Virus Entry through Cell Surface GRP78 on Monocytes—Evidence from a Translational Clinical and Experimental Approach. EBioMedicine 2023, 98, 104869. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, C.-M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.-H.; Sze, K.-H.; Yang, D.; Shuai, H.; et al. Middle East Respiratory Syndrome Coronavirus and Bat Coronavirus HKU9 Both Can Utilize GRP78 for Attachment onto Host Cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [PubMed]
- Köseler, A.; Sabirli, R.; Gören, T.; Türkçüer, İ.; Kurt, Ö. Endoplasmic Reticulum Stress Markers in SARS-CoV-2 Infection and Pneumonia: Case-Control Study. Vivo 2020, 34, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Fernandes, L.; Lazari, L.C.; Da Silva, J.M.; De Morais Gomes, V.; Machado, R.R.G.; Dos Santos, A.F.; Araujo, D.B.; Coutinho, J.V.P.; Arini, G.S.; Angeli, C.B.; et al. SARS-CoV-2 Activates ER Stress and Unfolded Protein Response. bioRxiv 2021. [Google Scholar] [CrossRef]
- Aoe, T. Pathological Aspects of COVID-19 as a Conformational Disease and the Use of Pharmacological Chaperones as a Potential Therapeutic Strategy. Front. Pharmacol. 2020, 11, 1095. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-P.; Siu, K.-L.; Chin, K.-T.; Yuen, K.-Y.; Zheng, B.; Jin, D.-Y. Modulation of the Unfolded Protein Response by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [PubMed]
- Sabirli, R.; Koseler, A.; Goren, T.; Turkcuer, I.; Kurt, O. High GRP78 Levels in COVID-19 Infection: A Case-Control Study. Life Sci. 2021, 265, 118781. [Google Scholar] [CrossRef]
- Tang, J.; Guo, Y.-S.; Zhang, Y.; Yu, X.-L.; Li, L.; Huang, W.; Li, Y.; Chen, B.; Jiang, J.-L.; Chen, Z.-N. CD147 Induces UPR to Inhibit Apoptosis and Chemosensitivity by Increasing the Transcription of Bip in Hepatocellular Carcinoma. Cell Death Differ. 2012, 19, 1779–1790. [Google Scholar] [CrossRef]
- Carlos, A.J.; Ha, D.P.; Yeh, D.-W.; Van Krieken, R.; Tseng, C.-C.; Zhang, P.; Gill, P.; Machida, K.; Lee, A.S. The Chaperone GRP78 Is a Host Auxiliary Factor for SARS-CoV-2 and GRP78 Depleting Antibody Blocks Viral Entry and Infection. J. Biol. Chem. 2021, 296, 100759. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Åkerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic Reticulum Stress Contributes to Beta Cell Apoptosis in Type 2 Diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, P.; Peng, J.-L.; Wu, S.; Zhao, X.-P.; Li, L.; Shen, G.-X. The Altered Expression of Glucose-Regulated Proteins 78 in Different Phase of Streptozotocin-Affected Pancreatic Beta-Cells. Cell Stress. Chaperones 2009, 14, 43–48. [Google Scholar] [CrossRef]
- Hull, R.L.; Zraika, S.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kahn, S.E. Amyloid Formation in Human IAPP Transgenic Mouse Islets and Pancreas, and Human Pancreas, Is Not Associated with Endoplasmic Reticulum Stress. Diabetologia 2009, 52, 1102–1111. [Google Scholar] [CrossRef]
- Nourbakhsh, M.; Sharifi, R.; Heydari, N.; Nourbakhsh, M.; Ezzati-Mobasser, S.; Zarrinnahad, H. Circulating TRB3 and GRP78 Levels in Type 2 Diabetes Patients: Crosstalk between Glucose Homeostasis and Endoplasmic Reticulum Stress. J. Endocrinol. Investig. 2022, 45, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Bugliani, M.; Lupi, R.; Marselli, L.; Masini, M.; Boggi, U.; Filipponi, F.; Weir, G.C.; Eizirik, D.L.; Cnop, M. The Endoplasmic Reticulum in Pancreatic Beta Cells of Type 2 Diabetes Patients. Diabetologia 2007, 50, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Marselli, L.; Piron, A.; Suleiman, M.; Colli, M.L.; Yi, X.; Khamis, A.; Carrat, G.R.; Rutter, G.A.; Bugliani, M.; Giusti, L.; et al. Persistent or Transient Human β Cell Dysfunction Induced by Metabolic Stress: Specific Signatures and Shared Gene Expression with Type 2 Diabetes. Cell Rep. 2020, 33, 108466. [Google Scholar] [CrossRef]
- Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef]
- Cao, Z.-H.; Wu, Z.; Hu, C.; Zhang, M.; Wang, W.-Z.; Hu, X.-B. Endoplasmic Reticulum Stress and Destruction of Pancreatic β Cells in Type 1 Diabetes. Chin. Med. J. 2020, 133, 68–73. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic β-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 4843. [Google Scholar] [CrossRef]
- Lin, B.; Ferguson, C.; White, J.T.; Wang, S.; Vessella, R.; True, L.D.; Hood, L.; Nelson, P.S. Prostate-Localized and Androgen-Regulated Expression of the Membrane-Bound Serine Protease TMPRSS2. Cancer Res. 1999, 59, 4180–4184. [Google Scholar] [PubMed]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG Gene Fusion in Prostate Cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Gioukaki, C.; Georgiou, A.; Gkaralea, L.E.; Kroupis, C.; Lazaris, A.C.; Alamanis, C.; Thomopoulou, G.E. Unravelling the Role of P300 and TMPRSS2 in Prostate Cancer: A Literature Review. Int. J. Mol. Sci. 2023, 24, 11299. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence That TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A Transmembrane Serine Protease Is Linked to the Severe Acute Respiratory Syndrome Coronavirus Receptor and Activates Virus Entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Deng, Q.; Rasool, R.U.; Russell, R.M.; Natesan, R.; Asangani, I.A. Targeting Androgen Regulation of TMPRSS2 and ACE2 as a Therapeutic Strategy to Combat COVID-19. iScience 2021, 24, 102254. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Tissue Expression of TMPRSS2—Summary. Available online: https://www.proteinatlas.org/ENSG00000184012-TMPRSS2/tissue (accessed on 2 June 2024).
- Piva, F.; Sabanovic, B.; Cecati, M.; Giulietti, M. Expression and Co-Expression Analyses of TMPRSS2, a Key Element in COVID-19. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 451–455. [Google Scholar] [CrossRef]
- Patel, K.P.; Patel, P.A.; Vunnam, R.R.; Hewlett, A.T.; Jain, R.; Jing, R.; Vunnam, S.R. Gastrointestinal, Hepatobiliary, and Pancreatic Manifestations of COVID-19. J. Clin. Virol. 2020, 128, 104386. [Google Scholar] [CrossRef]
- Zhang, J.; He, L.; Huang, R.; Alvarez, J.F.; Yang, D.H.; Sun, Q.; Wang, F.; Peng, Z.; Jiang, N.; Su, L. Synergistic Effect of Elevated Glucose Levels with SARS-CoV-2 Spike Protein Induced NOX-Dependent ROS Production in Endothelial Cells. Mol. Biol. Rep. 2023, 50, 6039–6047. [Google Scholar] [CrossRef] [PubMed]
- Jocher, G.; Grass, V.; Tschirner, S.K.; Riepler, L.; Breimann, S.; Kaya, T.; Oelsner, M.; Hamad, M.S.; Hofmann, L.I.; Blobel, C.P.; et al. ADAM10 and ADAM17 Promote SARS-CoV-2 Cell Entry and Spike Protein-mediated Lung Cell Fusion. EMBO Rep. 2022, 23, e54305. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Menghini, R.; Fiorentino, L.; Casagrande, V.; Lauro, R.; Federici, M. The Role of ADAM17 in Metabolic Inflammation. Atherosclerosis 2013, 228, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Villescas, S.; Herat, L.; Schlaich, M.; Matthews, V. Implications of ADAM17 Activation for Hyperglycaemia, Obesity and Type 2 Diabetes. Biosci. Rep. 2021, 41, BSR20210029. [Google Scholar] [CrossRef] [PubMed]
- Caescu, C.I.; Jeschke, G.R.; Turk, B.E. Active-Site Determinants of Substrate Recognition by the Metalloproteinases TACE and ADAM10. Biochem. J. 2009, 424, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Reiss, K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: Novel Drug Targets with Therapeutic Potential? Eur. J. Cell Biol. 2011, 90, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Dreymueller, D.; Pruessmeyer, J.; Groth, E.; Ludwig, A. The Role of ADAM-Mediated Shedding in Vascular Biology. Eur. J. Cell Biol. 2012, 91, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Hagiyama, M.; Ito, A. Renal ADAM10 and 17: Their Physiological and Medical Meanings. Front. Cell Dev. Biol. 2018, 6, 153. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Vanderkerken, M.; Hammad, H. The Emerging Role of ADAM Metalloproteinases in Immunity. Nat. Rev. Immunol. 2018, 18, 745–758. [Google Scholar] [CrossRef]
- Chou, C.-W.; Huang, Y.-K.; Kuo, T.-T.; Liu, J.-P.; Sher, Y.-P. An Overview of ADAM9: Structure, Activation, and Regulation in Human Diseases. Int. J. Mol. Sci. 2020, 21, 7790. [Google Scholar] [CrossRef] [PubMed]
- Melano, I.; Cheng, W.-C.; Kuo, L.-L.; Liu, Y.-M.; Chou, Y.C.; Hung, M.-C.; Lai, M.M.C.; Sher, Y.-P.; Su, W.-C. A Disintegrin and Metalloproteinase Domain 9 Facilitates SARS-CoV-2 Entry into Cells with Low ACE2 Expression. Microbiol. Spectr. 2023, 11, e0385422. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liu, Y.; Wang, J.; Long, T.; Yee Mok, B.W.; Huang, Y.; Peng, Z.; Jia, Q.; Liu, C.; So, P.-K.; et al. Identifications of Novel Host Cell Factors That Interact with the Receptor-Binding Domain of the SARS-CoV-2 Spike Protein. J. Biol. Chem. 2024, 300, 107390. [Google Scholar] [CrossRef] [PubMed]
- Carapito, R.; Li, R.; Helms, J.; Carapito, C.; Gujja, S.; Rolli, V.; Guimaraes, R.; Malagon-Lopez, J.; Spinnhirny, P.; Lederle, A.; et al. Identification of Driver Genes for Critical Forms of COVID-19 in a Deeply Phenotyped Young Patient Cohort. Sci. Transl. Med. 2022, 14, eabj7521. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Gohda, J.; Kobayashi, A.; Tomita, K.; Hirayama, Y.; Koshikawa, N.; Seiki, M.; Semba, K.; Akiyama, T.; Kawaguchi, Y.; et al. Metalloproteinase-Dependent and TMPRSS2-Independent Cell Surface Entry Pathway of SARS-CoV-2 Requires the Furin Cleavage Site and the S2 Domain of Spike Protein. mBio 2022, 13, e0051922. [Google Scholar] [CrossRef] [PubMed]
- Palacios, Y.; Ruiz, A.; Ramón-Luing, L.A.; Ocaña-Guzman, R.; Barreto-Rodriguez, O.; Sánchez-Monciváis, A.; Tecuatzi-Cadena, B.; Regalado-García, A.G.; Pineda-Gudiño, R.D.; García-Martínez, A.; et al. Severe COVID-19 Patients Show an Increase in Soluble TNFR1 and ADAM17, with a Relationship to Mortality. Int. J. Mol. Sci. 2021, 22, 8423. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor Necrosis Factor-α Convertase (ADAM17) Mediates Regulated Ectodomain Shedding of the Severe-Acute Respiratory Syndrome-Coronavirus (SARS-CoV) Receptor, Angiotensin-Converting Enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Silva Enciso, J.E.; Greenberg, B.H. Selective and Specific Regulation of Ectodomain Shedding of Angiotensin-Converting Enzyme 2 by Tumor Necrosis Factor Alpha-Converting Enzyme. Am. J. Physiol. Cell Physiol. 2009, 297, C1318–C1329. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B. Ectodomain Shedding of Angiotensin Converting Enzyme 2 in Human Airway Epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef] [PubMed]
- Niehues, R.V.; Wozniak, J.; Wiersch, F.; Lilienthal, E.; Tacken, N.; Schumertl, T.; Garbers, C.; Ludwig, A.; Düsterhöft, S. The Collectrin-like Part of the SARS-CoV-1 and -2 Receptor ACE2 Is Shed by the Metalloproteinases ADAM10 and ADAM17. FASEB J. 2022, 36, e22234. [Google Scholar] [CrossRef]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.-P.; Zhou, R.; Chan, K.-H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-Mediated Cell Entry of SARS-CoV-2 via Interaction with Proteins Related to the Renin-Angiotensin System. Cell 2021, 184, 2212–2228.e12. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zimpelmann, J.; Agaybi, S.; Gurley, S.B.; Puente, L.; Burns, K.D. Characterization of Angiotensin-Converting Enzyme 2 Ectodomain Shedding from Mouse Proximal Tubular Cells. PLoS ONE 2014, 9, e85958. [Google Scholar] [CrossRef] [PubMed]
- Conrad, N.; Schwager, S.L.U.; Carmona, A.K.; Sturrock, E.D. The Effect of Structural Motifs on the Ectodomain Shedding of Human Angiotensin-Converting Enzyme. Biochem. Biophys. Res. Commun. 2016, 481, 111–116. [Google Scholar] [CrossRef]
- Lambert, D.W.; Clarke, N.E.; Hooper, N.M.; Turner, A.J. Calmodulin Interacts with Angiotensin-Converting Enzyme-2 (ACE2) and Inhibits Shedding of Its Ectodomain. FEBS Lett. 2008, 582, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.-C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.B.; Chodavarapu, H.; Porretta, C.; Robinson, L.K.; Lazartigues, E. Dynamics of ADAM17-Mediated Shedding of ACE2 Applied to Pancreatic Islets of Male Db/Db Mice. Endocrinology 2015, 156, 4411–4425. [Google Scholar] [CrossRef] [PubMed]
- Grützmann, R.; Lüttges, J.; Sipos, B.; Ammerpohl, O.; Dobrowolski, F.; Alldinger, I.; Kersting, S.; Ockert, D.; Koch, R.; Kalthoff, H.; et al. ADAM9 Expression in Pancreatic Cancer Is Associated with Tumour Type and Is a Prognostic Factor in Ductal Adenocarcinoma. Br. J. Cancer 2004, 90, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Asayesh, A.; Alanentalo, T.; Khoo, N.K.S.; Ahlgren, U. Developmental Expression of Metalloproteases ADAM 9, 10, and 17 Becomes Restricted to Divergent Pancreatic Compartments. Dev. Dyn. 2005, 232, 1105–1114. [Google Scholar] [CrossRef]
- Abdel-Bakky, M.S.; Alqasoumi, A.; Altowayan, W.M.; Amin, E.; Darwish, M.A. Resveratrol Inhibited ADAM10 Mediated CXCL16-Cleavage and T-Cells Recruitment to Pancreatic β-Cells in Type 1 Diabetes Mellitus in Mice. Pharmaceutics 2022, 14, 594. [Google Scholar] [CrossRef]
- Puddu, A.; Ravera, S.; Panfoli, I.; Bertola, N.; Maggi, D. High Glucose Impairs Expression and Activation of MerTK in ARPE-19 Cells. Int. J. Mol. Sci. 2022, 23, 1144. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Soluble Neuropilin-1 Response to Hypoglycemia in Type 2 Diabetes: Increased Risk or Protection in SARS-CoV-2 Infection? Front. Endocrinol. 2021, 12, 665134. [Google Scholar] [CrossRef] [PubMed]
- Suresh Babu, S.; Thandavarayan, R.A.; Joladarashi, D.; Jeyabal, P.; Krishnamurthy, S.; Bhimaraj, A.; Youker, K.A.; Krishnamurthy, P. MicroRNA-126 Overexpression Rescues Diabetes-Induced Impairment in Efferocytosis of Apoptotic Cardiomyocytes. Sci. Rep. 2016, 6, 36207. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Henderson, F.C.; Yi, Q.; Lei, Q.; Li, Y.; Chen, N. Chemokine CXCL16 Expression Suppresses Migration and Invasiveness and Induces Apoptosis in Breast Cancer Cells. Mediat. Inflamm. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Wang, J.; Wang, K.; Zhu, X.; Pang, R.; Li, X.; Zhu, G.; Pan, X. Serum CXCL16 as a Novel Biomarker of Coronary Artery Disease in Type 2 Diabetes Mellitus: A Pilot Study. Ann. Clin. Lab. Sci. 2016, 46, 184–189. [Google Scholar] [PubMed]
- Tawfik, M.S.; Abdel-Messeih, P.L.; Nosseir, N.M.; Mansour, H.H. Circulating CXCL16 in Type 2 Diabetes Mellitus Egyptian Patients. J. Radiat. Res. Appl. Sci. 2021, 14, 9–15. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Guetta, T.; Barnea, A.; Waldman, M.; Ben-Dor, N.; Barac, Y.D.; Kornowski, R.; Arad, M.; Hochhauser, E.; Aravot, D. Expression of the SARS-CoV-2 receptorACE2 in Human Heart Is Associated with Uncontrolled Diabetes, Obesity, and Activation of the Renin Angiotensin System. Cardiovasc. Diabetol. 2021, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G. Furin at the Cutting Edge: From Protein Traffic to Embryogenesis and Disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Murgolo, N.; Therien, A.G.; Howell, B.; Klein, D.; Koeplinger, K.; Lieberman, L.A.; Adam, G.C.; Flynn, J.; McKenna, P.; Swaminathan, G.; et al. SARS-CoV-2 Tropism, Entry, Replication, and Propagation: Considerations for Drug Discovery and Development. PLoS Pathog. 2021, 17, e1009225. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin Cleavage of SARS-CoV-2 Spike Promotes but Is Not Essential for Infection and Cell-Cell Fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; De Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike Glycoprotein of the New Coronavirus 2019-nCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Claas, E.C.; Osterhaus, A.D.; Van Beek, R.; De Jong, J.C.; Rimmelzwaan, G.F.; Senne, D.A.; Krauss, S.; Shortridge, K.F.; Webster, R.G. Human Influenza A H5N1 Virus Related to a Highly Pathogenic Avian Influenza Virus. Lancet 1998, 351, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Puranik, A.; Aravamudan, M.; Venkatakrishnan, A.; Soundararajan, V. SARS-CoV-2 Strategically Mimics Proteolytic Activation of Human ENaC. eLife 2020, 9, e58603. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Xie, X.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; Zhang, L.; et al. Furin Cleavage Site Is Key to SARS-CoV-2 Pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sawada, Y.; Kameya, T.; Aizawa, T.; Izumi, T.; Takeuchi, T. Proprotein-Processing Endoprotease Furin and Its Substrate Parathyroid Hormone-Related Protein Are Coexpressed in Insulinoma Cells. Endocr. Pathol. 2000, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kayo, T.; Konda, Y.; Tanaka, S.; Takata, K.; Koizumi, A.; Takeuchi, T. Developmental Expression of Proprotein-Processing Endoprotease Furin in Rat Pancreatic Islets. Endocrinology 1996, 137, 5126–5134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Louagie, E.; Taylor, N.A.; Flamez, D.; Roebroek, A.J.M.; Bright, N.A.; Meulemans, S.; Quintens, R.; Herrera, P.L.; Schuit, F.; Van De Ven, W.J.M.; et al. Role of Furin in Granular Acidification in the Endocrine Pancreas: Identification of the V-ATPase Subunit Ac45 as a Candidate Substrate. Proc. Natl. Acad. Sci. USA 2008, 105, 12319–12324. [Google Scholar] [CrossRef]
- Fernandez, C.; Rysä, J.; Almgren, P.; Nilsson, J.; Engström, G.; Orho-Melander, M.; Ruskoaho, H.; Melander, O. Plasma Levels of the Proprotein Convertase Furin and Incidence of Diabetes and Mortality. J. Intern. Med. 2018, 284, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.; Khan, M.S.H.; Dhurandhar, N.V.; Hegde, V. The Triumvirate: Why Hypertension, Obesity, and Diabetes Are Risk Factors for Adverse Effects in Patients with COVID-19. Acta Diabetol. 2021, 58, 831–843. [Google Scholar] [CrossRef]
- Ando, W.; Horii, T.; Uematsu, T.; Hanaki, H.; Atsuda, K.; Otori, K. Impact of Overlapping Risks of Type 2 Diabetes and Obesity on Coronavirus Disease Severity in the United States. Sci. Rep. 2021, 11, 17968. [Google Scholar] [CrossRef]
- Adu-Agyeiwaah, Y.; Grant, M.B.; Obukhov, A.G. The Potential Role of Osteopontin and Furin in Worsening Disease Outcomes in COVID-19 Patients with Pre-Existing Diabetes. Cells 2020, 9, 2528. [Google Scholar] [CrossRef]
- Glebov, O.O. Understanding SARS-CoV-2 Endocytosis for COVID-19 Drug Repurposing. FEBS J. 2020, 287, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-Mediated Enhancement of Severe Acute Respiratory Syndrome Coronavirus Infection. Proc. Natl. Acad. Sci. USA 2005, 102, 12543–12547. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, P.; Desikan, R.; Dixit, N.M. Targeting TMPRSS2 and Cathepsin B/L Together May Be Synergistic against SARS-CoV-2 Infection. PLoS Comput. Biol. 2020, 16, e1008461. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS Coronavirus Entry into Host Cells through a Novel Clathrin- and Caveolae-Independent Endocytic Pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus into Target Cells Expressing ACE2 with the Cytoplasmic Tail Deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [PubMed]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 Infects Cells after Viral Entry via Clathrin-Mediated Endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef] [PubMed]
- Burkard, C.; Verheije, M.H.; Wicht, O.; van Kasteren, S.I.; van Kuppeveld, F.J.; Haagmans, B.L.; Pelkmans, L.; Rottier, P.J.M.; Bosch, B.J.; de Haan, C.A.M. Coronavirus Cell Entry Occurs through the Endo-/Lysosomal Pathway in a Proteolysis-Dependent Manner. PLoS Pathog. 2014, 10, e1004502. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Shang, J.; Yang, Y.; Liu, C.; Wan, Y.; Geng, Q.; Wang, M.; Baric, R.; Li, F. Lysosomal Proteases Are a Determinant of Coronavirus Tropism. J. Virol. 2018, 92, e01504-18. [Google Scholar] [CrossRef]
- Baer, G.S.; Ebert, D.H.; Chung, C.J.; Erickson, A.H.; Dermody, T.S. Mutant Cells Selected during Persistent Reovirus Infection Do Not Express Mature Cathepsin L and Do Not Support Reovirus Disassembly. J. Virol. 1999, 73, 9532–9543. [Google Scholar] [CrossRef]
- Ebert, D.H.; Deussing, J.; Peters, C.; Dermody, T.S. Cathepsin L and Cathepsin B Mediate Reovirus Disassembly in Murine Fibroblast Cells. J. Biol. Chem. 2002, 277, 24609–24617. [Google Scholar] [CrossRef]
- Golden, J.W.; Linke, J.; Schmechel, S.; Thoemke, K.; Schiff, L.A. Addition of Exogenous Protease Facilitates Reovirus Infection in Many Restrictive Cells. J. Virol. 2002, 76, 7430–7443. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.W.; Bahe, J.A.; Lucas, W.T.; Nibert, M.L.; Schiff, L.A. Cathepsin S Supports Acid-Independent Infection by Some Reoviruses. J. Biol. Chem. 2004, 279, 8547–8557. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-Based Unconventional Secretory Pathway for Extracellular Delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal Proteolysis of the Ebola Virus Glycoprotein Is Necessary for Infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J.M. Cathepsin L Functionally Cleaves the Severe Acute Respiratory Syndrome Coronavirus Class I Fusion Protein Upstream of Rather than Adjacent to the Fusion Peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar] [CrossRef]
- Ou, T.; Mou, H.; Zhang, L.; Ojha, A.; Choe, H.; Farzan, M. Hydroxychloroquine-Mediated Inhibition of SARS-CoV-2 Entry Is Attenuated by TMPRSS2. PLoS Pathog. 2021, 17, e1009212. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of Cathepsin L Prevent Severe Acute Respiratory Syndrome Coronavirus Entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.-C.; Bosch, B.J.; Li, F.; Li, W.; Lee, K.H.; Ghiran, S.; Vasilieva, N.; Dermody, T.S.; Harrison, S.C.; Dormitzer, P.R.; et al. SARS Coronavirus, but Not Human Coronavirus NL63, Utilizes Cathepsin L to Infect ACE2-Expressing Cells. J. Biol. Chem. 2006, 281, 3198–3203. [Google Scholar] [CrossRef]
- Ding, X.; Ye, N.; Qiu, M.; Guo, H.; Li, J.; Zhou, X.; Yang, M.; Xi, J.; Liang, Y.; Gong, Y.; et al. Cathepsin B Is a Potential Therapeutic Target for Coronavirus Disease 2019 Patients with Lung Adenocarcinoma. Chem. Biol. Interact. 2022, 353, 109796. [Google Scholar] [CrossRef]
- Milan Bonotto, R.; Mitrović, A.; Sosič, I.; Martínez-Orellana, P.; Dattola, F.; Gobec, S.; Kos, J.; Marcello, A. Cathepsin Inhibitors Nitroxoline and Its Derivatives Inhibit SARS-CoV-2 Infection. Antivir. Res. 2023, 216, 105655. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-M.; Yang, W.-L.; Yang, F.-Y.; Zhang, L.; Huang, W.-J.; Hou, W.; Fan, C.-F.; Jin, R.-H.; Feng, Y.-M.; Wang, Y.-C.; et al. Cathepsin L Plays a Key Role in SARS-CoV-2 Infection in Humans and Humanized Mice and Is a Promising Target for New Drug Development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.P.; Fernandes, D.E.; Casimiro, F.; Da Mata, G.F.; Passos, M.T.; Varela, P.; Mastroianni-Kirsztajn, G.; Pesquero, J.B. Cathepsin L in COVID-19: From Pharmacological Evidences to Genetics. Front. Cell. Infect. Microbiol. 2020, 10, 589505. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous Treatment of Human Bronchial Epithelial Cells with Serine and Cysteine Protease Inhibitors Prevents Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.-P. Cathepsin L-Selective Inhibitors: A Potentially Promising Treatment for COVID-19 Patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.B.; Anjaneyulu, K.; Kidwai, J.R.; Mohan Rao, V.K. In Vitro Conversion of Proinsulin to Insulin by Cathepsin B and Role of C-Peptide. Acta Diabetol. Lat. 1978, 15, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Watanabe, T.; Ishii, Y.; Matsuba, H.; Kimura, S.; Fujita, T.; Kominami, E.; Katunuma, N.; Uchiyama, Y. Immunocytochemical Localization of Cathepsins B, H, and Their Endogenous Inhibitor, Cystatin Beta, in Islet Endocrine Cells of Rat Pancreas. J. Histochem. Cytochem. 1988, 36, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Docherty, K.; Phillips, I.D. Molecular Forms of Cathepsin B in Rat Thyroid Cells (FRTL5): Comparison with Molecular Forms in Liver (Hep G2) and Insulin-Secreting Cells (HIT T15). Biochim. Biophys. Acta 1988, 964, 168–174. [Google Scholar] [CrossRef]
- Jung, M.; Lee, J.; Seo, H.-Y.; Lim, J.S.; Kim, E.-K. Cathepsin Inhibition-Induced Lysosomal Dysfunction Enhances Pancreatic Beta-Cell Apoptosis in High Glucose. PLoS ONE 2015, 10, e0116972. [Google Scholar] [CrossRef]
- San Segundo, B.; Chan, S.J.; Steiner, D.F. Differences in Cathepsin B mRNA Levels in Rat Tissues Suggest Specialized Functions. FEBS Lett. 1986, 201, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Vidotti, D.B.; Casarini, D.E.; Cristovam, P.C.; Leite, C.A.; Schor, N.; Boim, M.A. High Glucose Concentration Stimulates Intracellular Renin Activity and Angiotensin II Generation in Rat Mesangial Cells. Am. J. Physiol. Ren. Physiol. 2004, 286, F1039–F1045. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yao, Q.; Hu, T.; Cai, Z.; Xie, Q.; Zhao, J.; Yuan, Y.; Ni, J.; Wu, Q.Q. Cathepsin B Deteriorates Diabetic Cardiomyopathy Induced by Streptozotocin via Promoting NLRP3-Mediated Pyroptosis. Mol. Ther. Nucleic Acids 2022, 30, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Peres, G.B.; Schor, N.; Michelacci, Y.M. Impact of High Glucose and AGEs on Cultured Kidney-Derived Cells. Effects on Cell Viability, Lysosomal Enzymes and Effectors of Cell Signaling Pathways. Biochimie 2017, 135, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in Human Type 2 Diabetes Pancreatic Beta Cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Maehr, R.; Mintern, J.D.; Herman, A.E.; Lennon-Duménil, A.-M.; Mathis, D.; Benoist, C.; Ploegh, H.L. Cathepsin L Is Essential for Onset of Autoimmune Diabetes in NOD Mice. J. Clin. Investig. 2005, 115, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-W.; Kryvalap, Y.; Sheu, T.-J.; Chang, C.-H.; Czyzyk, J. Cellular Proliferation in Mouse and Human Pancreatic Islets Is Regulated by Serpin B13 Inhibition and Downstream Targeting of E-Cadherin by Cathepsin L. Diabetologia 2019, 62, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Zhang, Y.; DeCastro, R.; Guo, H.; Nakamura, T.; Kataoka, H.; Nabeshima, K. The Human Tumor Cell-Derived Collagenase Stimulatory Factor (Renamed EMMPRIN) Is a Member of the Immunoglobulin Superfamily. Cancer Res. 1995, 55, 434–439. [Google Scholar] [PubMed]
- Grass, G.D.; Toole, B.P. How, with Whom and When: An Overview of CD147-Mediated Regulatory Networks Influencing Matrix Metalloproteinase Activity. Biosci. Rep. 2015, 36, e00283. [Google Scholar] [CrossRef]
- Iacono, K.T.; Brown, A.L.; Greene, M.I.; Saouaf, S.J. CD147 Immunoglobulin Superfamily Receptor Function and Role in Pathology. Exp. Mol. Pathol. 2007, 83, 283–295. [Google Scholar] [CrossRef]
- Gabison, E.E.; Hoang-Xuan, T.; Mauviel, A.; Menashi, S. EMMPRIN/CD147, an MMP Modulator in Cancer, Development and Tissue Repair. Biochimie 2005, 87, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Edwards, C.; Zhou, L. The Biological Function and Clinical Utilization of CD147 in Human Diseases: A Review of the Current Scientific Literature. Int. J. Mol. Sci. 2014, 15, 17411. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.-Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.-X.; Gong, L.; et al. CD147-Spike Protein Is a Novel Route for SARS-CoV-2 Infection to Host Cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Ahmetaj-Shala, B.; Vaja, R.; Atanur, S.S.; George, P.M.; Kirkby, N.S.; Mitchell, J.A. Cardiorenal Tissues Express SARS-CoV-2 Entry Genes and Basigin (BSG/CD147) Increases With Age in Endothelial Cells. JACC Basic. Transl. Sci. 2020, 5, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Ilikci Sagkan, R.; Akin-Bali, D.F. Structural Variations and Expression Profiles of the SARS-CoV-2 Host Invasion Genes in Lung Cancer. J. Med. Virol. 2020, 92, 2637–2647. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced Isolation of SARS-CoV-2 by TMPRSS2-Expressing Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, M.; Schürch, C.M. Expression of SARS-CoV-2 Entry Receptors in the Respiratory Tract of Healthy Individuals, Smokers and Asthmatics. Respir. Res. 2020, 21, 252. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Faulkner, N.; Cornish, G.H.; Rosa, A.; Harvey, R.; Hussain, S.; Ulferts, R.; Earl, C.; Wrobel, A.G.; Benton, D.J.; et al. Preexisting and de Novo Humoral Immunity to SARS-CoV-2 in Humans. Science 2020, 370, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26, and Other SARS-CoV-2 Associated Molecules in Tissues and Immune Cells in Health and in Asthma, COPD, Obesity, Hypertension, and COVID-19 Risk Factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef]
- Singh, M.; Bansal, V.; Feschotte, C. A Single-Cell RNA Expression Map of Human Coronavirus Entry Factors. Cell Rep. 2020, 32, 108175. [Google Scholar] [CrossRef]
- Ulrich, H.; Pillat, M.M. CD147 as a Target for COVID-19 Treatment: Suggested Effects of Azithromycin and Stem Cell Engagement. Stem Cell Rev. Rep. 2020, 16, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Zamorano Cuervo, N.; Grandvaux, N. ACE2: Evidence of Role as Entry Receptor for SARS-CoV-2 and Implications in Comorbidities. eLife 2020, 9, e61390. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 Promote SARS-CoV-2 Infection of Human Small Intestinal Enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Fang, Y.; Xu, T.; Ni, W.-J.; Shen, A.-Z.; Meng, X.-M. Potential Therapeutic Targets and Promising Drugs for Combating SARS-CoV-2. Br. J. Pharmacol. 2020, 177, 3147–3161. [Google Scholar] [CrossRef] [PubMed]
- Herrera, N.G.; Morano, N.C.; Celikgil, A.; Georgiev, G.I.; Malonis, R.J.; Lee, J.H.; Tong, K.; Vergnolle, O.; Massimi, A.B.; Yen, L.Y.; et al. Characterization of the SARS-CoV-2 S Protein: Biophysical, Biochemical, Structural, and Antigenic Analysis. ACS Omega 2021, 6, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, A.; Geng, K.; Honnen, W.; Wang, X.; Bruiners, N.; Singh, S.; Ferrara, F.; D’Angelo, S.; Bradbury, A.R.M.; et al. Human Immunodeficiency Viruses Pseudotyped with SARS-CoV-2 Spike Proteins Infect a Broad Spectrum of Human Cell Lines through Multiple Entry Mechanisms. Viruses 2021, 13, 953. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Fei, F.; Chen, Y.; Xu, L.; Zhang, Z.; Huang, Q.; Zhang, H.; Yang, H.; Chen, Z.; Xing, J. Hypoxia Upregulates CD147 through a Combined Effect of HIF-1α and Sp1 to Promote Glycolysis and Tumor Progression in Epithelial Solid Tumors. Carcinogenesis 2012, 33, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, V.; Zybarth, G.; O’Connor, M.; Dai, W.W.; Franchin, G.; Hao, T.; Guo, H.; Hung, H.-C.; Toole, B.; Gallay, P.; et al. Active Site Residues of Cyclophilin A Are Crucial for Its Signaling Activity via CD147. J. Biol. Chem. 2002, 277, 22959–22965. [Google Scholar] [CrossRef]
- Pushkarsky, T.; Zybarth, G.; Dubrovsky, L.; Yurchenko, V.; Tang, H.; Guo, H.; Toole, B.; Sherry, B.; Bukrinsky, M. CD147 Facilitates HIV-1 Infection by Interacting with Virus-Associated Cyclophilin A. Proc. Natl. Acad. Sci. USA 2001, 98, 6360–6365. [Google Scholar] [CrossRef]
- Chen, Z.; Mi, L.; Xu, J.; Yu, J.; Wang, X.; Jiang, J.; Xing, J.; Shang, P.; Qian, A.; Li, Y.; et al. Function of HAb18G/CD147 in Invasion of Host Cells by Severe Acute Respiratory Syndrome Coronavirus. J. Infect. Dis. 2005, 191, 755–760. [Google Scholar] [CrossRef]
- Rangu, R.; Wander, P.L.; Barrow, B.M.; Zraika, S. Going Viral in the Islet: Mediators of SARS-CoV-2 Entry beyond ACE2. J. Mol. Endocrinol. 2022, 69, R63–R79. [Google Scholar] [CrossRef]
- Zhao, C.; Wilson, M.C.; Schuit, F.; Halestrap, A.P.; Rutter, G.A. Expression and Distribution of Lactate/Monocarboxylate Transporter Isoforms in Pancreatic Islets and the Exocrine Pancreas. Diabetes 2001, 50, 361–366. [Google Scholar] [CrossRef]
- Li, T.; Quan, H.; Zhang, H.; Lin, L.; Ou, Q.; Chen, K. Silencing Cyclophilin A Improves Insulin Secretion, Reduces Cell Apoptosis, and Alleviates Inflammation as Well as Oxidant Stress in High Glucose-Induced Pancreatic β-Cells via MAPK/NF-Kb Signaling Pathway. Bioengineered 2020, 11, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Huang, W.; Ma, L.-T.; Jiang, J.-L.; Chen, Z.-N. Importance of N-Glycosylation on CD147 for Its Biological Functions. Int. J. Mol. Sci. 2014, 15, 6356–6377. [Google Scholar] [CrossRef]
- Bao, W.; Min, D.; Twigg, S.M.; Shackel, N.A.; Warner, F.J.; Yue, D.K.; McLennan, S.V. Monocyte CD147 Is Induced by Advanced Glycation End Products and High Glucose Concentration: Possible Role in Diabetic Complications. Am. J. Physiol. Cell Physiol. 2010, 299, C1212–C1219. [Google Scholar] [CrossRef]
- Costa, L.B.; Perez, L.G.; Palmeira, V.A.; Macedo E Cordeiro, T.; Ribeiro, V.T.; Lanza, K.; Simões E Silva, A.C. Insights on SARS-CoV-2 Molecular Interactions With the Renin-Angiotensin System. Front. Cell Dev. Biol. 2020, 8, 559841. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Ali, M.M. High Glucose and Advanced Glycation End Products Induce CD147-Mediated MMP Activity in Human Adipocytes. Cells 2021, 10, 2098. [Google Scholar] [CrossRef] [PubMed]
- Ragotte, R.J.; Pulido, D.; Donnellan, F.R.; Hill, M.L.; Gorini, G.; Davies, H.; Brun, J.; McHugh, K.; King, L.D.W.; Skinner, K.; et al. Human Basigin (CD147) Does Not Directly Interact with SARS-CoV-2 Spike Glycoprotein. mSphere 2021, 6, e0064721. [Google Scholar] [CrossRef] [PubMed]
- Shilts, J.; Crozier, T.W.M.; Greenwood, E.J.D.; Lehner, P.J.; Wright, G.J. No Evidence for Basigin/CD147 as a Direct SARS-CoV-2 Spike Binding Receptor. Sci. Rep. 2021, 11, 413. [Google Scholar] [CrossRef]
- Mina-Osorio, P. The Moonlighting Enzyme CD13: Old and New Functions to Target. Trends Mol. Med. 2008, 14, 361–371. [Google Scholar] [CrossRef]
- Lendeckel, U.; Arndt, M.; Frank, K.; Wex, T.; Ansorge, S. Role of Alanyl Aminopeptidase in Growth and Function of Human T Cells (Review). Int. J. Mol. Med. 1999, 4, 17–44. [Google Scholar] [CrossRef]
- Wolke, C.; Tadje, J.; Bukowska, A.; Täger, M.; Bank, U.; Ittenson, A.; Ansorge, S.; Lendeckel, U. Assigning the Phenotype of a Natural Regulatory T-Cell to the Human T-Cell Line, KARPAS-299. Int. J. Mol. Med. 2006, 17, 275–278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bank, U.; Tadje, J.; Täger, M.; Wolke, C.; Bukowska, A.; Ittenson, A.; Reinhold, D.; Helmuth, M.; Ansorge, S.; Shakespeare, A.; et al. Inhibition of Alanyl-Aminopeptidase on CD4+CD25+ Regulatory T-Cells Enhances Expression of FoxP3 and TGF-Beta1 and Ameliorates Acute Colitis in Mice. Int. J. Mol. Med. 2007, 20, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Mouritsen, S.; Meldal, M.; Werdelin, O.; Hansen, A.S.; Buus, S. MHC Molecules Protect T Cell Epitopes against Proteolytic Destruction. J. Immunol. 1992, 149, 1987–1993. [Google Scholar] [CrossRef] [PubMed]
- Falk, K.; Rötzschke, O.; Stevanović, S.; Jung, G.; Rammensee, H.G. Pool Sequencing of Natural HLA-DR, DQ, and DP Ligands Reveals Detailed Peptide Motifs, Constraints of Processing, and General Rules. Immunogenetics 1994, 39, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Mina-Osorio, P.; Shapiro, L.H.; Ortega, E. CD13 in Cell Adhesion: Aminopeptidase N (CD13) Mediates Homotypic Aggregation of Monocytic Cells. J. Leukoc. Biol. 2006, 79, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.E.; Cronin, C.; Ghosh, M.; Zhou, S.-Y.; Agosto, M.; Subramani, J.; Wang, R.; Shen, J.-B.; Schacke, W.; Liang, B.; et al. CD13 Is Essential for Inflammatory Trafficking and Infarct Healing Following Permanent Coronary Artery Occlusion in Mice. Cardiovasc. Res. 2013, 100, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Gerber, C.; Rahman, M.M.; Vernier, K.M.; Pereira, F.E.; Subramani, J.; Caromile, L.A.; Shapiro, L.H. Molecular Mechanisms Regulating CD13-Mediated Adhesion. Immunology 2014, 142, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Lo, R.; Ivic, I.; Aguilera, B.; Qendro, V.; Devarakonda, C.; Shapiro, L.H. CD13 Tethers the IQGAP1-ARF6-EFA6 Complex to the Plasma Membrane to Promote ARF6 Activation, Β1 Integrin Recycling, and Cell Migration. Sci. Signal 2019, 12, eaav5938. [Google Scholar] [CrossRef]
- Wang, R.; Simoneau, C.R.; Kulsuptrakul, J.; Bouhaddou, M.; Travisano, K.A.; Hayashi, J.M.; Carlson-Stevermer, J.; Zengel, J.R.; Richards, C.M.; Fozouni, P.; et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 2021, 184, 106–119.e14. [Google Scholar] [CrossRef]
- Kolb, A.F.; Hegyi, A.; Maile, J.; Heister, A.; Hagemann, M.; Siddell, S.G. Molecular Analysis of the Coronavirus-Receptor Function of Aminopeptidase N. Adv. Exp. Med. Biol. 1998, 440, 61–67. [Google Scholar] [CrossRef]
- Nomura, R.; Kiyota, A.; Suzaki, E.; Kataoka, K.; Ohe, Y.; Miyamoto, K.; Senda, T.; Fujimoto, T. Human Coronavirus 229E Binds to CD13 in Rafts and Enters the Cell through Caveolae. J. Virol. 2004, 78, 8701–8708. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Walls, A.C.; Joshi, A.; Park, Y.-J.; Eguia, R.T.; Miranda, M.C.; Kepl, E.; Dosey, A.; Stevens-Ayers, T.; Boeckh, M.J.; et al. Structure, Receptor Recognition, and Antigenicity of the Human Coronavirus CCoV-HuPn-2018 Spike Glycoprotein. Cell 2022, 185, 2279–2291.e17. [Google Scholar] [CrossRef]
- Fuochi, V.; Floresta, G.; Emma, R.; Patamia, V.; Caruso, M.; Zagni, C.; Ronchi, F.; Ronchi, C.; Drago, F.; Rescifina, A.; et al. Heparan Sulfate and Enoxaparin Interact at the Interface of the Spike Protein of HCoV-229E but Not with HCoV-OC43. Viruses 2023, 15, 663. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; Matsuyama, S.; Taguchi, F. Protease-Mediated Entry via the Endosome of Human Coronavirus 229E. J. Virol. 2009, 83, 712–721. [Google Scholar] [CrossRef]
- Albiston, A.L.; McDowall, S.G.; Matsacos, D.; Sim, P.; Clune, E.; Mustafa, T.; Lee, J.; Mendelsohn, F.A.; Simpson, R.J.; Connolly, L.M.; et al. Evidence That the Angiotensin IV (AT4) Receptor Is the Enzyme Insulin-Regulated Aminopeptidase. J. Biol. Chem. 2001, 276, 48623–48626. [Google Scholar] [CrossRef]
- Jordens, I.; Molle, D.; Xiong, W.; Keller, S.R.; McGraw, T.E. Insulin-Regulated Aminopeptidase Is a Key Regulator of GLUT4 Trafficking by Controlling the Sorting of GLUT4 from Endosomes to Specialized Insulin-Regulated Vesicles. Mol. Biol. Cell 2010, 21, 2034–2044. [Google Scholar] [CrossRef]
- Takeuchi, M.; Itakura, A.; Okada, M.; Mizutani, S.; Kikkawa, F. Impaired Insulin-Regulated Membrane Aminopeptidase Translocation to the Plasma Membrane in Adipocytes of Otsuka Long Evans Tokushima Fatty Rats. Nagoya J. Med. Sci. 2006, 68, 155–163. [Google Scholar]
- Keller, S.R.; Davis, A.C.; Clairmont, K.B. Mice Deficient in the Insulin-Regulated Membrane Aminopeptidase Show Substantial Decreases in Glucose Transporter GLUT4 Levels but Maintain Normal Glucose Homeostasis. J. Biol. Chem. 2002, 277, 17677–17686. [Google Scholar] [CrossRef]
- Githens, S. Glutathione Metabolism in the Pancreas Compared with That in the Liver, Kidney, and Small Intestine. Int. J. Pancreatol. 1991, 8, 97–109. [Google Scholar] [CrossRef]
- Bordessoule, D.; Jones, M.; Gatter, K.C.; Mason, D.Y. Immunohistological Patterns of Myeloid Antigens: Tissue Distribution of CD13, CD14, CD16, CD31, CD36, CD65, CD66 and CD67. Br. J. Haematol. 1993, 83, 370–383. [Google Scholar] [CrossRef]
- Nagasaka, T.; Nomura, S.; Okamura, M.; Tsujimoto, M.; Nakazato, H.; Oiso, Y.; Nakashima, N.; Mizutani, S. Immunohistochemical Localization of Placental Leucine Aminopeptidase/Oxytocinase in Normal Human Placental, Fetal and Adult Tissues. Reprod. Fertil. Dev. 1997, 9, 747–753. [Google Scholar] [CrossRef]
- Kobayashi, H.; Mitsui, T.; Nomura, S.; Ohno, Y.; Kadomatsu, K.; Muramatsu, T.; Nagasaka, T.; Mizutani, S. Expression of Glucose Transporter 4 in the Human Pancreatic Islet of Langerhans. Biochem. Biophys. Res. Commun. 2004, 314, 1121–1125. [Google Scholar] [CrossRef]
- Siebelmann, M.; Wensing, J.; Verspohl, E.J. The Impact of ANG II and IV on INS-1 Cells and on Blood Glucose and Plasma Insulin. J. Recept. Signal Transduct. Res. 2010, 30, 234–245. [Google Scholar] [CrossRef]
- Wong, Y.-C.; Sim, M.-K.; Lee, K.-O. Des-Aspartate-Angiotensin-I and Angiotensin IV Improve Glucose Tolerance and Insulin Signalling in Diet-Induced Hyperglycaemic Mice. Biochem. Pharmacol. 2011, 82, 1198–1208. [Google Scholar] [CrossRef]
- Simonsen, A.; Lippé, R.; Christoforidis, S.; Gaullier, J.M.; Brech, A.; Callaghan, J.; Toh, B.H.; Murphy, C.; Zerial, M.; Stenmark, H. EEA1 Links PI(3)K Function to Rab5 Regulation of Endosome Fusion. Nature 1998, 394, 494–498. [Google Scholar] [CrossRef]
- Christoforidis, S.; McBride, H.M.; Burgoyne, R.D.; Zerial, M. The Rab5 Effector EEA1 Is a Core Component of Endosome Docking. Nature 1999, 397, 621–625. [Google Scholar] [CrossRef]
- Jordens, I.; Marsman, M.; Kuijl, C.; Neefjes, J. Rab Proteins, Connecting Transport and Vesicle Fusion. Traffic 2005, 6, 1070–1077. [Google Scholar] [CrossRef]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab Conversion as a Mechanism of Progression from Early to Late Endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zheng, C. When Rab GTPases Meet Innate Immune Signaling Pathways. Cytokine Growth Factor. Rev. 2021, 59, 95–100. [Google Scholar] [CrossRef]
- Mendoza, P.; Díaz, J.; Torres, V.A. On the Role of Rab5 in Cell Migration. Curr. Mol. Med. 2014, 14, 235–245. [Google Scholar] [CrossRef]
- Callaghan, J.; Nixon, S.; Bucci, C.; Toh, B.H.; Stenmark, H. Direct Interaction of EEA1 with Rab5b. Eur. J. Biochem. 1999, 265, 361–366. [Google Scholar] [CrossRef]
- Zerial, M.; McBride, H. Rab Proteins as Membrane Organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as Coordinators of Vesicle Traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Wandinger-Ness, A.; Zerial, M. Rab Proteins and the Compartmentalization of the Endosomal System. Cold Spring Harb. Perspect. Biol. 2014, 6, a022616. [Google Scholar] [CrossRef]
- Inpanathan, S.; Botelho, R.J. The Lysosome Signaling Platform: Adapting With the Times. Front. Cell Dev. Biol. 2019, 7, 113. [Google Scholar] [CrossRef]
- Vonderheit, A.; Helenius, A. Rab7 Associates with Early Endosomes to Mediate Sorting and Transport of Semliki Forest Virus to Late Endosomes. PLoS Biol. 2005, 3, e233. [Google Scholar] [CrossRef]
- Murray, J.L.; Mavrakis, M.; McDonald, N.J.; Yilla, M.; Sheng, J.; Bellini, W.J.; Zhao, L.; Le Doux, J.M.; Shaw, M.W.; Luo, C.-C.; et al. Rab9 GTPase Is Required for Replication of Human Immunodeficiency Virus Type 1, Filoviruses, and Measles Virus. J. Virol. 2005, 79, 11742–11751. [Google Scholar] [CrossRef]
- Wilcke, M.; Johannes, L.; Galli, T.; Mayau, V.; Goud, B.; Salamero, J. Rab11 Regulates the Compartmentalization of Early Endosomes Required for Efficient Transport from Early Endosomes to the Trans-Golgi Network. J. Cell Biol. 2000, 151, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Hollinshead, M.; Johns, H.L.; Sayers, C.L.; Gonzalez-Lopez, C.; Smith, G.L.; Elliott, G. Endocytic Tubules Regulated by Rab GTPases 5 and 11 Are Used for Envelopment of Herpes Simplex Virus. EMBO J. 2012, 31, 4204–4220. [Google Scholar] [CrossRef] [PubMed]
- Niazy, N.; Temme, S.; Bocuk, D.; Giesen, C.; König, A.; Temme, N.; Ziegfeld, A.; Gregers, T.F.; Bakke, O.; Lang, T.; et al. Misdirection of Endosomal Trafficking Mediated by Herpes Simplex Virus-Encoded Glycoprotein B. FASEB J. 2017, 31, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-N.; Liu, Y.-Y.; Xiao, F.-C.; Liu, C.-C.; Liang, X.-D.; Chen, J.; Zhou, J.; Baloch, A.S.; Kan, L.; Zhou, B.; et al. Rab5, Rab7, and Rab11 Are Required for Caveola-Dependent Endocytosis of Classical Swine Fever Virus in Porcine Alveolar Macrophages. J. Virol. 2018, 92, e00797-18. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Zhang, J.; Zhang, H.; Xia, S.; Ren, J.; Tian, L.; Bai, D.; Fang, L.; Xiao, S. Porcine Deltacoronavirus Enters Porcine IPI-2I Intestinal Epithelial Cells via Macropinocytosis and Clathrin-Mediated Endocytosis Dependent on pH and Dynamin. J. Virol. 2021, 95, e0134521. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Hou, W.; Shan, Y.; Wang, S.; Liu, F. Dynamic Dissection of the Endocytosis of Porcine Epidemic Diarrhea Coronavirus Cooperatively Mediated by Clathrin and Caveolae as Visualized by Single-Virus Tracking. mBio 2021, 12, e00256-21. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e14. [Google Scholar] [CrossRef] [PubMed]
- Atik, N.; Wirawan, F.; Amalia, R.; Khairani, A.F.; Pradini, G.W. Differences in Endosomal Rab Gene Expression between Positive and Negative COVID-19 Patients. BMC Res. Notes 2022, 15, 252. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Mishra, A.R.; Gupta, A.; Nayak, D. Structural Similarity-Based Prediction of Host Factors Associated with SARS-CoV-2 Infection and Pathogenesis. J. Biomol. Struct. Dyn. 2022, 40, 5868–5879. [Google Scholar] [CrossRef] [PubMed]
- Chamnanphon, M.; Pongpanich, M.; Suttichet, T.B.; Jantarabenjakul, W.; Torvorapanit, P.; Putcharoen, O.; Sodsai, P.; Phokaew, C.; Hirankarn, N.; Chariyavilaskul, P.; et al. Host Genetic Factors of COVID-19 Susceptibility and Disease Severity in a Thai Population. J. Hum. Genet. 2022, 67, 295–301. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Jung, E.-M.; Joo, S.-S.; Yoo, Y.-M. Nanomolar Melatonin Influences Insulin Synthesis and Secretion in Rat Insulinoma INS-1E Cells. J. Physiol. Pharmacol. 2020, 71, 705–716. [Google Scholar] [CrossRef]
- Sugawara, K.; Shibasaki, T.; Mizoguchi, A.; Saito, T.; Seino, S. Rab11 and Its Effector Rip11 Participate in Regulation of Insulin Granule Exocytosis. Genes. Cells 2009, 14, 445–456. [Google Scholar] [CrossRef]
- Hewawasam, N.V.; Lhaf, F.; Taylor, H.A.; Viloria, K.; Austin, A.; King, A.; Jones, P.; Jones, L.; Turner, M.D.; Hill, N.J. Modulation of Rab7a-Mediated Growth Factor Receptor Trafficking Inhibits Islet Beta Cell Apoptosis and Autophagy under Conditions of Metabolic Stress. Sci. Rep. 2020, 10, 15741. [Google Scholar] [CrossRef] [PubMed]
- da Silva, D.R.; Gonzalez, C.F.; Lorca, G.L. Internalization of Extracellular Vesicles from Lactobacillus Johnsonii N6.2 Elicit an RNA Sensory Response in Human Pancreatic Cell Lines. J. Extracell. Biol. 2023, 2, e101. [Google Scholar] [CrossRef] [PubMed]
- Girada, S.B.; Kuna, R.S.; Bele, S.; Zhu, Z.; Chakravarthi, N.R.; DiMarchi, R.D.; Mitra, P. Gαs Regulates Glucagon-Like Peptide 1 Receptor-Mediated Cyclic AMP Generation at Rab5 Endosomal Compartment. Mol. Metab. 2017, 6, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Fehmann, H.C.; Habener, J.F. Insulinotropic Hormone Glucagon-like Peptide-I(7-37) Stimulation of Proinsulin Gene Expression and Proinsulin Biosynthesis in Insulinoma Beta TC-1 Cells. Endocrinology 1992, 130, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Incretin Action in the Pancreas: Potential Promise, Possible Perils, and Pathological Pitfalls. Diabetes 2013, 62, 3316–3323. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-Interacting Lysosomal Protein (RILP): The Rab7 Effector Required for Transport to Lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 Effector Protein RILP Controls Lysosomal Transport by Inducing the Recruitment of Dynein-Dynactin Motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, Z.; Zhang, S.; Zhuang, R.; Liu, H.; Liu, X.; Qiu, X.; Zhang, M.; Zheng, Y.; Li, L.; et al. RILP Restricts Insulin Secretion Through Mediating Lysosomal Degradation of Proinsulin. Diabetes 2020, 69, 67–82. [Google Scholar] [CrossRef]
- Kessler, A.; Tomas, E.; Immler, D.; Meyer, H.E.; Zorzano, A.; Eckel, J. Rab11 Is Associated with GLUT4-Containing Vesicles and Redistributes in Response to Insulin. Diabetologia 2000, 43, 1518–1527. [Google Scholar] [CrossRef]
- Tapia-Limonchi, R.; Díaz, I.; Cahuana, G.M.; Bautista, M.; Martín, F.; Soria, B.; Tejedo, J.R.; Bedoya, F.J. Impact of Exposure to Low Concentrations of Nitric Oxide on Protein Profile in Murine and Human Pancreatic Islet Cells. Islets 2014, 6, e995997. [Google Scholar] [CrossRef]
- Vandermoere, F.; El Yazidi-Belkoura, I.; Slomianny, C.; Demont, Y.; Bidaux, G.; Adriaenssens, E.; Lemoine, J.; Hondermarck, H. The Valosin-Containing Protein (VCP) Is a Target of Akt Signaling Required for Cell Survival. J. Biol. Chem. 2006, 281, 14307–14313. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.B.; Rutter, J. Mitochondrial Quality Control by the Ubiquitin-Proteasome System. Biochem. Soc. Trans. 2011, 39, 1509–1513. [Google Scholar] [CrossRef]
- Guo, X.; Qi, X. VCP Cooperates with UBXD1 to Degrade Mitochondrial Outer Membrane Protein MCL1 in Model of Huntington’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Bug, M.; Bremer, S. Emerging Functions of the VCP/P97 AAA-ATPase in the Ubiquitin System. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- van den Boom, J.; Meyer, H. VCP/P97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, K.; Halder, S.; Wiseman, K.; Vaz, B. Strategic Role of the Ubiquitin-Dependent Segregase P97 (VCP or Cdc48) in DNA Replication. Chromosoma 2017, 126, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, I.; Oehler, J.; Ramadan, K. The Role of Ubiquitin-Dependent Segregase P97 (VCP or Cdc48) in Chromatin Dynamics after DNA Double Strand Breaks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160282. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Tomita, Y.; Nakatsuka, S.; Hoshida, Y.; Myoui, A.; Yoshikawa, H.; Aozasa, K. VCP (P97) Regulates NFkappaB Signaling Pathway, Which Is Important for Metastasis of Osteosarcoma Cell Line. Jpn. J. Cancer Res. 2002, 93, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Bozko, P.M.; Dubiel, W.; Naumann, M. CSN Controls NF-kappaB by Deubiquitinylation of IkappaBalpha. EMBO J. 2007, 26, 1532–1541. [Google Scholar] [CrossRef]
- Ramanathan, H.N.; Ye, Y. The P97 ATPase Associates with EEA1 to Regulate the Size of Early Endosomes. Cell Res. 2012, 22, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Bug, M.; Meyer, H. Expanding into New Markets--VCP/P97 in Endocytosis and Autophagy. J. Struct. Biol. 2012, 179, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, P.; Bug, M.; Meyer, H. Ubiquitination of the N-Terminal Region of Caveolin-1 Regulates Endosomal Sorting by the VCP/P97 AAA-ATPase. J. Biol. Chem. 2013, 288, 7363–7372. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A Mighty “Protein Extractor” of the Cell: Structure and Function of the P97/CDC48 ATPase. Front. Mol. Biosci. 2017, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Wakita, T.; Shimizu, H. Valosin-Containing Protein (VCP/P97) Is Required for Poliovirus Replication and Is Involved in Cellular Protein Secretion Pathway in Poliovirus Infection. J. Virol. 2012, 86, 5541–5553. [Google Scholar] [CrossRef] [PubMed]
- McEwan, W.A.; James, L.C. TRIM21-Dependent Intracellular Antibody Neutralization of Virus Infection. Prog. Mol. Biol. Transl. Sci. 2015, 129, 167–187. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, N.; Rapoport, T. Toward an Understanding of the Cdc48/P97 ATPase. F1000Res. 2017, 6, 1318. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Prendergast, J.; Grey, F. The Host Ubiquitin-Dependent Segregase VCP/P97 Is Required for the Onset of Human Cytomegalovirus Replication. PLoS Pathog. 2017, 13, e1006329. [Google Scholar] [CrossRef] [PubMed]
- Carissimo, G.; Chan, Y.-H.; Utt, A.; Chua, T.-K.; Bakar, F.A.; Merits, A.; Ng, L.F.P. VCP/P97 Is a Proviral Host Factor for Replication of Chikungunya Virus and Other Alphaviruses. Front. Microbiol. 2019, 10, 2236. [Google Scholar] [CrossRef]
- Huryn, D.M.; Kornfilt, D.J.P.; Wipf, P. P97: An Emerging Target for Cancer, Neurodegenerative Diseases, and Viral Infections. J. Med. Chem. 2020, 63, 1892–1907. [Google Scholar] [CrossRef]
- Wong, H.H.; Kumar, P.; Tay, F.P.L.; Moreau, D.; Liu, D.X.; Bard, F. Genome-Wide Screen Reveals Valosin-Containing Protein Requirement for Coronavirus Exit from Endosomes. J. Virol. 2015, 89, 11116–11128. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.-W.; Li, S.; Wang, F.; Ruiz-Lopez, N.M.; Houerbi, N.; Chou, T.-F. Impacts of P97 on Proteome Changes in Human Cells during Coronaviral Replication. Cells 2021, 10, 2953. [Google Scholar] [CrossRef] [PubMed]
- Nicolls, M.R.; D’Antonio, J.M.; Hutton, J.C.; Gill, R.G.; Czwornog, J.L.; Duncan, M.W. Proteomics as a Tool for Discovery: Proteins Implicated in Alzheimer’s Disease Are Highly Expressed in Normal Pancreatic Islets. J. Proteome Res. 2003, 2, 199–205. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Tissue Expression of VCP—Staining in Pancreas. Available online: https://www.proteinatlas.org/ENSG00000165280-VCP/tissue/pancreas (accessed on 24 June 2024).
- Liu, M.; Wright, J.; Guo, H.; Xiong, Y.; Arvan, P. Proinsulin Entry and Transit through the Endoplasmic Reticulum in Pancreatic Beta Cells. Vitam. Horm. 2014, 95, 35–62. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Hoelen, H.; van de Weijer, M.L.; Janssen, G.M.; Costa, A.I.; van Veelen, P.A.; Lebbink, R.J.; Wiertz, E.J.H.J. Proinsulin Degradation and Presentation of a Proinsulin B-Chain Autoantigen Involves ER-Associated Protein Degradation (ERAD)-Enzyme UBE2G2. PLoS ONE 2024, 19, e0287877. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Burcin, M.; Gromada, J.; Urano, F. Endoplasmic Reticulum Stress in β-Cells and Development of Diabetes. Curr. Opin. Pharmacol. 2009, 9, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, C.; Rowicka, M.; Kudlicki, A.; Nowis, D.; McConnell, E.; Kujawa, M.; DeMartino, G.N. Valosin-Containing Protein (P97) Is a Regulator of Endoplasmic Reticulum Stress and of the Degradation of N-End Rule and Ubiquitin-Fusion Degradation Pathway Substrates in Mammalian Cells. MBoC 2006, 17, 4606–4618. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yang, Y.; Allister, E.M.; Wijesekara, N.; Wheeler, M.B. The Identification of Potential Factors Associated with the Development of Type 2 Diabetes: A Quantitative Proteomics Approach. Mol. Cell Proteom. 2008, 7, 1434–1451. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.C.; Williams, D.; Vicogne, J.; Pessin, J.E. The Glucose Transporter 2 Undergoes Plasma Membrane Endocytosis and Lysosomal Degradation in a Secretagogue-Dependent Manner. Endocrinology 2009, 150, 4056–4064. [Google Scholar] [CrossRef]
- Thorens, B.; Wu, Y.J.; Leahy, J.L.; Weir, G.C. The Loss of GLUT2 Expression by Glucose-Unresponsive Beta Cells of Db/Db Mice Is Reversible and Is Induced by the Diabetic Environment. J. Clin. Investig. 1992, 90, 77–85. [Google Scholar] [CrossRef]
- Guillam, M.-T.; Hümmler, E.; Schaerer, E.; Wu, J.-Y.; Birnbaum, M.J.; Beermann, F.; Schmidt, A.; Dériaz, N.; Thorens, B. Early Diabetes and Abnormal Postnatal Pancreatic Islet Development in Mice Lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Yan, F.; Shimamura, S.; Barg, S.; Shyng, S.-L. Membrane Phosphoinositides Control Insulin Secretion Through Their Effects on ATP-Sensitive K+ Channel Activity. Diabetes 2005, 54, 2852–2858. [Google Scholar] [CrossRef]
- Gremlich, S.; Roduit, R.; Thorens, B. Dexamethasone Induces Posttranslational Degradation of GLUT2 and Inhibition of Insulin Secretion in Isolated Pancreatic β Cells. J. Biol. Chem. 1997, 272, 3216–3222. [Google Scholar] [CrossRef] [PubMed]
- Mills, I.G.; Jones, A.T.; Clague, M.J. Involvement of the Endosomal Autoantigen EEA1 in Homotypic Fusion of Early Endosomes. Curr. Biol. 1998, 8, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, O.; Oral, O.; Kocaturk, N.M.; Akkoc, Y.; Eberhart, K.; Kosar, A.; Gozuacik, D. IBMPFD Disease-Causing Mutant VCP/P97 Proteins Are Targets of Autophagic-Lysosomal Degradation. PLoS ONE 2016, 11, e0164864. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).