Abstract
The innate immune system serves as the first line of defense against β-coronaviruses (β-CoVs), a family of viruses that includes SARS-CoV-2. Viral sensing via pattern recognition receptors triggers inflammation and cell death, which are essential components of the innate immune response that facilitate viral clearance. However, excessive activation of the innate immune system and inflammatory cell death can result in uncontrolled release of proinflammatory cytokines, resulting in cytokine storm and pathology. PANoptosis, innate immune, inflammatory cell death initiated by innate immune sensors and driven by caspases and RIPKs through PANoptosome complexes, has been implicated in the pathology of viral infections. Therefore, understanding the molecular mechanisms regulating PANoptosis in response to β-CoV infection is critical for identifying new therapeutic targets that can mitigate disease severity. In the current study, we analyzed findings from a cell death-based CRISPR screen with archetypal β-CoV mouse hepatitis virus (MHV) as the trigger to characterize host molecules required for inflammatory cell death. As a result, we identified SMARCA4, a chromatin regulator, as a putative host factor required for PANoptosis in response to MHV. Furthermore, we observed that gRNA-mediated deletion of Smarca4 inhibited MHV-induced PANoptotic cell death in macrophages. These findings have potential translational and clinical implications for the advancement of treatment strategies for β-CoVs and other infections.
1. Introduction
The innate immune system serves as the first line of defense against infections, using pattern recognition receptors (PRRs) to detect pathogens. In the context of β-coronavirus (β-CoV) infections, such as SARS-CoV-2, the causative agent of coronavirus disease 2019 (COVID-19), these genetically encoded PRRs activate the innate immune response, which can initiate inflammatory responses and cell death [1,2,3]. The innate immune system is a key factor in defining the severity of β-CoV and SARS-CoV-2 infections [4,5,6,7]. While innate immune-mediated cell death is meant to provide host protection and impede viral replication, uncontrolled cell death can result in disrupted immunological responses, cytokine storm, organ damage, and mortality [8]. One particular inflammatory cell death pathway, PANoptosis, has been widely implicated in the pathophysiology of β-CoVs and SARS-CoV-2 [9,10,11,12]. PANoptosis is an innate immune, inflammatory cell death pathway initiated by innate immune sensors and driven by caspases and receptor-interacting protein (RIP) kinases (RIPKs) via PANoptosomes. PANoptosomes are multiprotein complexes assembled by innate immune sensor(s) in response to pathogens, pathogen- or damage-associated molecular patterns (PAMPs or DAMPs), cytokines, or homeostatic changes that drive PANoptosis [13,14,15]. In the context of SARS-CoV-2, PANoptosis can result in cytokine storm and multiorgan damage [9,10,11]. Patients with severe COVID-19 present with increased levels of TNF and IFN-γ, which can drive PANoptosis [9,10]. Additionally, the lethality of IFN therapy in murine β-CoV infection models is driven by PANoptosis [12], suggesting that IFN-mediated pathology during SARS-CoV-2 infection occurs through PANoptosis. Furthermore, a recent RNA-seq dataset analysis established a strong association between PANoptosis and COVID-19 severity [11]. In addition to β-CoV pathophysiology, PANoptosis is linked to several other infections, inflammatory diseases, and cancers [15,16,17,18,19,20,21]. Despite the established role of PANoptosis in β-CoV, and specifically SARS-CoV-2, pathogenesis, the mechanistic understanding of how β-CoVs induce PANoptosis remains incomplete.
To define the molecular mechanisms underlying innate immune inflammatory PANoptotic cell death in β-CoV infections, we investigated putative regulators identified in a prior genome-wide host CRISPR knockout screen for cell death molecules that used MHV as a trigger [22]. MHV is a prototypical murine β-CoV that is commonly used as a model due to its ability to infect organs, induce acute and chronic illness, and promote PANoptosis in bone marrow-derived macrophages (BMDMs) [2,23,24], mimicking several key aspects of human β-CoV biology. Here, we re-evaluated the MHV-induced cell death CRISPR screen and identified the chromatin regulator SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 4 (SMARCA4) as a critical regulator. Moreover, the loss of Smarca4 led to a reduction in the cell death and decreased the activation of PANoptosis molecules, suggesting that SMARCA4 plays a critical role in β-CoV-mediated PANoptosis.
Overall, our work highlights the power of using genome-wide screens in macrophage cell lines to identify critical host components involved in innate immune responses. Moreover, addressing the molecular mechanisms that lead to inflammatory cell death in β-CoV infections offers valuable knowledge for identifying potential therapeutic targets in infections.
2. Materials and Methods
2.1. Mouse Hepatitis Virus (MHV) Culture
The mouse hepatitis virus (A59 strain) was propagated in 17Cl-1 cells as previously described [25]. The viral titer was measured using a plaque assay in 17Cl-1 cells.
2.2. CRISPR Screen Analysis
The data generated previously from the MHV CRISPR screen in Brie library-containing immortalized BMDMs (iBMDMs) were used for the analyses, following the same procedures described previously (BioProject: PRJNA1009133) [22]. Briefly, calc_auc_v1.1.py (https://github.com/mhegde/, accessed on 21 September 2020) and count_spacers.py [26] were used to validate and verify the presence of gRNA, followed by the analysis of CRISPR KO screens using MAGeCK-VISPR (version 0.5.7) [27]. The MAGeCK pipeline was used to estimate the log2 (fold change) with significance levels for each gene in the CRISPR screen. The CRISPR screen leveraged the intrinsic MHV-mediated cell death to eliminate susceptible cells, leaving behind only those in which gene deletion protected from MHV-induced cell death. Therefore, genes with a positive fold change were expected to be required for cell death. The enriched genes that were significant in the CRISPR screen were visualized using a volcano plot and an RRA score plot using MAGeCKFlute v2.0.0 [28]. The expression profiles of individual gRNAs for the control and treated samples were visualized using a scatter plot.
2.3. Cas9-iBMDM Culture and Infection
For in vitro validation studies, Cas9-expressing iBMDMs were simultaneously electroporated with two gRNAs targeting Smarca4 (Smarca4-RNA1: CCCTGTGAAGGTGATCCACG, Smarca4-RNA2: GAGGTATGTGATGAGCGCGA); the resulting pool of cells was referred to as the Smarca4-g sample. The gRNA-electroporated cells were allowed to rest for 5 days to achieve CRISPR-based deletion of the targeted gene. The cells were then expanded and seeded in 12- or 24-well plates at a seeding density of 1 × 106 cells/well and 0.5 × 106 cells/well, respectively, in complete DMEM (DMEM supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS; S1620, Biowest, Lakewood Ranch, FL, USA) and 1% penicillin-streptomycin (15070-063, Thermo Fisher Scientific, Waltham, MA, USA), and rested for approximately 24 h before the experiments. The cells were then washed with PBS and infected with MHV at an MOI of 0.1 in DMEM plain media (Sigma, D6171, St. Louis, MI, USA) with 1% penicillin/streptomycin. After 1 h of incubation with MHV, the cells were supplemented with HI-FBS to a final concentration of 10% and used for IncuCyte imaging or incubated for the indicated times before collecting samples for downstream analyses.
2.4. Cell Death Analysis
The IncuCyte® S3 and/or SX5 Live-Cell Analysis System was used to image and analyze cell death in real time, as previously described [29]. In brief, the control and Cas9-iBMDMs electroporated with Smarca4 gRNA were seeded in 24-well plates (0.5 × 106 cells/well) and infected with MHV as described above. After 1 h of incubation, propidium iodide (PI; Life Technologies, P3566, Carlsbad, CA, USA) was added to the cells together with HI-FBS. The plate was scanned for fluorescent and phase-contrast images (4 image fields/well) in real time for a total of 18 h at intervals of 1 or 2 h. Cell death was quantified as the percentage of PI+ cells.
2.5. Western Blotting
To blot for caspases, Cas9-iBMDMs were lysed along with the supernatant using 125 μL of 4× SDS loading buffer containing 50 μL of caspase lysis buffer (1× protease inhibitors, 1× phosphatase inhibitors, 10% NP-40, and 25 mM DTT). To detect other proteins in the cell lysates alone, the supernatants were aspirated, and the cells were lysed using RIPA buffer. Caspase and RIPA lysates were separated on 10–12% polyacrylamide gels and then transferred onto PVDF membranes. After blocking the membranes with 5% skim milk for 1 h at room temperature, the blots were incubated with their respective primary antibodies overnight at 4 °C. Subsequently, the blots were washed with 1× TBST and incubated with secondary antibodies conjugated with horseradish peroxidase (HRP) for 1 h at room temperature. The GE Amersham Imager 600 was used to develop the blots. In certain cases, the immunoblots were stripped using stripping buffer (Restore Western Blot Stripping Buffer, 21059, Thermo Fisher Scientific) and reprobed for a different protein of interest. The antibodies used were as follows: anti–caspase-1 (AdipoGen, AG-20B-0042, 1:1000, San Diego, CA, USA), anti–caspase-3 (#9662, Cell Signaling Technology [CST], 1:1000, Danvers, MA, USA), anti-cleaved caspase-3 (#9661, CST, 1:1000), anti–caspase-8 (CST, #4927, 1:1000), anti-cleaved caspase-8 (CST, #8592, 1:1000), anti-GSDMD (Abcam, ab209845, 1:1000, Waltham, MA, USA), anti-pMLKL (CST, #37333, 1:1000), anti-tMLKL (Abcepta, AP14272B, 1:1000, San Diego, CA, USA), anti-β-actin (66009–1-IG, Proteintech, 1:5000, Rosemont, IL, USA), and HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, anti-rabbit [111-035-047], 1:5000; anti-mouse [315-035-047], 1:5000).
2.6. Quantitative PCR
Total RNA was isolated using TRIzol® reagent (Ambion, Waltham, MA, USA, #15596018). A total of 500 ng of RNA was reverse transcribed to cDNA using a high-capacity cDNA synthesis reverse transcription kit (Applied Biosystems, Waltham, MA, USA, #4368813), as per the manufacturer’s instructions. cDNA was used to measure Smarca4 gene expression in Cas9-expressing iBMDMs with or without Smarca4 depletion by gRNA treatment (described above). mRNA levels were quantified in real time using Sybr® Green (Applied Biosystems, #4367659) and the Quant Studio™ 7 Flex Real-Time PCR System (Applied Biosystems). The expression levels were normalized to Actb for host cellular Smarca4 gene expression and are presented as % Smarca4 expression. Forward and reverse primer sequences used in the study were as follows: Smarca4, Forward: 5-CAAAGACAAGCATATCCTAGCCA-3, Reverse: 5-CACGTAGTGTGTGTTAAGGACC-3; and Actb, Forward: 5-CAGCTTCTTTGCAGCTCCTT-3, Reverse: 5-CACGATGGAGGGGAATACAG-3.
2.7. Statistical Analysis
Data analysis was performed using the GraphPad Prism 10.0 software. Data are presented as the mean ± SEM of two or three independent repeats. The Student’s t-test was used to determine statistical significance. p values < 0.05 were considered significant, where ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
3. Results
3.1. A Genome-Wide CRISPR Screen for Regulators of MHV-Induced Cell Death Identifies Critical Host Genes in β-CoV Infection
β-CoVs continue to pose a significant risk to global health. Identifying the host genes and cell death pathways implicated in β-CoV pathogenesis is critical for understanding the underlying mechanisms that drive pathogenesis and identifying treatment targets. Therefore, we evaluated a previous genome-wide cell death CRISPR knockout screen to identify potential host factors that regulate cell death during β-CoV infection [22]. This screen utilized the archetypal β-CoV MHV, along with iBMDMs derived from Cas9-transgenic mice and the mouse whole genome pooled Brie library (Addgene, 73633) [30]. The screen identified several host genes that may be involved in MHV-induced cell death, based on the enrichment of their respective gRNA counts (Figure 1A). Cells that are susceptible to MHV-mediated cell death are eliminated by infection during the CRISPR screen; therefore, cells in which critical cell death molecules have been deleted will be retained, resulting in a relative increase in the gRNA counts for these genes. To validate our analysis pipeline, we confirmed that we could detect the previously identified and characterized host gene Ceacam1 as a top hit (Figure 1A). CEACAM1 is a known MHV receptor, and its deletion is known to inhibit MHV-mediated cell death [22]. Based on this validated pipeline, we leveraged this dataset and identified the enrichment of individual gRNAs that corresponded to the SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 4 (SMARCA4) (Figure 1A). Furthermore, analysis of individual gRNAs demonstrated that all four distinct gRNAs targeting Smarca4 were enriched in the MHV-infected pool of cells, although the specific level of enrichment varied (Figure 1B,C). Together, these results suggest that SMARCA4 plays a key role in MHV infection-induced cell death.
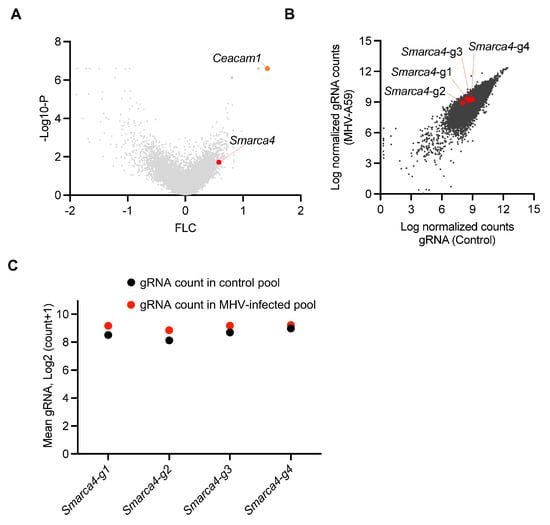
Figure 1.
CRISPR screen identifies host factors that facilitate β-CoV-induced cell death. (A) Volcano plot depicting the log2 mean fold change (FLC) for the gRNAs in the CRISPR screen based on an earlier genome-wide CRISPR screen in immortalized bone marrow-derived macrophages (iBMDMs) infected with mouse hepatitis virus (MHV; MOI 0.1) for 24 h. The newly identified enriched host gene Smarca4 is labeled alongside the previously identified gene Ceacam1. (B) Scatter plot illustrating the enrichment of all four gRNAs targeting Smarca4 in the pool of iBMDMs carrying gRNAs from the whole genome CRISPR screen following MHV infection. (C) Scatter plot illustrating the logarithmic distribution of the normalized gRNA counts in the control (uninfected) and MHV-infected pools of cells for each individual Smarca4 gRNA, as shown in panel B.
3.2. SMARCA4 Is a Regulator of MHV-Induced Inflammatory Cell Death
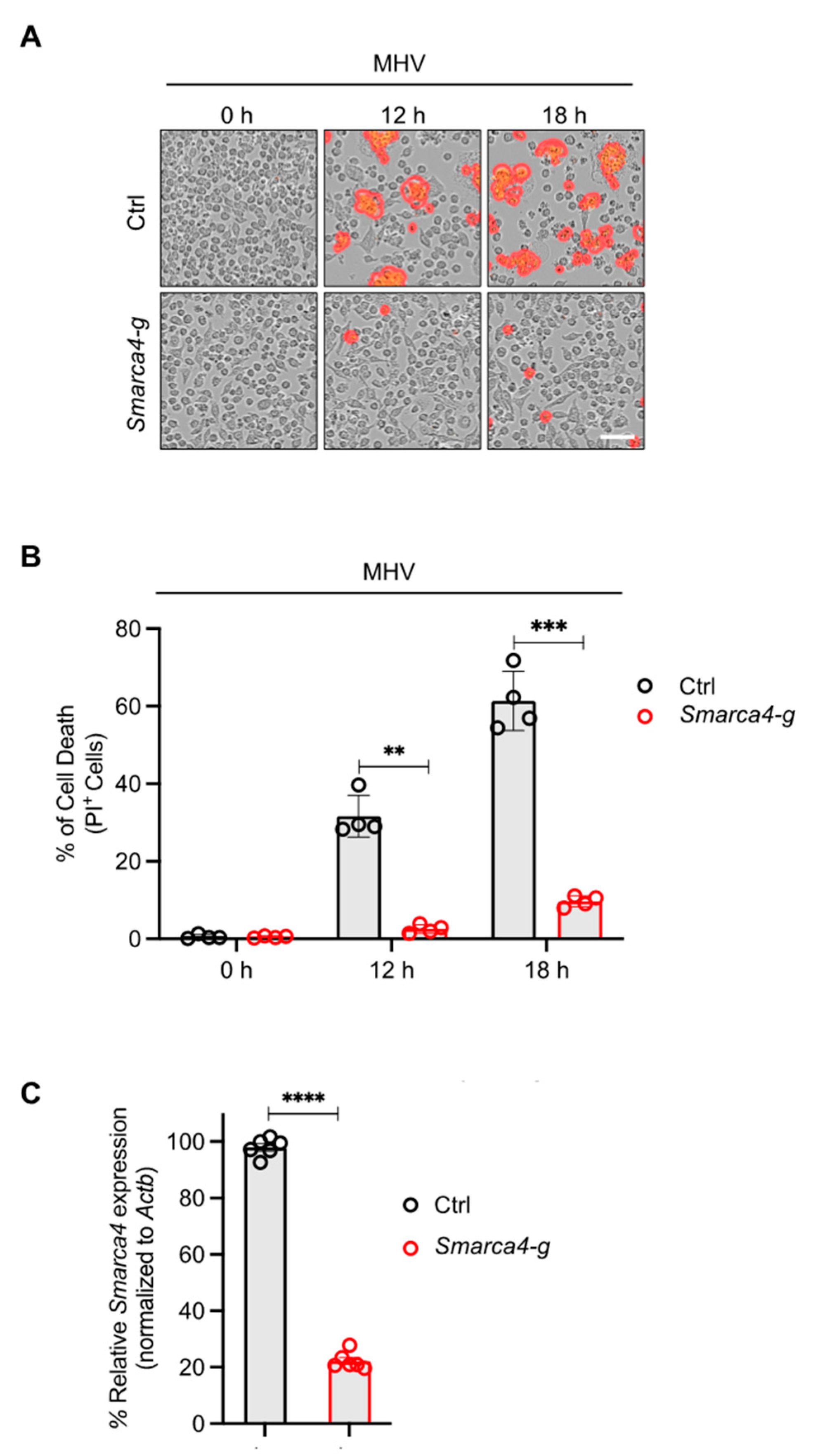
The results of the CRISPR screen suggested a potential role for SMARCA4 in cell death in response to MHV infection (Figure 1). Therefore, we sought to confirm these CRISPR screen results through functional validation. We evaluated cell death kinetics in MHV-infected iBMDMs and observed a significant time-dependent increase in lytic cell death in wild-type (WT) iBMDMs infected with MHV (Figure 2A,B). Additionally, gRNA-mediated deletion of Smarca4 inhibited MHV-induced cell death (Figure 2A–C).
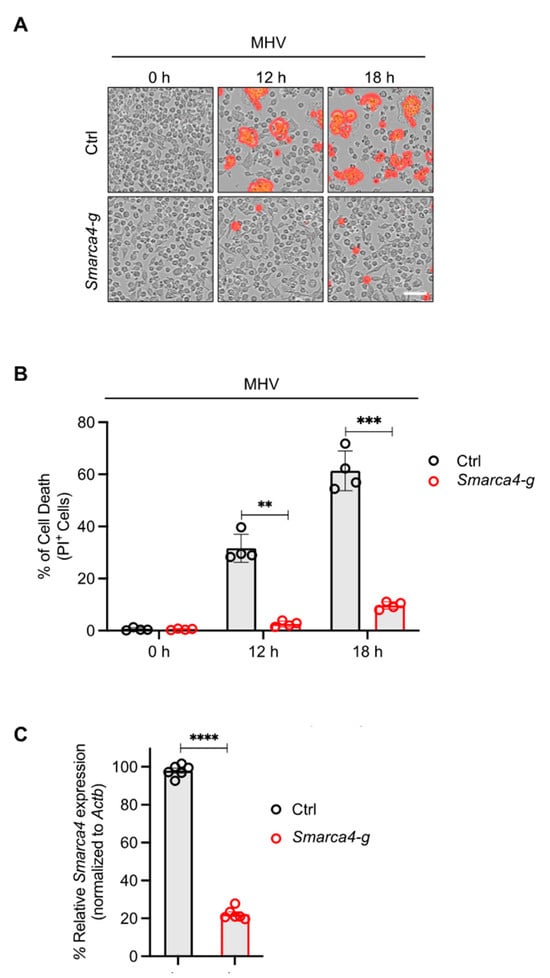
Figure 2.
SMARCA4 is required for MHV-induced inflammatory cell death. (A) Representative images of cell death in mouse hepatitis virus (MHV; MOI 0.1)-infected immortalized bone marrow-derived macrophages (iBMDMs) with and without Smarca4 gRNA treatment at the indicated time points. The red mask denotes dead cells, and the scale bar represents 50 μm. (B) Quantification of the percentage of cells undergoing lytic cell death at specified time points following MHV infection in iBMDMs treated with or without Smarca4 gRNA. (C) Smarca4 expression in uninfected iBMDMs treated with and without Smarca4 gRNA. Actb was used to normalize Smarca4 expression. The reported data are representative of two independent experiments with 4–6 technical replicates (A–C). Similar results were obtained for each experiment. The data are shown as the mean ± SEM (B,C). The Student’s t-test was used to determine statistical significance. ** p < 0.01; *** p < 0.001; and **** p < 0.0001. Ctrl: Control with no gRNA; Smarca4-g: Smarca4 gRNA-treated.
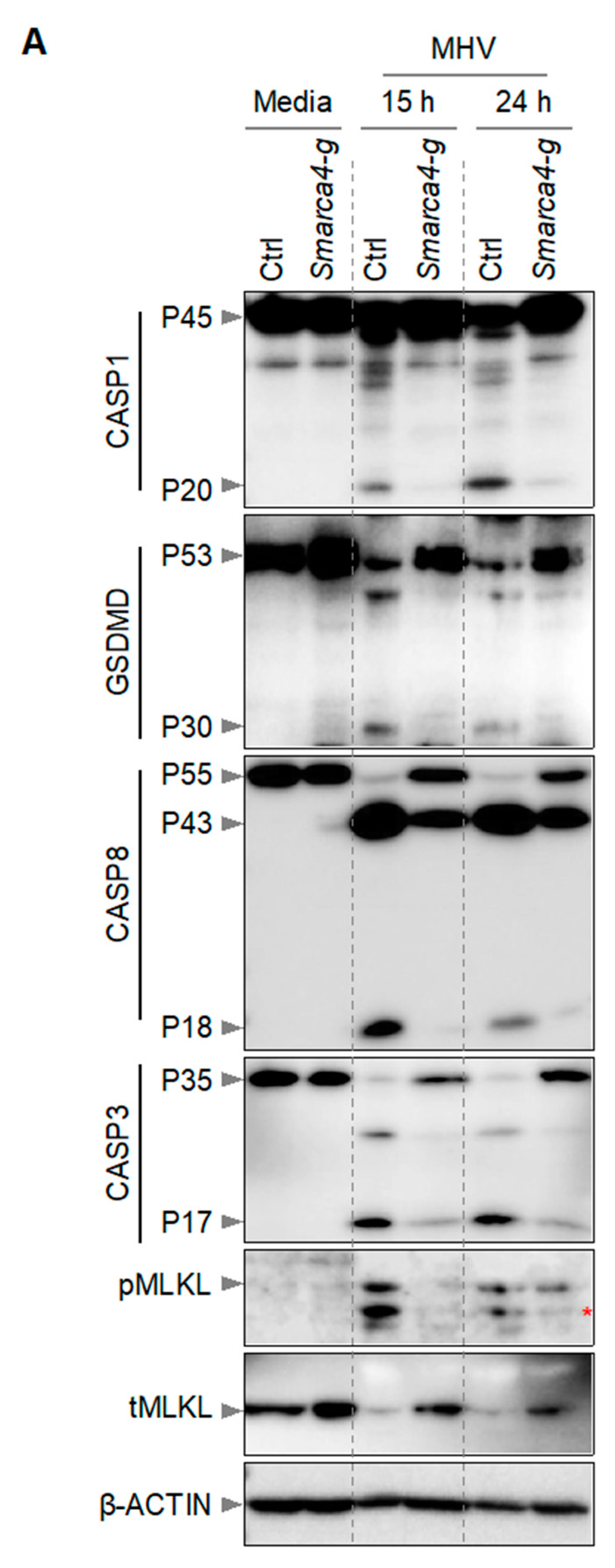
It has been previously reported that macrophages infected with MHV undergo PANoptosis, which involves the activation of multiple caspases, RIPKs, and other effectors and executioners of inflammatory cell death [2,12,22]. Therefore, we next investigated the effect of Smarca4 loss on the activation of PANoptosis molecules in iBMDMs. Following MHV infection, we detected cleaved and active caspase-1 (indicating inflammasome activation) in WT iBMDMs (Figure 3). Caspase-1 cleaves GSDMD into its P30 N-terminal domain fragment (GSDMD-N), which promotes the formation of cell membrane pores and results in cell death [31,32,33,34,35]. Consistently, we also observed the P30 fragment of GSDMD in WT iBMDMs following MHV infection (Figure 3). In addition, we observed cleavage of caspases-8 and -3 in WT iBMDMs in response to MHV infection (Figure 3). However, gRNA-based deletion of Smarca4 diminished the cleavage of these caspases and GSDMD. Furthermore, we observed phosphorylation of mixed lineage kinase domain-like pseudokinase (MLKL) in MHV-infected WT iBMDMs, which was reduced in Smarca4-depleted cells (Figure 3). Overall, the gRNA-mediated loss of Smarca4 reduced the activation of caspases-1, -3, and -8, GSDMD, and pMLKL in response to MHV infection (Figure 3). These data collectively validated the role of SMARCA4 in MHV-induced PANoptotic cell death.
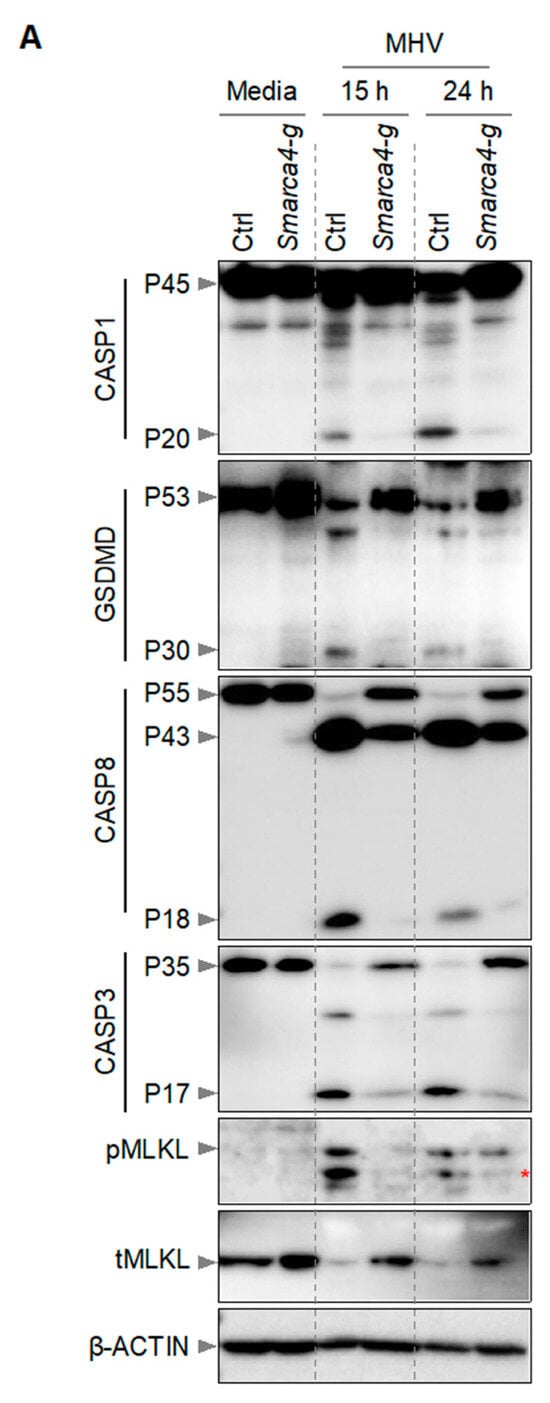
Figure 3.
SMARCA4 is required for MHV-induced PANoptosis. (A) Immunoblot analysis of pro- (P45) and cleaved caspase-1 (P20; CASP1), pro- (P53) and activated (P30) gasdermin D (GSDMD), pro- (P55) and cleaved caspase-8 (P43, P18; CASP8), pro- (P35) and cleaved caspase-3 (P17; CASP3), and phospho-mixed lineage kinase domain-like pseudokinase (pMLKL) and total MLKL (tMLKL) from mouse hepatitis virus (MHV; MOI 0.1)-infected immortalized bone marrow-derived macrophages (iBMDMs) with and without Smarca4 gRNA treatment at the indicated time points. Blots were reprobed using β-ACTIN antibody as an internal control. The reported data are representative of two independent experiments. Similar observations were obtained in each experiment. Ctrl: Control with no gRNA; Smarca4-g: Smarca4 gRNA-treated. The red asterisk (*) denotes a non-specific band.
4. Discussion
The recognition of viruses or viral PAMPs via innate immune PRRs activates signaling mechanisms that result in inflammatory cell death and the release of proinflammatory cytokines. These innate immune mechanisms are the key first lines of defense against infections [1,2]. Furthermore, cell death pathways play a critical role in the host’s ability to limit the spread of viruses. While inflammatory cell death is beneficial for combating viral infections, dysregulated innate immune responses can aggravate inflammation and drive pathology, which can be detrimental to the host [8,9,12,36].
In recent years, β-CoVs, such as SARS-CoV-2, have posed significant challenges to human health. Growing evidence suggests an association between inflammatory cell death pathways and disease severity in SARS-CoV-2 infection [9,10,11,12,37,38]. In particular, the PANoptosis pathway is induced in viral infections, including β-CoVs and SARS-CoV-2 [9,10,11,12,21,22,36,39]. PANoptosis has also been linked to a wide range of infections, inflammatory diseases, immune disorders, and cancers [13,14,15,16,17,18,19,20,21,29,40,41,42]. Innate immune sensors (including ZBP1, RIPK1, AIM2, and NLRC5 and NLRP12) that drive the assembly of PANoptosome cell death complexes are known for their central role in the immune response to pathogens, PAMPs, DAMPs, and sterile triggers [18,20,21,39,43,44,45]. In the context of β-CoVs, ZBP1 plays a key role in mediating PANoptosis [12]. Furthermore, the cytokines TNF and IFN-γ serve as triggers for initiating PANoptosis and are known to exacerbate COVID-19 [9,10,46]. Neutralizing TNF and IFN-γ decreases mortality in SARS-CoV-2 murine models [9], indicating that cytokine-induced PANoptosis plays a critical role in disease pathogenesis.
Increasing evidence suggests that PANoptosis is a key driver of β-CoV infection severity and outcomes, particularly in the context of SARS-CoV-2 [9,10,11], and additional research is required to fully understand the upstream molecular mechanisms that initiate PANoptosis to identify new therapeutic targets. Our results identified Smarca4 as a significantly enriched gRNA in a genome-wide CRISPR screen of host regulators of MHV-induced cell death, suggesting its role as a key upstream host factor in β-CoV-mediated PANoptosis. SMARCA4 is a critical regulator of chromatin organization and transcription [47,48]. RNA viruses are known to require host transcriptional machinery to enhance their replication [49], and it is possible that SMARCA4 is one of the key host factors required for β-CoV replication. SMARCA4 has also been previously identified as a top hit in other genome-wide CRISPR–Cas9-based screens and as an enriched molecule in several human cell lines and cell types in the context of SARS-CoV-2 viral infection [50,51,52]. However, in these previous studies, SMARCA4 was identified as a regulator of ACE2 expression [51], the key SARS-CoV-2 receptor [53,54,55]. As CEACAM1, and not ACE2, is the key receptor for MHV invasion [56], our results suggest that SMARCA4 acts through an alternative mechanism to regulate MHV-induced PANoptosis. However, future studies should address whether SMARCA4 regulates the expression of CEACAM1 or is required for viral replication during MHV infection in myeloid and non-myeloid cell types.
Overall, our findings substantiate and expand the importance of genome-wide screens for identifying host factors, such as SMARCA4, that regulate PANoptosis in response to β-CoV. These results contribute to our understanding of the role of the innate immune response in viral pathogenesis and cell death. Moreover, these findings suggest that targeting SMARCA4 may have therapeutic applications in limiting viral pathogenesis.
Author Contributions
R.K.S.M. and T.-D.K. designed the study. R.K.S.M. performed the experiments. R.K.S.M. analyzed the data. T.-D.K. oversaw the project. All authors have read and agreed to the published version of the manuscript.
Funding
Work from our laboratory was supported by the US National Institutes of Health (AI101935, AI124346, AI160179, AR056296, and CA253095 to T.-D.K.) and the American Lebanese Syrian Associated Charities (to T.-D.K.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Next-generation sequencing results from the CRISPR screen have been deposited in the BioProject: PRJNA1009133. All other datasets are included in the published article and the supplementary information.
Acknowledgments
We thank all the members of the Kanneganti laboratory for their scientific input, comments, and suggestions during the development of the manuscript. We thank Anu Sharma, Sara Resende, and Rebecca Tweedell, for scientific writing and editing, and Katie Thorne and Lauren Kneeland for mouse colony support. The content is the sole responsibility of the authors, and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kanneganti, T.D. Intracellular innate immune receptors: Life inside the cell. Immunol. Rev. 2020, 297, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Kanneganti, T.D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef] [PubMed]
- van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; Asselta, R.; et al. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e23. [Google Scholar] [CrossRef] [PubMed]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kanneganti, T.D. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Palacios, Y.; Ramón-Luing, L.A.; Ruiz, A.; García-Martínez, A.; Sánchez-Monciváis, A.; Barreto-Rodríguez, O.; Falfán-Valencia, R.; Pérez-Rubio, G.; Medina-Quero, K.; Buendia-Roldan, I.; et al. COVID-19 patients with high TNF/IFN-γ levels show hallmarks of PANoptosis, an inflammatory cell death. Microbes Infect. 2023, 25, 105179. [Google Scholar] [CrossRef]
- Yang, Q.; Song, W.; Reheman, H.; Wang, D.; Qu, J.; Li, Y. PANoptosis, an indicator of COVID-19 severity and outcomes. Brief. Bioinform. 2024, 25, bbae124. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Lee, S.; Mall, R.; Pandian, N.; Wang, Y.; Sharma, B.R.; Malireddi, R.S.; Yang, D.; Trifkovic, S.; Steele, J.A.; et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci. Immunol. 2022, 7, eabo6294. [Google Scholar] [CrossRef] [PubMed]
- Pandeya, A.; Kanneganti, T.D. Therapeutic potential of PANoptosis: Innate sensors, inflammasomes, and RIPKs in PANoptosomes. Trends Mol. Med. 2024, 30, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Pandian, N.; Kanneganti, T.D. PANoptosis: A Unique Innate Immune Inflammatory Cell Death Modality. J. Immunol. 2022, 209, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gullett, J.M.; Tweedell, R.E.; Kanneganti, T.D. Innate immune inflammatory cell death: PANoptosis and PANoptosomes in host defense and disease. Eur. J. Immunol. 2023, 53, e2250235. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Gurung, P.; Vogel, P.; Johnson, G.R.; Carter, R.A.; McGoldrick, D.J.; Bandi, S.R.; Calabrese, C.R.; Vande Walle, L.; Lamkanfi, M.; et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 2014, 516, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Gullett, J.M.; Tweedell, R.E.; Kanneganti, T.D. It’s All in the PAN: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by PANoptosis Underlying the Totality of Cell Death-Associated Biological Effects. Cells 2022, 11, 1495. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Kesavardhana, S.; Karki, R.; Kancharana, B.; Burton, A.R.; Kanneganti, T.D. RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons 2020, 4, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sundaram, B.; Sharma, B.R.; Lee, S.; Malireddi, R.K.S.; Nguyen, L.N.; Christgen, S.; Zheng, M.; Wang, Y.; Samir, P.; et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021, 37, 109858. [Google Scholar] [CrossRef]
- Sundaram, B.; Pandian, N.; Mall, R.; Wang, Y.; Sarkar, R.; Kim, H.J.; Malireddi, R.K.S.; Karki, R.; Janke, L.J.; Vogel, P.; et al. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell 2023, 186, 2783–2801.e20. [Google Scholar] [CrossRef]
- Lee, S.; Karki, R.; Wang, Y.Q.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Bynigeri, R.R.; Mall, R.; Connelly, J.P.; Pruett-Miller, S.M.; Kanneganti, T.D. Inflammatory cell death, PANoptosis, screen identifies host factors in coronavirus innate immune response as therapeutic targets. Commun. Biol. 2023, 6, 1071. [Google Scholar] [CrossRef]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [PubMed]
- Roth-Cross, J.K.; Bender, S.J.; Weiss, S.R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 2008, 82, 9829–9838. [Google Scholar] [CrossRef]
- Schickli, J.H.; Zelus, B.D.; Wentworth, D.E.; Sawicki, S.G.; Holmes, K.V. The murine coronavirus mouse hepatitis virus strain A59 from persistently infected murine cells exhibits an extended host range. J. Virol. 1997, 71, 9499–9507. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef]
- Li, W.; Köster, J.; Xu, H.; Chen, C.H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015, 16, 281. [Google Scholar] [CrossRef]
- Wang, B.; Wang, M.; Zhang, W.; Xiao, T.; Chen, C.H.; Wu, A.; Wu, F.; Traugh, N.; Wang, X.; Li, Z.; et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019, 14, 756–780. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Gurung, P.; Mavuluri, J.; Dasari, T.K.; Klco, J.M.; Chi, H.; Kanneganti, T.D. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 2018, 215, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O‘Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Sborgi, L.; Ruhl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Kanneganti, T.D. PANoptosis in Viral Infection: The Missing Puzzle Piece in the Cell Death Field. J. Mol. Biol. 2022, 434, 167249. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Ma, Z.; Xie, J.; Li, W.; Su, L.; Zhang, G.; Xu, J.; Wu, Y.; Zhang, M.; Liu, W. The role of cell death in SARS-CoV-2 infection. Signal Transduct. Target. Ther. 2023, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Riegler, A.N.; Benson, P.; Long, K.; Leal, S.M., Jr. Differential activation of programmed cell death in patients with severe SARS-CoV-2 infection. Cell Death Discov. 2023, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, 2. [Google Scholar] [CrossRef]
- Han, J.H.; Karki, R.; Malireddi, R.K.S.; Mall, R.; Sarkar, R.; Sharma, B.R.; Klein, J.; Berns, H.; Pisharath, H.; Pruett-Miller, S.M.; et al. NINJ1 mediates inflammatory cell death, PANoptosis, and lethality during infection conditions and heat stress. Nat. Commun. 2024, 15, 1739. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.-D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity–independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217, e20191644. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Karki, R.; Sundaram, B.; Kancharana, B.; Lee, S.; Samir, P.; Kanneganti, T.D. Inflammatory Cell Death, PANoptosis, Mediated by Cytokines in Diverse Cancer Lineages Inhibits Tumor Growth. Immunohorizons 2021, 5, 568–580. [Google Scholar] [CrossRef]
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, R.K.S.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S.; et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell Infect. Microbiol. 2020, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687 e613. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, B.; Pandian, N.; Kim, H.J.; Abdelaal, H.M.; Mall, R.; Indari, O.; Sarkar, R.; Tweedell, R.E.; Alonzo, E.Q.; Klein, J.; et al. NLRC5 senses NAD+ depletion, forming a PANoptosome and driving PANoptosis and inflammation. Cell 2024, 187, 4061–4077.e17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Mears, J.R.; Shakib, L.; Beynor, J.I.; Shanaj, S.; Korsunsky, I.; Nathan, A.; Donlin, L.T.; Raychaudhuri, S. IFN- γ and TNF- α drive a CXCL10 + CCL2 + macrophage phenotype expanded in severe COVID-19 and other diseases with tissue inflammation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Euskirchen, G.M.; Auerbach, R.K.; Davidov, E.; Gianoulis, T.A.; Zhong, G.; Rozowsky, J.; Bhardwaj, N.; Gerstein, M.B.; Snyder, M. Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet. 2011, 7, e1002008. [Google Scholar] [CrossRef]
- Trotter, K.W.; Archer, T.K. The BRG1 transcriptional coregulator. Nucl. Recept. Signal 2008, 6, e004. [Google Scholar] [CrossRef]
- Nagy, P.D.; Pogany, J. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 2011, 10, 137–149. [Google Scholar] [CrossRef]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184, 76–91.e13. [Google Scholar] [CrossRef]
- Wei, J.; Patil, A.; Collings, C.K.; Alfajaro, M.M.; Liang, Y.; Cai, W.L.; Strine, M.S.; Filler, R.B.; DeWeirdt, P.C.; Hanna, R.E.; et al. Pharmacological disruption of mSWI/SNF complex activity restricts SARS-CoV-2 infection. Nat. Genet. 2023, 55, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Rebendenne, A.; Roy, P.; Bonaventure, B.; Chaves Valadão, A.L.; Desmarets, L.; Arnaud-Arnould, M.; Rouillé, Y.; Tauziet, M.; Giovannini, D.; Touhami, J.; et al. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal HCoVs. Nat. Genet. 2022, 54, 1090–1102. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Devaux, C.A.; Rolain, J.M.; Raoult, D. ACE2 receptor polymorphism: Susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J. Microbiol. Immunol. Infect. 2020, 53, 425–435. [Google Scholar] [CrossRef]
- Williams, R.K.; Jiang, G.S.; Holmes, K.V. Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc. Natl. Acad. Sci. USA 1991, 88, 5533–5536. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).