
The Dual Role of TRIM7 in Viral Infections


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
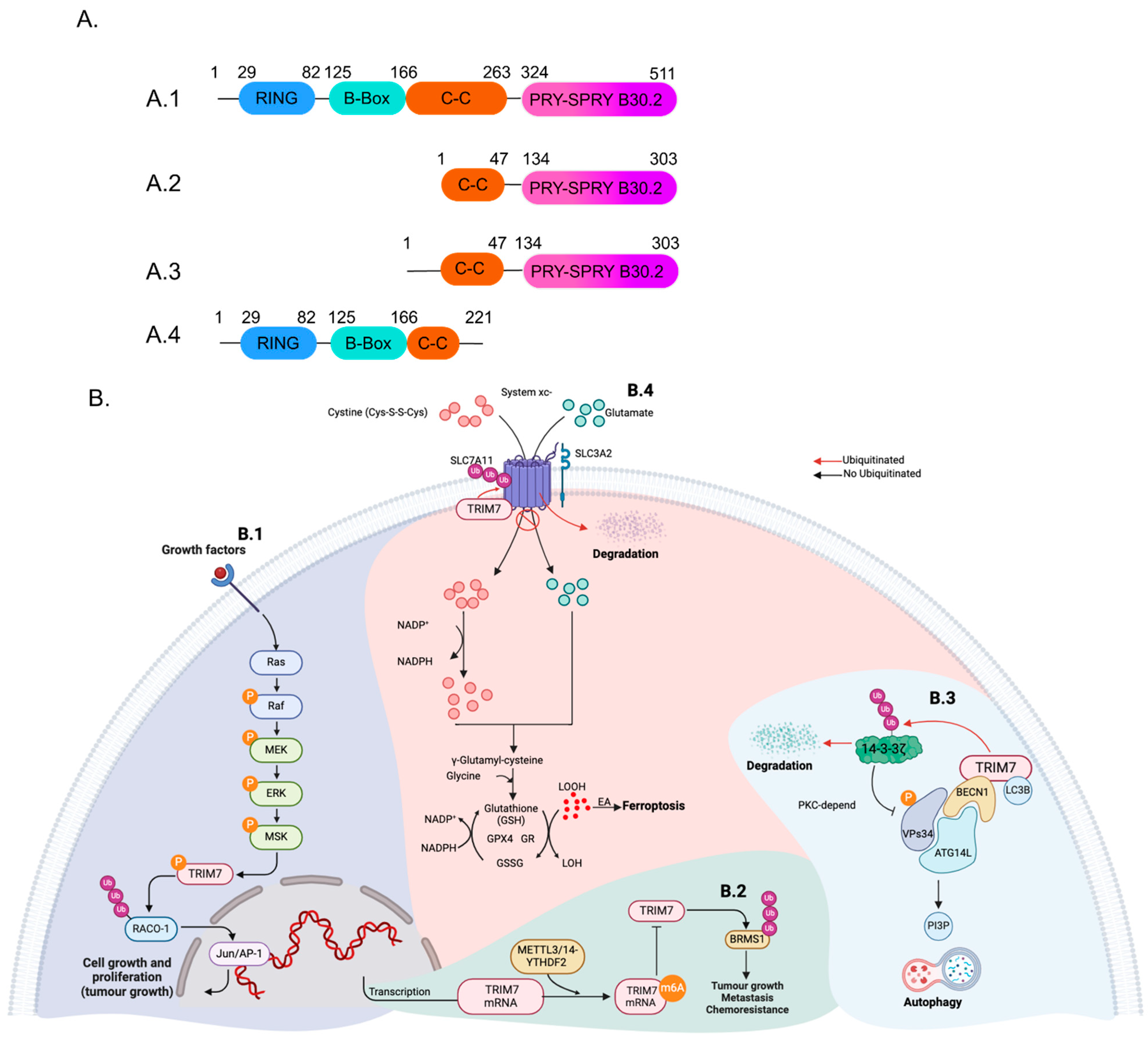
2. TRIM7 Structure and Isoforms
3. TRIM7 in Non-Viral Conditions
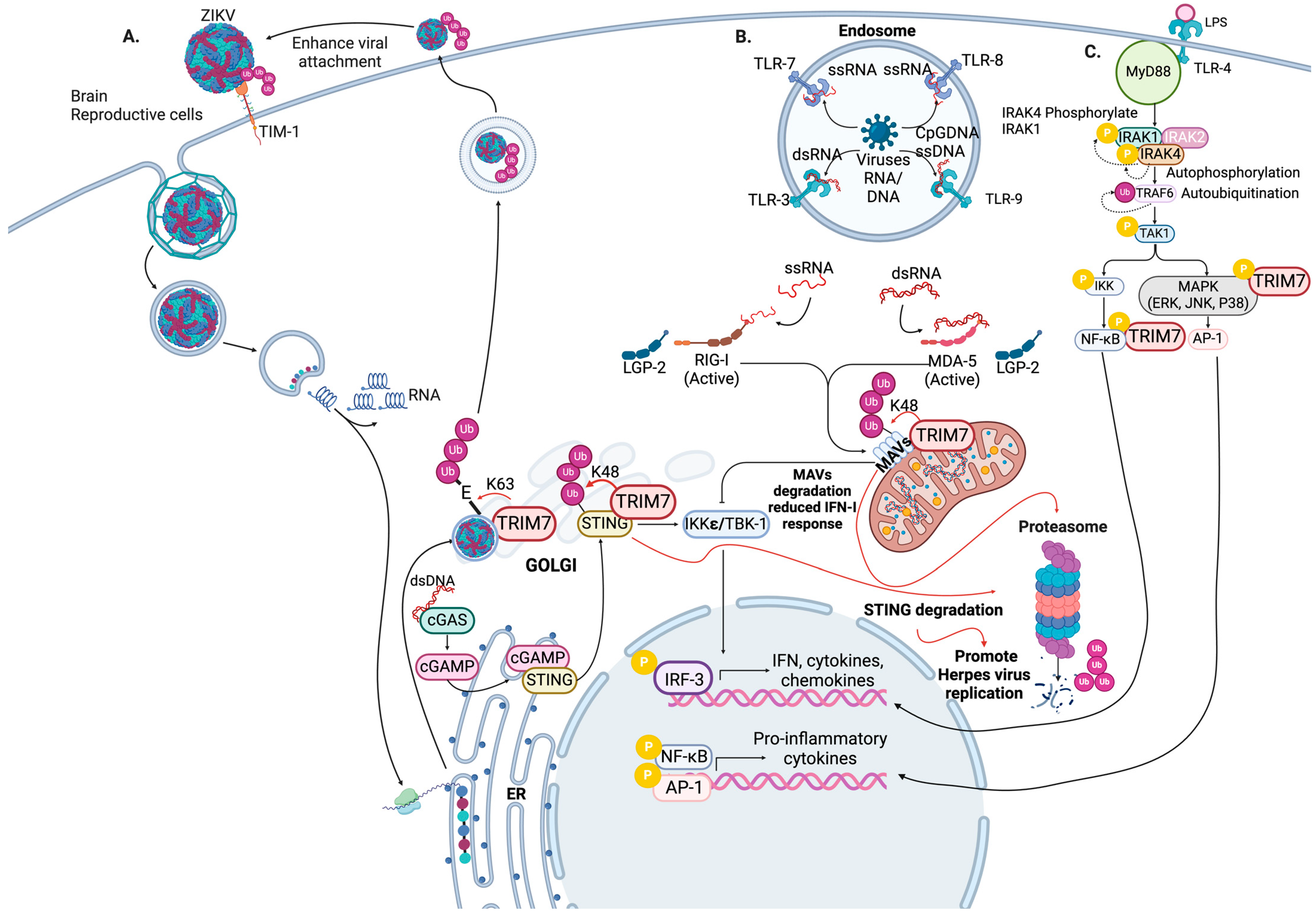
4. TRIM7 Regulates the Antiviral Innate Immune Response
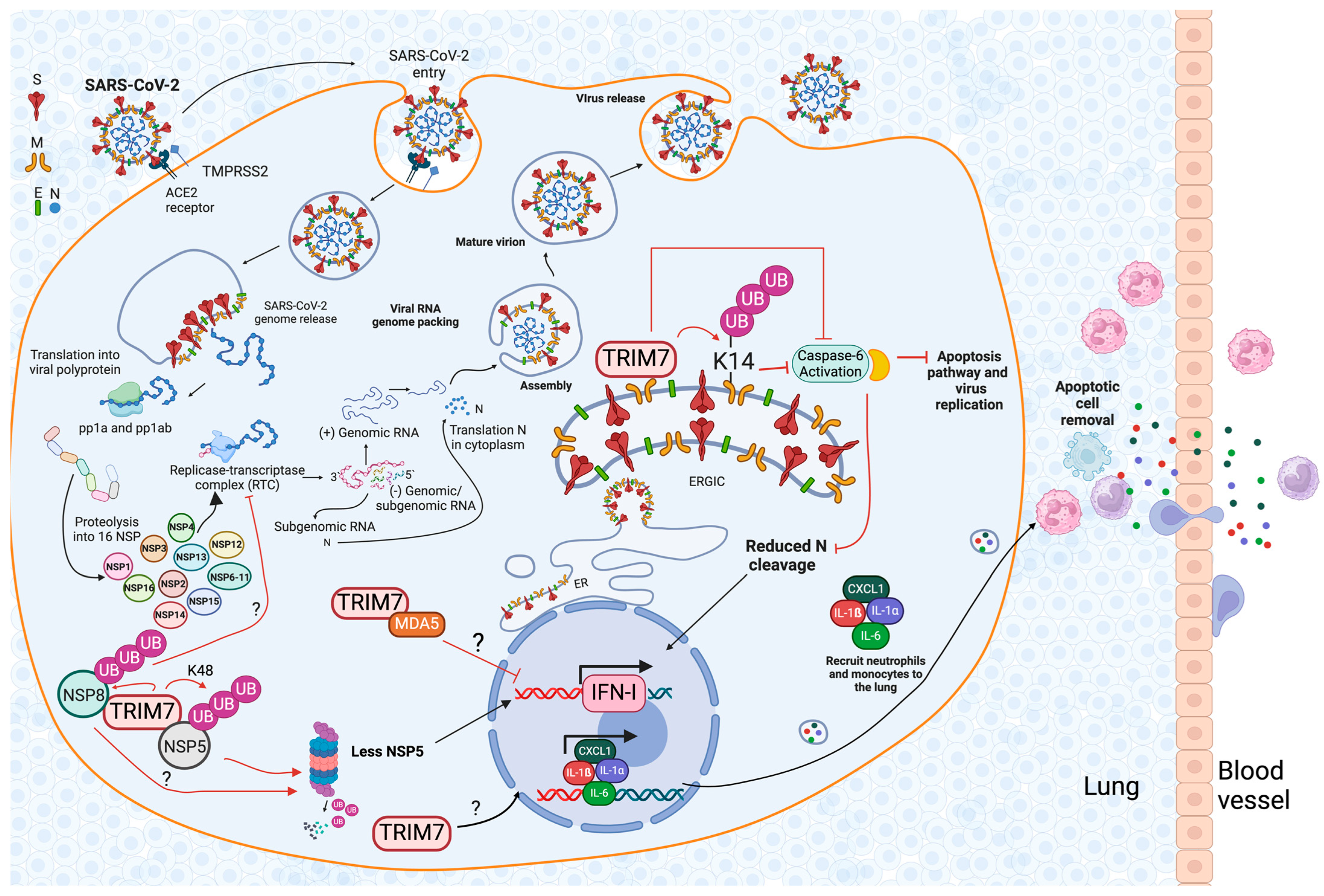
5. Proviral Role of TRIM7 during Virus Replication
6. Antiviral Roles of TRIM7
6.1. TRIM7 in Enteric Virus Infection
6.2. The Role of TRIM7 during SARS-CoV-2 Infection
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Lee, J.M.; Hammaren, H.M.; Savitski, M.M.; Baek, S.H. Control of protein stability by post-translational modifications. Nat. Commun. 2023, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenen-Stass, A.; Pertel, T.; Luban, J.; Mothes, W. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 2013, 87, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines 2017, 5, 23. [Google Scholar] [CrossRef]
- Damgaard, R.B. The ubiquitin system: From cell signalling to disease biology and new therapeutic opportunities. Cell Death Differ. 2021, 28, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Hage, A.; Rajsbaum, R. To TRIM or not to TRIM: The balance of host-virus interactions mediated by the ubiquitin system. J. Gen. Virol. 2019, 100, 1641–1662. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, J.; Chen, D.; Wang, Y. E3 ubiquitin ligases: Styles, structures and functions. Mol. Biomed. 2021, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Valerdi, K.M.; Hage, A.; van Tol, S.; Rajsbaum, R.; Giraldo, M.I. The Role of the Host Ubiquitin System in Promoting Replication of Emergent Viruses. Viruses 2021, 13, 369. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Hage, A.; van Tol, S.; Rajsbaum, R. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr. Clin. Microbiol. Rep. 2020, 7, 101–114. [Google Scholar] [CrossRef]
- Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur. J. Immunol. 2008, 38, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Versteeg, G.A.; Schmid, S.; Maestre, A.M.; Belicha-Villanueva, A.; Martinez-Romero, C.; Patel, J.R.; Morrison, J.; Pisanelli, G.; Miorin, L.; et al. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKKepsilon kinase-mediated antiviral response. Immunity 2014, 40, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; Rajsbaum, R.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Valdiviezo, J.; Shi, M.; Inn, K.S.; Fernandez-Sesma, A.; Jung, J.; Garcia-Sastre, A. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 2013, 38, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zou, C.; Wang, X.; Huang, C.; Feng, T.; Pan, W.; Wu, Q.; Wang, P.; Dai, J. Interferon-stimulated TRIM69 interrupts dengue virus replication by ubiquitinating viral nonstructural protein 3. PLoS Pathog. 2018, 14, e1007287. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, J.; Wang, S.; Wu, F.; Chen, Z.; Li, C.; Cheng, G.; Qin, F.X. Inhibition of Influenza A Virus Replication by TRIM14 via Its Multifaceted Protein-Protein Interaction With NP. Front. Microbiol. 2019, 10, 344. [Google Scholar] [CrossRef] [PubMed]
- Bharaj, P.; Atkins, C.; Luthra, P.; Giraldo, M.I.; Dawes, B.E.; Miorin, L.; Johnson, J.R.; Krogan, N.J.; Basler, C.F.; Freiberg, A.N.; et al. The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication. J. Virol. 2017, 91, e00833-17. [Google Scholar] [CrossRef] [PubMed]
- van Tol, S.; Kalveram, B.; Ilinykh, P.A.; Ronk, A.; Huang, K.; Aguilera-Aguirre, L.; Bharaj, P.; Hage, A.; Atkins, C.; Giraldo, M.I.; et al. Ubiquitination of Ebola virus VP35 at lysine 309 regulates viral transcription and assembly. PLoS Pathog. 2022, 18, e1010532. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Salazar, C.A.; van Tol, S.; Mailhot, O.; Gonzalez-Orozco, M.; Galdino, G.T.; Warren, A.N.; Teruel, N.; Behera, P.; Afreen, K.S.; Zhang, L.; et al. Ebola virus VP35 interacts non-covalently with ubiquitin chains to promote viral replication. PLoS Biol. 2024, 22, e3002544. [Google Scholar] [CrossRef]
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.G.; Gao, N.; Yu, H.; et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014, 24, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Skurat, A.V.; Dietrich, A.D.; Zhai, L.; Roach, P.J. GNIP, a novel protein that binds and activates glycogenin, the self-glucosylating initiator of glycogen biosynthesis. J. Biol. Chem. 2002, 277, 19331–19338. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Dietrich, A.; Skurat, A.V.; Roach, P.J. Structure-function analysis of GNIP, the glycogenin-interacting protein. Arch. Biochem. Biophys. 2004, 421, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Munoz Sosa, C.J.; Issoglio, F.M.; Carrizo, M.E. Crystal structure and mutational analysis of the human TRIM7 B30.2 domain provide insights into the molecular basis of its binding to glycogenin-1. J. Biol. Chem. 2021, 296, 100772. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Lu, Z.; Wang, X.; Liu, Y.; Han, T.; Wang, Y.; Wang, T.; Gan, M.; Xie, C.; Wang, J.; et al. E3 ubiquitin ligase TRIM7 negatively regulates NF-kappa B signaling pathway by degrading p65 in lung cancer. Cell Signal 2020, 69, 109543. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhu, X.; Yang, Z.; Zhang, W.; Sun, Z.; Ji, Q.; Chen, X.; Zhu, J.; Wang, C.; Nie, S. E3 ubiquitin ligase tripartite motif 7 positively regulates the TLR4-mediated immune response via its E3 ligase domain in macrophages. Mol. Immunol. 2019, 109, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ma, A.; Liu, J.; Zhou, W.; Cao, P.; Chu, T.; Fan, L. Study on the expression of TRIM7 in peripheral blood mononuclear cells of patients with sepsis and its early diagnostic value. BMC Infect. Dis. 2022, 22, 865. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Diefenbacher, M.E.; Mylona, A.; Kassel, O.; Behrens, A. The E3 ubiquitin ligase Trim7 mediates c-Jun/AP-1 activation by Ras signalling. Nat. Commun. 2015, 6, 6782. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhang, Z.; Zhu, X.; Qian, G.; Zhou, Y.; Sun, Y.; Yu, W.; Wang, J.; Lu, H.; Lin, F.; et al. N6-Methyladenosine modification of the TRIM7 positively regulates tumorigenesis and chemoresistance in osteosarcoma through ubiquitination of BRMS1. EBioMedicine 2020, 59, 102955. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, Y.; Ai, W.; Wang, Y.; Gan, M.; Jiang, Q.; Han, T.; Wang, J.B. GNIP1 functions both as a scaffold protein and an E3 ubiquitin ligase to regulate autophagy in lung cancer. Cell Commun. Signal 2022, 20, 133. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, T.; Zeng, R.; Zhang, K.; Li, B.; Zhu, Z.; Ma, X.; Zhang, Y.; Li, L.; Zhu, J.; et al. The E3 ligase TRIM7 suppresses the tumorigenesis of gastric cancer by targeting SLC7A11. Sci. Rep. 2024, 14, 6655. [Google Scholar] [CrossRef]
- Zhu, L.; Qin, C.; Li, T.; Ma, X.; Qiu, Y.; Lin, Y.; Ma, D.; Qin, Z.; Sun, C.; Shen, X.; et al. The E3 ubiquitin ligase TRIM7 suppressed hepatocellular carcinoma progression by directly targeting Src protein. Cell Death Differ. 2020, 27, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qin, X.; Huang, Y.; Zhang, Q.; Pei, J.; Wang, Y.; Goren, I.; Ma, S.; Song, Z.; Liu, Y.; et al. TRIM7/RNF90 promotes autophagy via regulation of ATG7 ubiquitination during L. monocytogenes infection. Autophagy 2023, 19, 1844–1862. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, G.; Qin, X.; Huang, Y.; Ren, X.; Sun, J.; Ma, S.; Liu, Y.; Song, D.; Liu, Y.; et al. Negative Regulation of RNF90 on RNA Virus-Triggered Antiviral Immune Responses Targeting MAVS. Front. Immunol. 2021, 12, 730483. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Liu, Y.; Cui, Y.; Song, D.; Zhang, G.; Ma, S.; Liu, Y.; Chen, M.; Chen, F.; Wang, H.; et al. RNF90 negatively regulates cellular antiviral responses by targeting MITA for degradation. PLoS Pathog. 2020, 16, e1008387. [Google Scholar] [CrossRef]
- Gonzalez-Orozco, M.; Tseng, H.C.; Hage, A.; Xia, H.; Behera, P.; Afreen, K.; Penaflor-Tellez, Y.; Giraldo, M.I.; Huante, M.; Puebla-Clark, L.; et al. TRIM7 ubiquitinates SARS-CoV-2 membrane protein to limit apoptosis and viral replication. bioRxiv 2024. [Google Scholar] [CrossRef]
- Li, M.; Yan, J.; Zhu, H.; Guo, C.; Jiang, X.; Gao, Y.; Liu, X.; Jiang, P.; Bai, J. TRIM7 inhibits encephalomyocarditis virus replication by activating interferon-beta signaling pathway. Vet. Microbiol. 2023, 281, 109729. [Google Scholar] [CrossRef]
- Ma, X.; Yuan, Z.; Yi, Z. Identification and characterization of key residues in Zika virus envelope protein for virus assembly and entry. Emerg. Microbes Infect. 2022, 11, 1604–1620. [Google Scholar] [CrossRef]
- Fan, W.; Mar, K.B.; Sari, L.; Gaszek, I.K.; Cheng, Q.; Evers, B.M.; Shelton, J.M.; Wight-Carter, M.; Siegwart, D.J.; Lin, M.M.; et al. TRIM7 inhibits enterovirus replication and promotes emergence of a viral variant with increased pathogenicity. Cell 2021, 184, 3410–3425.e3417. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Thibaut, H.J.; Strating, J.; van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16, 368–381. [Google Scholar] [CrossRef]
- Liang, X.; Xiao, J.; Li, X.; Liu, Y.; Lu, Y.; Wen, Y.; Li, Z.; Che, X.; Ma, Y.; Zhang, X.; et al. A C-terminal glutamine recognition mechanism revealed by E3 ligase TRIM7 structures. Nat. Chem. Biol. 2022, 18, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Ru, Y.; Yan, X.; Zhang, B.; Song, L.; Feng, Q.; Ye, C.; Zhou, Z.; Yang, Z.; Li, Y.; Zhang, Z.; et al. C-terminal glutamine acts as a C-degron targeted by E3 ubiquitin ligase TRIM7. Proc. Natl. Acad. Sci. USA 2022, 119, e2203218119. [Google Scholar] [CrossRef] [PubMed]
- Luptak, J.; Mallery, D.L.; Jahun, A.S.; Albecka, A.; Clift, D.; Ather, O.; Slodkowicz, G.; Goodfellow, I.; James, L.C. TRIM7 Restricts Coxsackievirus and Norovirus Infection by Detecting the C-Terminal Glutamine Generated by 3C Protease Processing. Viruses 2022, 14, 1610. [Google Scholar] [CrossRef] [PubMed]
- Orchard, R.C.; Sullender, M.E.; Dunlap, B.F.; Balce, D.R.; Doench, J.G.; Virgin, H.W. Identification of Antinorovirus Genes in Human Cells Using Genome-Wide CRISPR Activation Screening. J. Virol. 2019, 93, e01324-18. [Google Scholar] [CrossRef] [PubMed]
- Sullender, M.E.; Pierce, L.R.; Annaswamy Srinivas, M.; Crockett, S.L.; Dunlap, B.F.; Rodgers, R.; Schriefer, L.A.; Kennedy, E.A.; Stewart, B.M.; Doench, J.G.; et al. Selective Polyprotein Processing Determines Norovirus Sensitivity to Trim7. J. Virol. 2022, 96, e0070722. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; McDougal, M.B.; Schoggins, J.W. Enterovirus 3C Protease Cleaves TRIM7 To Dampen Its Antiviral Activity. J. Virol. 2022, 96, e0133222. [Google Scholar] [CrossRef]
- Chen, J.; Li, Z.; Guo, J.; Xu, S.; Zhou, J.; Chen, Q.; Tong, X.; Wang, D.; Peng, G.; Fang, L.; et al. SARS-CoV-2 nsp5 Exhibits Stronger Catalytic Activity and Interferon Antagonism than Its SARS-CoV Ortholog. J. Virol. 2022, 96, e0003722. [Google Scholar] [CrossRef]
- Liang, W.; Gu, M.; Zhu, L.; Yan, Z.; Schenten, D.; Herrick, S.; Li, H.; Samrat, S.K.; Zhu, J.; Chen, Y. The main protease of SARS-CoV-2 downregulates innate immunity via a translational repression. Signal Transduct. Target. Ther. 2023, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, M.; Aktepe, T.E.; Deerain, J.M.; McAuley, J.L.; Audsley, M.D.; David, C.T.; Purcell, D.F.J.; Urin, V.; Hartmann, R.; Moseley, G.W.; et al. SARS-CoV-2 suppresses IFNbeta production mediated by NSP1, 5, 6, 15, ORF6 and ORF7b but does not suppress the effects of added interferon. PLoS Pathog. 2021, 17, e1009800. [Google Scholar] [CrossRef]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.W.; Xu, Y.; Zhang, J.; Nan, M.L.; Xiao, Y.; Zhan, P.; Liu, X.; et al. SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal Transduct. Target. Ther. 2022, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, C.; Rao, Y.; Ngo, C.; Feng, J.J.; Zhao, J.; Zhang, S.; Wang, T.Y.; Carriere, J.; Savas, A.C.; et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. mBio 2021, 12, e0233521. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Hu, B.; Xiao, H.; Tan, X.; Li, Y.; Tang, K.; Zhang, Y.; Cai, K.; Ding, B. The E3 Ubiquitin Ligase RNF5 Facilitates SARS-CoV-2 Membrane Protein-Mediated Virion Release. mBio 2021, 13, e0316821. [Google Scholar] [CrossRef]
- Chu, H.; Hou, Y.; Yang, D.; Wen, L.; Shuai, H.; Yoon, C.; Shi, J.; Chai, Y.; Yuen, T.T.; Hu, B.; et al. Coronaviruses exploit a host cysteine-aspartic protease for replication. Nature 2022, 609, 785–792. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Orozco, M.; Rodriguez-Salazar, C.A.; Giraldo, M.I. The Dual Role of TRIM7 in Viral Infections. Viruses 2024, 16, 1285. https://doi.org/10.3390/v16081285
Gonzalez-Orozco M, Rodriguez-Salazar CA, Giraldo MI. The Dual Role of TRIM7 in Viral Infections. Viruses. 2024; 16(8):1285. https://doi.org/10.3390/v16081285
Chicago/Turabian StyleGonzalez-Orozco, Maria, Carlos A. Rodriguez-Salazar, and Maria I. Giraldo. 2024. "The Dual Role of TRIM7 in Viral Infections" Viruses 16, no. 8: 1285. https://doi.org/10.3390/v16081285
APA StyleGonzalez-Orozco, M., Rodriguez-Salazar, C. A., & Giraldo, M. I. (2024). The Dual Role of TRIM7 in Viral Infections. Viruses, 16(8), 1285. https://doi.org/10.3390/v16081285