
A Standardized Pipeline for Assembly and Annotation of African Swine Fever Virus Genome

 , , , , , , , ,
, , , , , , , ,  ,
,  , , ,
, , ,  , ,
, , 
 , , add
Show full author list
, , add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Pipeline Overview
2.1.1. Illumina Reads QC (Both Pipelines)
2.1.2. De Novo Assembly (Both Pipelines)
2.1.3. Reference Prediction and Mapping (Illumina Only)
2.1.4. Contig Extension (De Novo Only)
2.1.5. Error Correction/Variant Identification and Consensus Extraction (Both Pipelines)
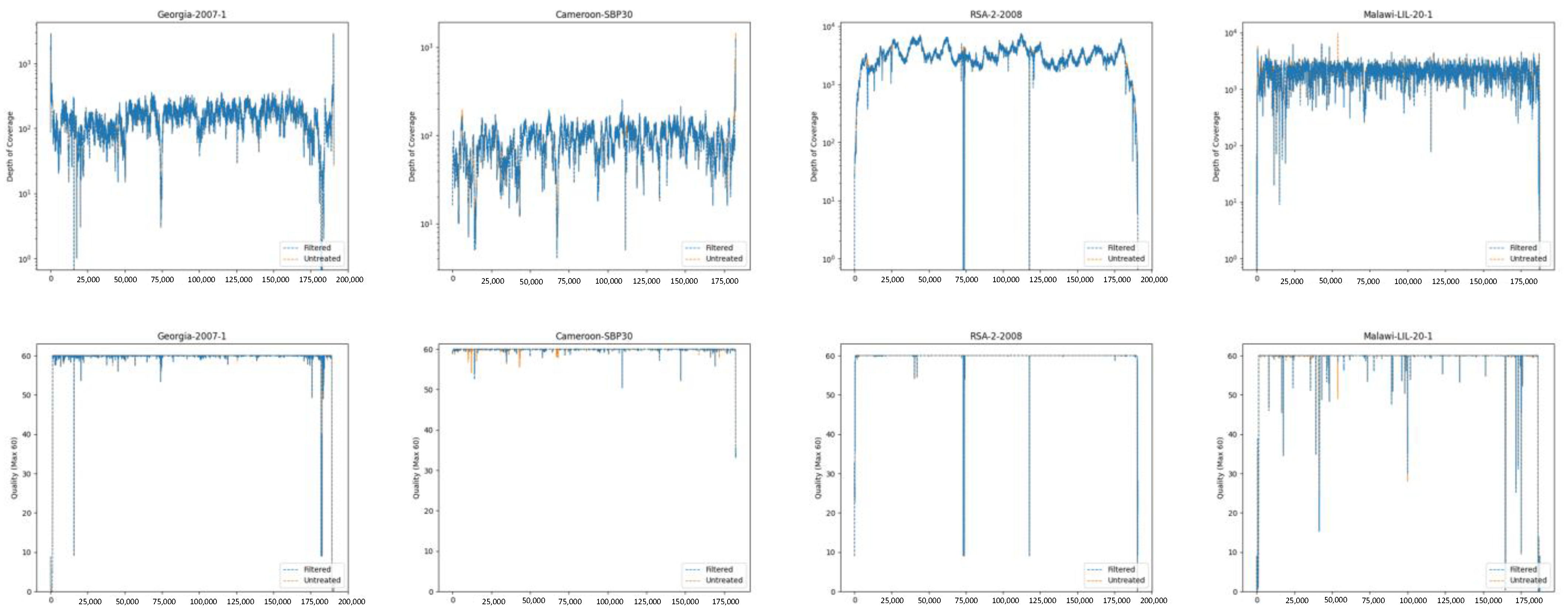
2.1.6. Developing Statistics and Coverage/Quality Map Images (Both Pipelines)
2.1.7. Biotyping, Genotyping, and Annotation (Both Pipelines)
2.2. Usage
2.3. Interpretation of Outputs
3. Results and Discussion
3.1. Filtering the Illumina Reads for Host Sequence Does Not Result in a Large Loss of Reads
3.2. De Novo Assembly ASFV DR-1980
3.3. Reference-Based Assembly of DR-1980
3.4. Shared Unresolvable Variants DR-1980
3.5. De Novo Assembly ASFV Ghana2022-35, a Less Than Perfect Isolate
3.6. Reference-Based Assembly Should Only Be Used for Highly Similar Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chapman, D.A.; Darby, A.C.; Da Silva, M.; Upton, C.; Radford, A.D.; Dixon, L.K. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg. Infect. Dis. 2011, 17, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Le, V.P.; Nguyen, V.T.; Le, T.B.; Mai, N.T.A.; Nguyen, V.D.; Than, T.T.; Lai, T.N.H.; Cho, K.H.; Hong, S.K.; Kim, Y.H.; et al. Detection of Recombinant African Swine Fever Virus Strains of p72 Genotypes I and II in Domestic Pigs, Vietnam, 2023. Emerg. Infect. Dis. 2024, 30, 991–994. [Google Scholar] [CrossRef]
- Zhao, D.; Sun, E.; Huang, L.; Ding, L.; Zhu, Y.; Zhang, J.; Shen, D.; Zhang, X.; Zhang, Z.; Ren, T.; et al. Highly lethal genotype I and II recombinant African swine fever viruses detected in pigs. Nat. Commun. 2023, 14, 3096. [Google Scholar] [CrossRef] [PubMed]
- Carriquiry, M.; Elobeid, A.; Swenson, D.; Hayes, D. Impacts of African Swine Fever in Iowa and the United States; Center for Agricultural and Rural Development: Ames, IA, USA, 2020. [Google Scholar]
- Bastos, A.D.; Penrith, M.L.; Cruciere, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef]
- Spinard, E.; Dinhobl, M.; Tesler, N.; Birtley, H.; Signore, A.V.; Ambagala, A.; Masembe, C.; Borca, M.V.; Gladue, D.P. A Re-Evaluation of African Swine Fever Genotypes Based on p72 Sequences Reveals the Existence of Only Six Distinct p72 Groups. Viruses 2023, 15, 2246. [Google Scholar] [CrossRef] [PubMed]
- Dinhobl, M.; Spinard, E.; Tesler, N.; Birtley, H.; Signore, A.; Ambagala, A.; Masembe, C.; Borca, M.V.; Gladue, D.P. Reclassification of ASFV into 7 Biotypes Using Unsupervised Machine Learning. Viruses 2023, 16, 67. [Google Scholar] [CrossRef]
- Kabuuka, T.; Mulindwa, H.; Bastos, A.D.S.; van Heerden, J.; Heath, L.; Fasina, F.O. Retrospective Multi-Locus Sequence Analysis of African Swine Fever Viruses by “PACT” Confirms Co-Circulation of Multiple Outbreak Strains in Uganda. Animals 2023, 14, 71. [Google Scholar] [CrossRef]
- Mazloum, A.; van Schalkwyk, A.; Chernyshev, R.; Igolkin, A.; Heath, L.; Sprygin, A. A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers. Microorganisms 2023, 11, 642. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Casado, N.; Soler, A.; Djadjovski, I.; Krivko, L.; Madueño, E.; Nieto, R.; Perez, C.; Simon, A.; Ivanova, E.; et al. A multi gene-approach genotyping method identifies 24 genetic clusters within the genotype II-European African swine fever viruses circulating from 2007 to 2022. Front. Vet. Sci. 2023, 10, 1112850. [Google Scholar] [CrossRef] [PubMed]
- Onzere, C.K.; Bastos, A.D.; Okoth, E.A.; Lichoti, J.K.; Bochere, E.N.; Owido, M.G.; Ndambuki, G.; Bronsvoort, M.; Bishop, R.P. Multi-locus sequence typing of African swine fever viruses from endemic regions of Kenya and Eastern Uganda (2011–2013) reveals rapid B602L central variable region evolution. Virus Genes 2018, 54, 111–123. [Google Scholar] [CrossRef]
- Yoo, D.; Kim, H.; Lee, J.Y.; Yoo, H.S. African swine fever: Etiology, epidemiological status in Korea, and perspective on control. J. Vet. Sci. 2020, 21, e38. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Jiang, M.; Wu, K.; Hu, J.; Wang, Y.; Quan, W.; Hao, M.; Liu, H.; Wei, H.; Fan, W.; et al. Nanopore sequencing of African swine fever virus. Sci. China Life Sci. 2020, 63, 160–164. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.K.; Grau, F.R.; Mayr, G.A.; Sturgill Samayoa, T.L.; Dodd, K.A.; Barrette, R.W. Rapid Sequence-Based Characterization of African Swine Fever Virus by Use of the Oxford Nanopore MinION Sequence Sensing Device and a Companion Analysis Software Tool. J. Clin. Microbiol. 2019, 58. [Google Scholar] [CrossRef] [PubMed]
- Olasz, F.; Tombacz, D.; Torma, G.; Csabai, Z.; Moldovan, N.; Dormo, A.; Prazsak, I.; Meszaros, I.; Magyar, T.; Tamas, V.; et al. Short and Long-Read Sequencing Survey of the Dynamic Transcriptomes of African Swine Fever Virus and the Host Cells. Front. Genet. 2020, 11, 758. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Filipe, A.; Vattipally, S.B.; Mair, D.; Ogweng, P.; Francis, M.J.; Muwanika, V.B.; Palmarini, M.; Biek, R.; Masembe, C. Host genome depletion to determine the evolution, genetic diversity and transmission patterns of full genome sequences of African swine fever genotype IX from Uganda. Access Microbiol. 2019, 1. [Google Scholar] [CrossRef]
- Ambagala, A.; Goonewardene, K.; Lamboo, L.; Goolia, M.; Erdelyan, C.; Fisher, M.; Handel, K.; Lung, O.; Blome, S.; King, J.; et al. Characterization of a Novel African Swine Fever Virus p72 Genotype II from Nigeria. Viruses 2023, 15, 915. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.H.; Forth, L.F.; King, J.; Groza, O.; Hubner, A.; Olesen, A.S.; Hoper, D.; Dixon, L.K.; Netherton, C.L.; Rasmussen, T.B.; et al. A Deep-Sequencing Workflow for the Fast and Efficient Generation of High-Quality African Swine Fever Virus Whole-Genome Sequences. Viruses 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- Farlow, J.; Donduashvili, M.; Kokhreidze, M.; Kotorashvili, A.; Vepkhvadze, N.G.; Kotaria, N.; Gulbani, A. Intra-epidemic genome variation in highly pathogenic African swine fever virus (ASFV) from the country of Georgia. Virol. J. 2018, 15, 190. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D.A.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 2008, 89, 397–408. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Salas, M.L.; Vinuela, E. Genes homologous to ubiquitin-conjugating proteins and eukaryotic transcription factor SII in African swine fever virus. Virology 1992, 186, 40–52. [Google Scholar] [CrossRef]
- Qi, X.; Feng, T.; Ma, Z.; Zheng, L.; Liu, H.; Shi, Z.; Shen, C.; Li, P.; Wu, P.; Ru, Y.; et al. Deletion of DP148R, DP71L, and DP96R Attenuates African Swine Fever Virus, and the Mutant Strain Confers Complete Protection against Homologous Challenges in Pigs. J. Virol. 2023, 97, e0024723. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. imeta 2023, 2, e107. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Warr, A.; Affara, N.; Aken, B.; Beiki, H.; Bickhart, D.M.; Billis, K.; Chow, W.; Eory, L.; Finlayson, H.A.; Flicek, P.; et al. An improved pig reference genome sequence to enable pig genetics and genomics research. Gigascience 2020, 9, giaa051. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Dinhobl, M.; Spinard, E.; Birtley, H.; Tesler, N.; Borca, M.V.; Gladue, D.P. African swine fever virus P72 genotyping tool. Microbiol. Resour. Announc. 2024, 13, e0089123. [Google Scholar] [CrossRef] [PubMed]
- Ribeca, P. The Transporter. Available online: https://zenodo.org/records/13361139 (accessed on 15 January 2024).
- Ndlovu, S.; Williamson, A.L.; Heath, L.; Carulei, O. Genome Sequences of Three African Swine Fever Viruses of Genotypes IV and XX from Zaire and South Africa, Isolated from a Domestic Pig (Sus scrofa domesticus), a Warthog (Phacochoerus africanus), and a European Wild Boar (Sus scrofa). Microbiol. Resour. Announc. 2020, 9, e00341-20. [Google Scholar] [CrossRef] [PubMed]
- Spinard, E.; Wade, A.; Unger, H.; Robert, N.; Mayega, F.J.; Sreenu, V.B.; Da Silva Filpe, A.; Mair, D.; Borca, M.V.; Gladue, D.P.; et al. Near-complete genome sequences of multiple genotype 1 African swine fever virus isolates from 2016 to 2018 in Cameroon. Microbiol. Resour. Announc. 2024, 13, e0097823. [Google Scholar] [CrossRef] [PubMed]
- Spinard, E.; O’Donnell, V.; Vuono, E.; Rai, A.; Davis, C.; Ramirez-Medina, E.; Espinoza, N.; Valladares, A.; Borca, M.V.; Gladue, D.P. Full genome sequence for the African swine fever virus outbreak in the Dominican Republic in 1980. Sci. Rep. 2023, 13, 1024. [Google Scholar] [CrossRef]
- Spinard, E.; Rai, A.; Osei-Bonsu, J.; O’Donnell, V.; Ababio, P.T.; Tawiah-Yingar, D.; Arthur, D.; Baah, D.; Ramirez-Medina, E.; Espinoza, N.; et al. The 2022 Outbreaks of African Swine Fever Virus Demonstrate the First Report of Genotype II in Ghana. Viruses 2023, 15, 1722. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| GenBank Accession | Isolate |
|---|---|
| U18466 | BA71V |
| AM712239 | Benin |
| AM712240 | OURT_88_3 |
| AY261360 | Kenya_1950 |
| AY261361 | Malawi_Lil_20_1 |
| AY261362 | Mkuzi_1979 |
| AY261363 | Pret_96_4 |
| AY261364 | Tengani62 |
| AY261365 | Warmbaths |
| AY261366 | Warthog |
| FN557520 | E75 |
| KM111294 | Ken05 |
| KM111295 | Ken06Bus |
| KM262844 | L60 |
| KM262845 | NHV |
| KP055815 | BA71 |
| FR682468 | Georgia_2007 |
| LR899131 | Ken_rie1 |
| LS478113 | Estonia2014 |
| MN318203 | LIV_5_40 |
| MN394630 | SPEC_57 |
| MN630494 | Zaire |
| MN641877 | RSA_2_2004 |
| MT956648 | Uvira_B53 |
| MW856067 | BUR_18_Rutana |
| MW856068 | MAL_19_Karonga |
| MZ202520 | K49 |
| OM249788 | KK262 |
| ON400500 | YNFN202103 |
| ON409981 | TAN_08_Mazimbu |
| ON409982 | TAN_17_Mbagala |
| OP672342 | Nigeria-RV502 |
| OQ504954 | Henan/123014/2022 |
| Isolate (Biotype) | Reference Genome and Reference | Total Number of Illumina Paired-Reads (% Compared to Post Trim) | Average Coverage | |||
|---|---|---|---|---|---|---|
| Post-Trim | Trimmed, Mapped to Reference, and Extracted | Trimmed, Filtered, Mapped to Reference, and Extracted | Trimmed, Mapped to Reference | Trimmed, Filtered, and Mapped to Reference | ||
| ASFV Georgia 2007/1 (2) | FR682468, [1,18] | 6,708,139 | 173,922 (2.59%) | 173,886 (2.59%) | 158 | 158 |
| RSA/2/2008 a (3) | MN336500, [38] | 4,575,230 | 3,256,629 (71.18%) | 3,212,202 (70.21%) | 3183 | 3140 |
| Cameroon/2023/SBP30 (1) | PP592890, [39] | 1,231,886 | 50,526 (4.10%) | 49796 (4.04%) | 91 | 89 |
| Malawi Lil 80 (6) | AY261361 | 75,762,615.00 | 2,631,494 (3.47%) | 2,586,133 (3.41%) | 2070 | 2034 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinard, E.; Dinhobl, M.; Erdelyan, C.N.G.; O’Dwyer, J.; Fenster, J.; Birtley, H.; Tesler, N.; Calvelage, S.; Leijon, M.; Steinaa, L.; et al. A Standardized Pipeline for Assembly and Annotation of African Swine Fever Virus Genome. Viruses 2024, 16, 1293. https://doi.org/10.3390/v16081293
Spinard E, Dinhobl M, Erdelyan CNG, O’Dwyer J, Fenster J, Birtley H, Tesler N, Calvelage S, Leijon M, Steinaa L, et al. A Standardized Pipeline for Assembly and Annotation of African Swine Fever Virus Genome. Viruses. 2024; 16(8):1293. https://doi.org/10.3390/v16081293
Chicago/Turabian StyleSpinard, Edward, Mark Dinhobl, Cassidy N. G. Erdelyan, James O’Dwyer, Jacob Fenster, Hillary Birtley, Nicolas Tesler, Sten Calvelage, Mikael Leijon, Lucilla Steinaa, and et al. 2024. "A Standardized Pipeline for Assembly and Annotation of African Swine Fever Virus Genome" Viruses 16, no. 8: 1293. https://doi.org/10.3390/v16081293
APA StyleSpinard, E., Dinhobl, M., Erdelyan, C. N. G., O’Dwyer, J., Fenster, J., Birtley, H., Tesler, N., Calvelage, S., Leijon, M., Steinaa, L., O’Donnell, V., Blome, S., Bastos, A., Ramirez-Medina, E., Lacasta, A., Ståhl, K., Qiu, H., Nilubol, D., Tennakoon, C., ... Gladue, D. P. (2024). A Standardized Pipeline for Assembly and Annotation of African Swine Fever Virus Genome. Viruses, 16(8), 1293. https://doi.org/10.3390/v16081293