
Discovery and Genomic Analysis of Three Novel Viruses in the Order Mononegavirales in Leafhoppers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and RNA Extraction
2.2. Bioinformatic Analysis
2.3. Obtaining the Full-Length Virus Genomes through RACE and Validation of Virus Genome
2.4. Sequence and Phylogenetic Analyses
3. Results
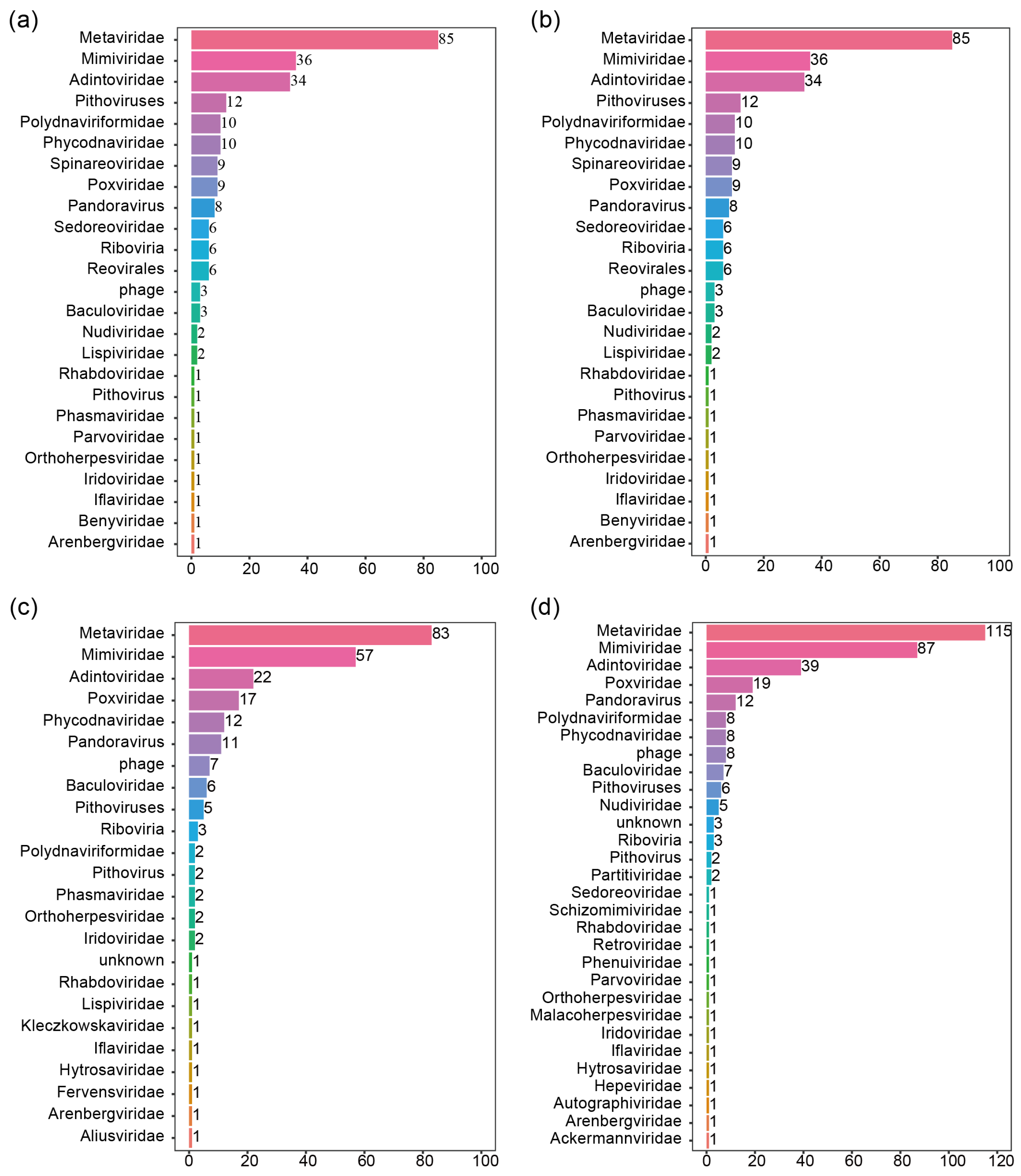
3.1. Transcriptome Assembly and Virus Discovery after Sample Collection
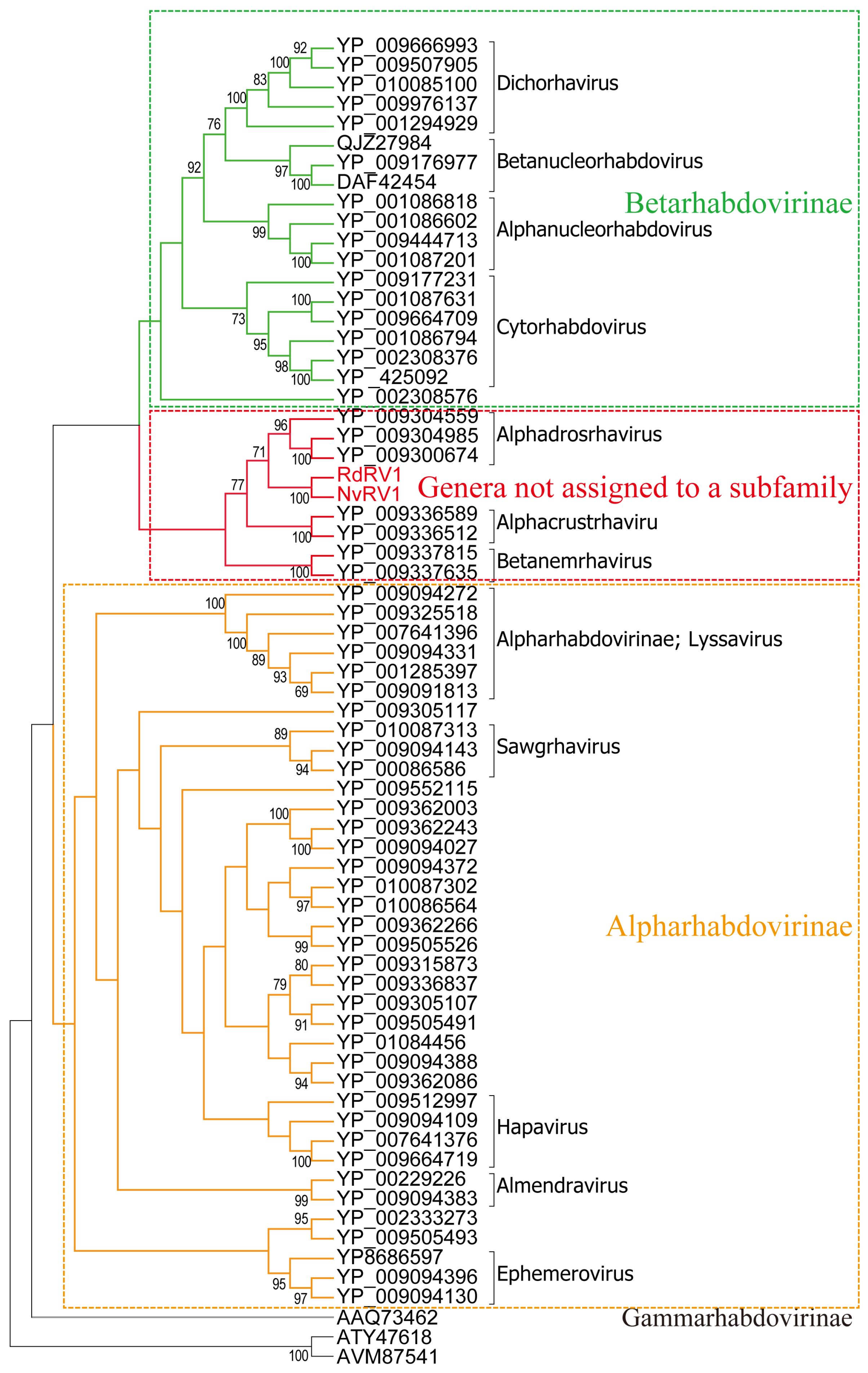
3.2. Characterization of Two Novel Viruses of the Family Rhabdoviridae
3.3. Characterization of a Novel Member of the Family Lispiviridae
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, R.R.; Montoya, V.; Gardy, J.L.; Patrick, D.M.; Tang, P. Metagenomics for pathogen detection in public health. Genome Med. 2013, 5, 81. [Google Scholar] [CrossRef]
- Simon, H.Y.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking metagenomics tools for taxonomic classification. Cell 2019, 178, 779–794. [Google Scholar]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Lauber, C. Bioinformatics of virus taxonomy: Foundations and tools for developing sequence-based hierarchical classification. Curr. Opin. Virol. 2022, 52, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Fujita, D.; Kohli, A.; Horgan, F.G. Rice resistance to planthoppers and leafhoppers. CRC Crit. Rev. Plant Sci. 2013, 32, 162–191. [Google Scholar] [CrossRef]
- Hogenhout, S.A.; Ammar, E.-D.; Whitfield, A.E.; Redinbaugh, M.G. Insect vector interactions with persistently transmitted viruses. Annu. Rev. Phytopathol. 2008, 46, 327–359. [Google Scholar] [CrossRef]
- Todd, J.C.; Ammar, E.-D.; Redinbaugh, M.G.; Hoy, C.; Hogenhout, S.A. Plant host range and leafhopper transmission of maize fine streak virus. Phytopathology 2010, 100, 1138–1145. [Google Scholar] [CrossRef]
- Ane, N.U.; Hussain, M. Diversity of insect pests in major rice growing areas of the world. J. Entomol. Zool. Stud. 2016, 4, 36–41. [Google Scholar]
- Wei, T.; Li, Y. Rice reoviruses in insect vectors. Annu. Rev. Phytopathol. 2016, 54, 99–120. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, C.; Li, M.; Wu, W.; Zhou, G.; Wei, T. Adverse effects of rice gall dwarf virus upon its insect vector Recilia dorsalis (Hemiptera: Cicadellidae). Plant Dis. 2016, 100, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, T.; Chen, B.; Zhou, G. Transmission biology of rice stripe mosaic virus by an efficient insect vector Recilia dorsalis (Hemiptera: Cicadellidae). Front. Microbiol. 2017, 8, 2457. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Adkins, S.; Alkhovsky, S.V.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; Ballinger, M.J.; Bandte, M.; Beer, M. 2022 taxonomic update of phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch. Virol. 2022, 167, 2857–2906. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Nie, B.; Chen, J.; Lv, K.; Xiao, J.; Liu, R. Full genome sequence of a novel iflavirus from the leafhopper Recilia dorsalis. Arch. Virol. 2022, 167, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Liang, Q.; Zhang, R.; Cheng, Y.; Wang, X.; Wang, H.; Zhang, J.; Jia, D.; Du, Y.; Zheng, W. Arboviruses and symbiotic viruses cooperatively hijack insect sperm-specific proteins for paternal transmission. Nat. Commun. 2023, 14, 1289. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Kimura, I.; Tsuchizaki, T.; Saito, Y. Infection by rice gall dwarf virus of cultured monolayers of leafhopper cells. J. Gen. Virol. 1988, 69, 429–432. [Google Scholar] [CrossRef]
- Wei, J.; Jia, D.; Mao, Q.; Zhang, X.; Chen, Q.; Wu, W.; Chen, H.; Wei, T. Complex interactions between insect-borne rice viruses and their vectors. Curr. Opin. Virol. 2018, 33, 18–23. [Google Scholar] [CrossRef]
- Jia, D.; Luo, G.; Shi, W.; Liu, Y.; Liu, H.; Zhang, X.; Wei, T. Rice gall dwarf virus promotes the propagation and transmission of rice stripe mosaic virus by co-infected insect vectors. Front. Microbiol. 2022, 13, 834712. [Google Scholar] [CrossRef]
- Ye, Z.-X.; Wang, S.-M.; Lu, G.; Chen, J.-P.; Zhang, C.-X.; Xu, L.-Y.; Li, J.-M. Complete genome sequence of a novel arlivirus from a yellow spotted stink bug (Erthesina fullo (Thunberg, 1783)). Arch. Virol. 2022, 167, 1205–1209. [Google Scholar] [CrossRef]
- Ammar, E.-D.; Tsai, C.-W.; Whitfield, A.E.; Redinbaugh, M.G.; Hogenhout, S.A. Cellular and molecular aspects of rhabdovirus interactions with insect and plant hosts. Annu. Rev. Entomol. 2009, 54, 447–468. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Kondo, H.; Goodin, M.M.; Kurath, G.; Vasilakis, N. The family Rhabdoviridae: Mono-and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 2017, 227, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Blasdell, K.R.; Calisher, C.H.; Dietzgen, R.G.; Kondo, H.; Kurath, G.; Longdon, B.; Stone, D.M.; Tesh, R.B.; Tordo, N. ICTV virus taxonomy profile: Rhabdoviridae. J. Gen. Virol. 2018, 99, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, A.E.; Huot, O.B.; Martin, K.M.; Kondo, H.; Dietzgen, R.G. Plant rhabdoviruses—Their origins and vector interactions. Curr. Opin. Virol. 2018, 33, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Vasilakis, N.; Gubler, D.J. Arboviruses: Molecular Biology, Evolution and Control; Caister Academic Press: Poole, UK, 2016. [Google Scholar]
- Maes, P.; Amarasinghe, G.K.; Ayllón, M.A.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A. Taxonomy of the order Mononegavirales: Second update 2018. Arch. Virol. 2019, 164, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Wang, F.; Ye, G.; Paraskevopoulou, S. ICTV virus taxonomy profile: Lispiviridae 2023. J. Gen. Virol. 2023, 104, 001869. [Google Scholar] [CrossRef] [PubMed]
- Käfer, S.; Paraskevopoulou, S.; Zirkel, F.; Wieseke, N.; Donath, A.; Petersen, M.; Jones, T.C.; Liu, S.; Zhou, X.; Middendorf, M. Re-assessing the diversity of negative strand RNA viruses in insects. PLoS Pathog. 2019, 15, e1008224. [Google Scholar] [CrossRef] [PubMed]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Jia, W.; Wang, F.; Xiao, S.; Yang, Y.; Chen, L.; Li, J.; Bao, Y.; Song, Q.; Ye, G. Identification and characterization of a novel rhabdovirus in green rice leafhopper, Nephotettix cincticeps. Virus Res. 2021, 296, 198281. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, V. Crop immunity against viruses: Outcomes and future challenges. Front. Plant Sci. 2014, 5, 118474. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.L.; Pauszek, S.J.; Bunch, T.A.; Schumann, K.R. Full-length genome analysis of natural isolates of vesicular stomatitis virus (Indiana 1 serotype) from North, Central and South America. J. Gen. Virol. 2002, 83, 2475–2483. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, J.; Nie, B.; Chen, M.-E.; Ge, D.; Liu, R. Discovery and Genomic Analysis of Three Novel Viruses in the Order Mononegavirales in Leafhoppers. Viruses 2024, 16, 1321. https://doi.org/10.3390/v16081321
Xiao J, Nie B, Chen M-E, Ge D, Liu R. Discovery and Genomic Analysis of Three Novel Viruses in the Order Mononegavirales in Leafhoppers. Viruses. 2024; 16(8):1321. https://doi.org/10.3390/v16081321
Chicago/Turabian StyleXiao, Jiajing, Binghua Nie, Meng-En Chen, Danfeng Ge, and Renyi Liu. 2024. "Discovery and Genomic Analysis of Three Novel Viruses in the Order Mononegavirales in Leafhoppers" Viruses 16, no. 8: 1321. https://doi.org/10.3390/v16081321
APA StyleXiao, J., Nie, B., Chen, M.-E., Ge, D., & Liu, R. (2024). Discovery and Genomic Analysis of Three Novel Viruses in the Order Mononegavirales in Leafhoppers. Viruses, 16(8), 1321. https://doi.org/10.3390/v16081321