
AP3B1 Has Type I Interferon-Independent Antiviral Function against SARS-CoV-2

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Declaration for SARS-CoV
2.3. siRNA Treatment
2.4. Plasmids and Transfection
2.5. Immunofluorescence Assay and Quantification
2.6. Interferon Treatment
2.7. Western Blot Analysis
2.8. qRT-PCR Primers
2.9. Statistical Analysis
3. Results
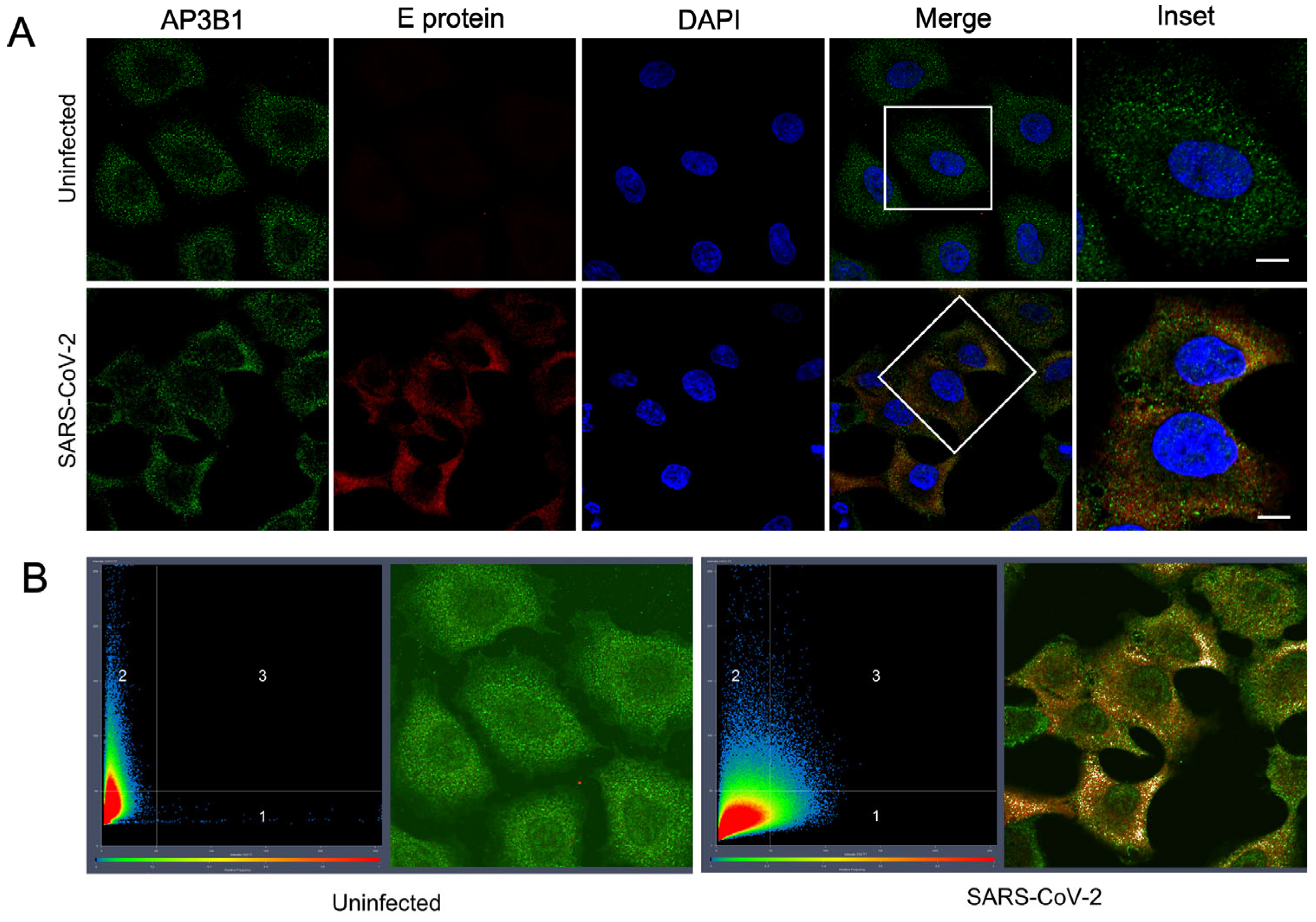
3.1. The Host Protein AP3B1 Interacts with SARS-CoV-2 Envelope (E) Protein
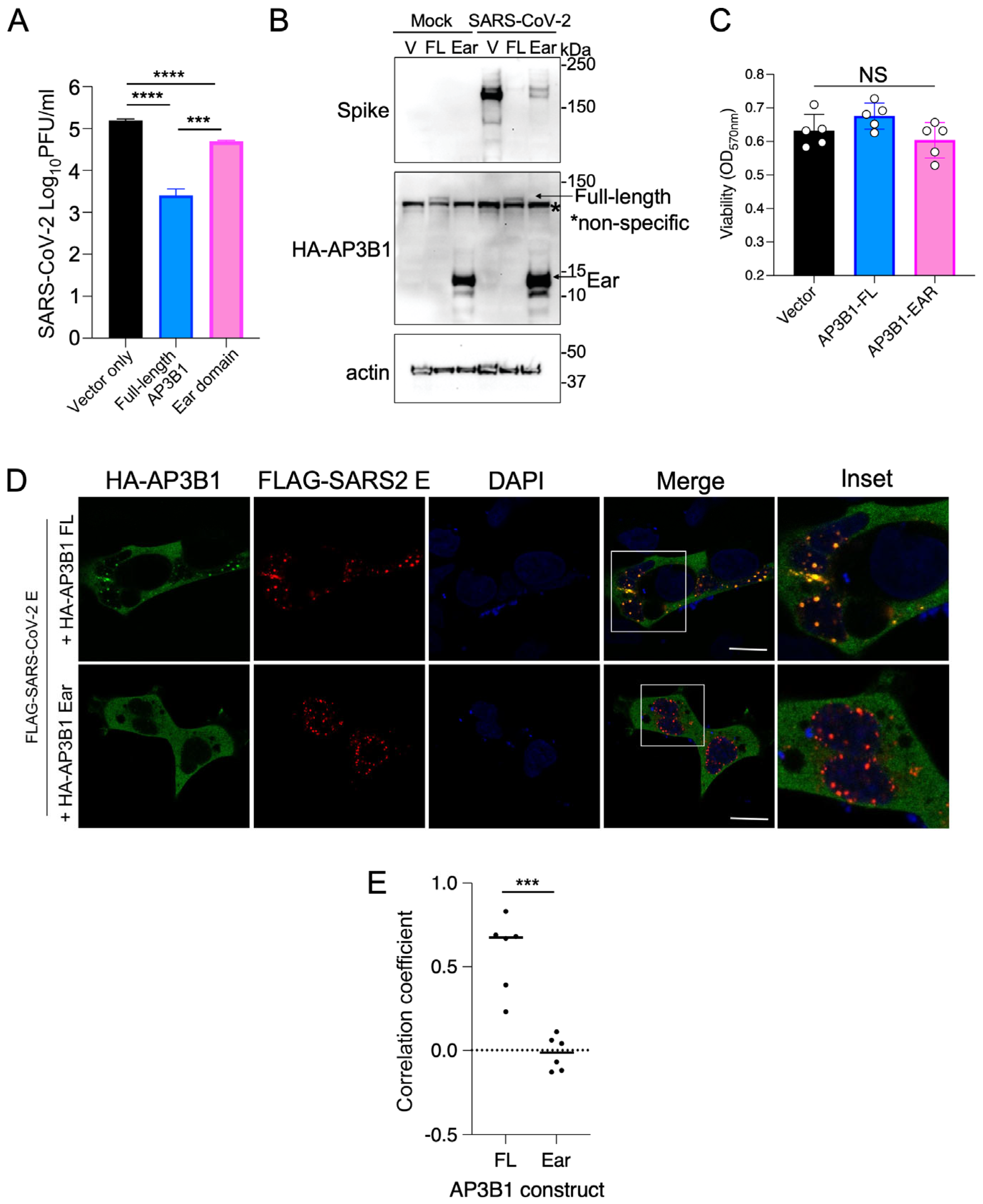
3.2. Overexpression of AP3B1 Inhibits SARS-CoV-2 Replication
3.3. Endogenous AP3B1 Inhibits Replication of SARS-CoV-2 but Not SARS-CoV
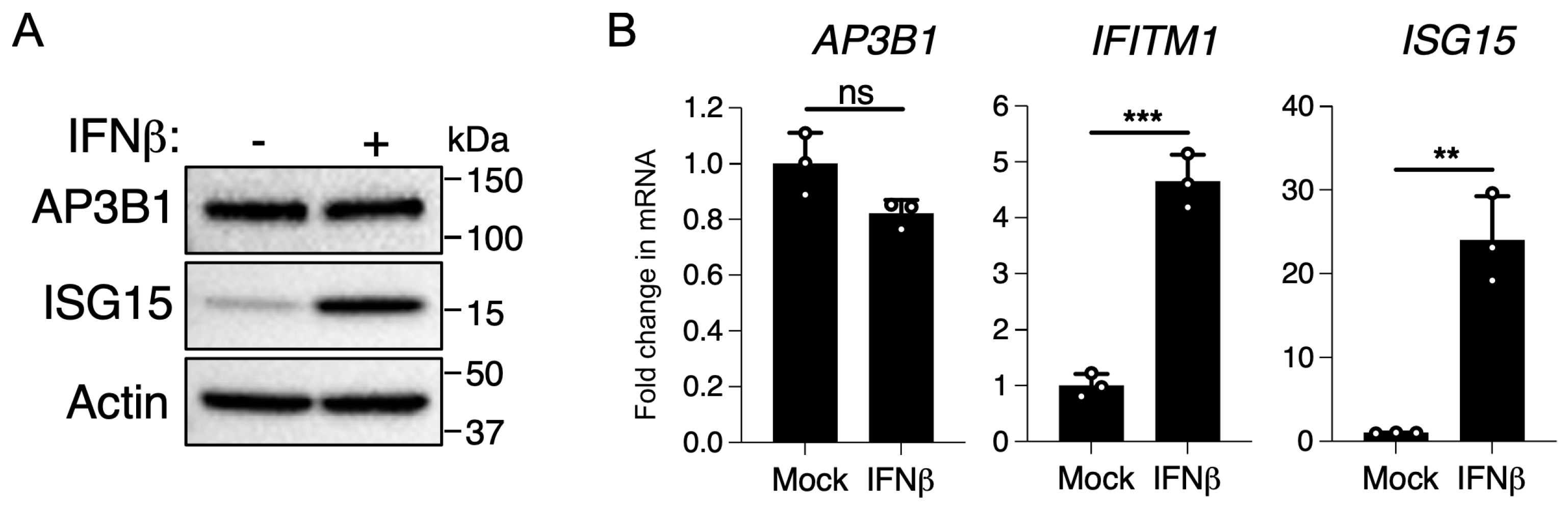
3.4. AP3B1 Is Not an Interferon-Stimulated Gene (ISG)
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.H.; Sanchez-Rivera, F.J.; Schneider, W.M.; Luna, J.M.; Soto-Feliciano, Y.M.; Ashbrook, A.W.; Le Pen, J.; Leal, A.A.; Ricardo-Lax, I.; Michailidis, E.; et al. Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe 2021, 29, 267–280.e265. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e1514. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Guo, X. Adaptor protein complexes and intracellular transport. Biosci. Rep. 2014, 34, e00123. [Google Scholar] [CrossRef]
- Sanger, A.; Hirst, J.; Davies, A.K.; Robinson, M.S. Adaptor protein complexes and disease at a glance. J. Cell Sci. 2019, 132, jcs222992. [Google Scholar] [CrossRef]
- Banushi, B.; Simpson, F. Overlapping Machinery in Lysosome-Related Organelle Trafficking: A Lesson from Rare Multisystem Disorders. Cells 2022, 11, 3702. [Google Scholar] [CrossRef]
- Karampini, E.; Schillemans, M.; Hofman, M.; van Alphen, F.; de Boer, M.; Kuijpers, T.W.; van den Biggelaar, M.; Voorberg, J.; Bierings, R. Defective AP-3-dependent VAMP8 trafficking impairs Weibel-Palade body exocytosis in Hermansky-Pudlak Syndrome type 2 blood outgrowth endothelial cells. Haematologica 2019, 104, 2091–2099. [Google Scholar] [CrossRef]
- Gao, L.; Zhu, L.; Huang, L.; Zhou, J. Synergistic defects of UNC13D and AP3B1 leading to adult hemophagocytic lymphohistiocytosis. Int. J. Hematol. 2015, 102, 488–492. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; AlRasheed, M.R.; et al. Pathophysiology of SARS-CoV-2: The Mount Sinai COVID-19 autopsy experience. Mod. Pathol. 2021, 34, 1456–1467. [Google Scholar] [CrossRef]
- Strazic Geljic, I.; Kucan Brlic, P.; Musak, L.; Karner, D.; Ambriovic-Ristov, A.; Jonjic, S.; Schu, P.; Rovis, T.L. Viral Interactions with Adaptor-Protein Complexes: A Ubiquitous Trait among Viral Species. Int. J. Mol. Sci. 2021, 22, 5274. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Burton, A.; Ruiz-Mateos, E.; Marsh, M.; Saiardi, A. Inositol pyrophosphate mediated pyrophosphorylation of AP3B1 regulates HIV-1 Gag release. Proc. Natl. Acad. Sci. USA 2009, 106, 21161–21166. [Google Scholar] [CrossRef]
- Sun, W.; McCrory, T.S.; Khaw, W.Y.; Petzing, S.; Myers, T.; Schmitt, A.P. Matrix proteins of Nipah and Hendra viruses interact with beta subunits of AP-3 complexes. J. Virol. 2014, 88, 13099–13110. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, T.; Schu, P.; Medigeshi, G.R. Adaptor protein complexes-1 and 3 are involved at distinct stages of flavivirus life-cycle. Sci. Rep. 2013, 3, 1813. [Google Scholar] [CrossRef] [PubMed]
- Scroggs, S.L.P.; Offerdahl, D.K.; Flather, D.P.; Morris, C.N.; Kendall, B.L.; Broeckel, R.M.; Beare, P.A.; Bloom, M.E. Fluoroquinolone Antibiotics Exhibit Low Antiviral Activity against SARS-CoV-2 and MERS-CoV. Viruses 2020, 13, 8. [Google Scholar] [CrossRef]
- Miura, K.; Suzuki, Y.; Ishida, K.; Arakawa, M.; Wu, H.; Fujioka, Y.; Emi, A.; Maeda, K.; Hamajima, R.; Nakano, T.; et al. Distinct motifs in the E protein are required for SARS-CoV-2 virus particle formation and lysosomal deacidification in host cells. J. Virol. 2023, 97, e0042623. [Google Scholar] [CrossRef]
- Chen, D.; Zheng, Q.; Sun, L.; Ji, M.; Li, Y.; Deng, H.; Zhang, H. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell 2021, 56, 3250–3263.e3255. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.J.; Mears, H.V.; Broncel, M.; Snijders, A.P.; Bauer, D.L.V.; Carlton, J.G. ER-export and ARFRP1/AP-1-dependent delivery of SARS-CoV-2 Envelope to lysosomes controls late stages of viral replication. Sci. Adv. 2024, 10, eadl5012. [Google Scholar] [CrossRef]
- Liu, L.; Sutton, J.; Woodruff, E.; Villalta, F.; Spearman, P.; Dong, X. Defective HIV-1 particle assembly in AP-3-deficient cells derived from patients with Hermansky-Pudlak syndrome type 2. J. Virol. 2012, 86, 11242–11253. [Google Scholar] [CrossRef]
- Dong, X.; Li, H.; Derdowski, A.; Ding, L.; Burnett, A.; Chen, X.; Peters, T.R.; Dermody, T.S.; Woodruff, E.; Wang, J.J.; et al. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell 2005, 120, 663–674. [Google Scholar] [CrossRef]
- Shevchenko, A.K.; Zhernakova, D.V.; Malov, S.V.; Komissarov, A.; Kolchanova, S.M.; Tamazian, G.; Antonik, A.; Cherkasov, N.; Kliver, S.; Turenko, A.; et al. Genome-wide association study reveals genetic variants associated with HIV-1C infection in a Botswana study population. Proc. Natl. Acad. Sci. USA 2021, 118, e2107830118. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.; Wang, P.; Meng, S.; Jetter, C.S.; Sucre, J.M.S.; Benjamin, J.T.; Gokey, J.J.; Hanby, H.A.; Jaume, A.; Goetzl, L.; et al. AP-3-dependent targeting of flippase ATP8A1 to lamellar bodies suppresses activation of YAP in alveolar epithelial type 2 cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2025208118. [Google Scholar] [CrossRef]
- Fortmann, S.D.; Patton, M.J.; Frey, B.F.; Tipper, J.L.; Reddy, S.B.; Vieira, C.P.; Hanumanthu, V.S.; Sterrett, S.; Floyd, J.L.; Prasad, R.; et al. Circulating SARS-CoV-2+ megakaryocytes are associated with severe viral infection in COVID-19. Blood Adv. 2023, 7, 4200–4214. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.W.; Ilyas, I.; Weng, J.P. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Liu, D.; Liu, W.; Wang, G.; Chen, L.; Cao, Y.; Wei, J.; Xiao, M.; Liu, X.; Huang, G.; et al. Germline variants in UNC13D and AP3B1 are enriched in COVID-19 patients experiencing severe cytokine storms. Eur. J. Hum. Genet. 2021, 29, 1312–1315. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramanian, G.; Hage, A.; Feldmann, F.; Chiramel, A.I.; McNally, K.L.; Sturdevant, G.L.; Beare, P.A.; Best, S.M. AP3B1 Has Type I Interferon-Independent Antiviral Function against SARS-CoV-2. Viruses 2024, 16, 1377. https://doi.org/10.3390/v16091377
Subramanian G, Hage A, Feldmann F, Chiramel AI, McNally KL, Sturdevant GL, Beare PA, Best SM. AP3B1 Has Type I Interferon-Independent Antiviral Function against SARS-CoV-2. Viruses. 2024; 16(9):1377. https://doi.org/10.3390/v16091377
Chicago/Turabian StyleSubramanian, Gayatri, Adam Hage, Friederike Feldmann, Abhilash I. Chiramel, Kristin L. McNally, Gail L. Sturdevant, Paul A. Beare, and Sonja M. Best. 2024. "AP3B1 Has Type I Interferon-Independent Antiviral Function against SARS-CoV-2" Viruses 16, no. 9: 1377. https://doi.org/10.3390/v16091377