
Combining RNA Interference and RIG-I Activation to Inhibit Hepatitis E Virus Replication

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
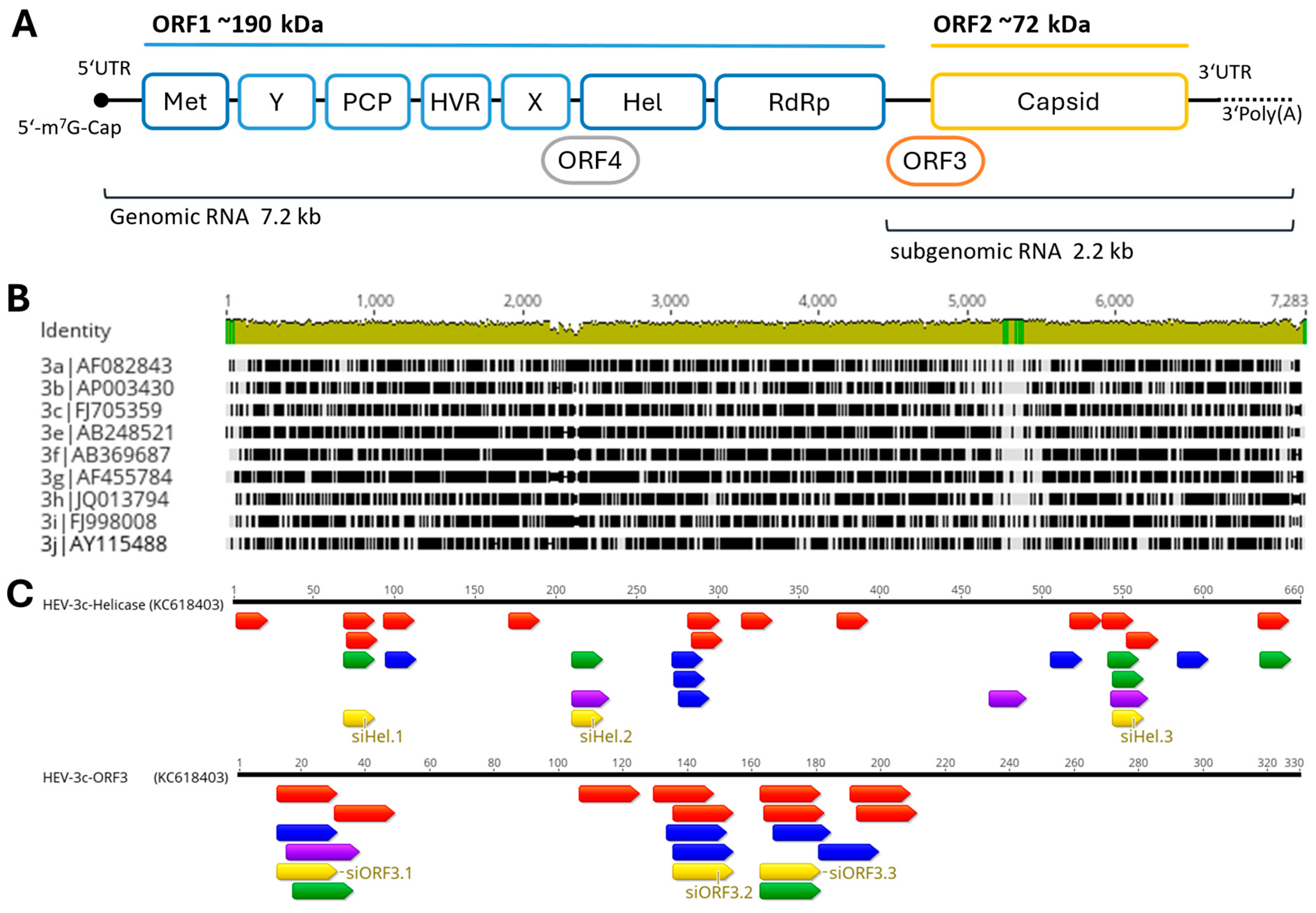
2.1. Design of siRNAs and Plasmid Construction
2.2. Cell Culture
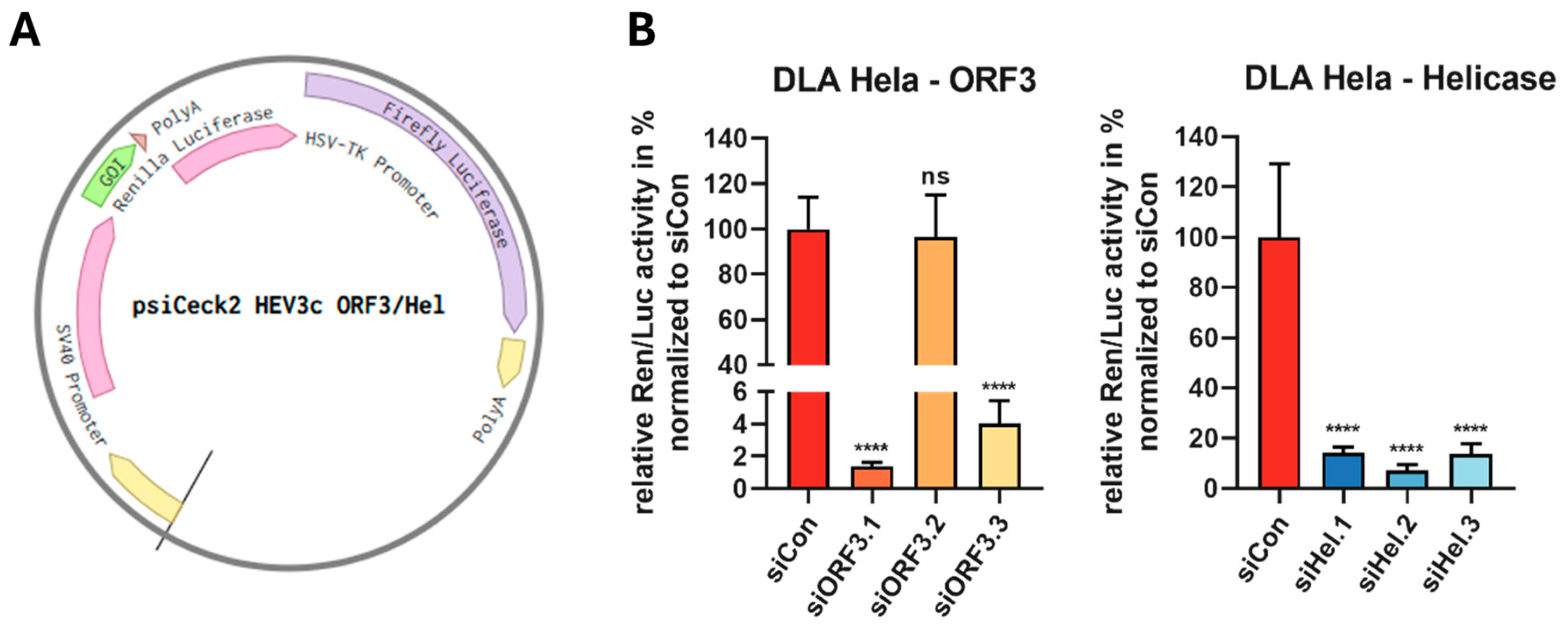
2.3. Dual Luciferase Reporter Assay
2.4. Synthesis of 5′-Triphosphorylated siRNA
2.5. Transfection of siRNA and 3p-siRNA
2.6. HEV-3c Production Using A549/Pers-HEV Cells
2.7. siRNA Antiviral Replication Assay
2.8. Reverse Transcription Quantitative PCR of HEV Samples
2.9. Immunoblotting
2.10. Statistical Analysis
3. Results
3.1. Design of HEV-3c-ORF3 and HEV-3c-Helicase Sequence Targeting siRNAs
3.2. RNAi-Mediated Targeting of the ORF3 and Helicase of HEV in Reporter Assays
3.3. Inhibition of HEV-3c Replication Using ORF3- and Helicase-Targeting siRNAs in Persistently Infected A549/Pers-HEV Cells
3.4. Inhibition of HEV-3c Replication with Antiorf3-siRNAs Followed by HEV Infection
3.5. RIG-I Activation and Inhibition of HEV Replication with 5′ Triphosphate siRNA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO-Hepatitis, E. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-e (accessed on 9 July 2024).
- Nelson, K.E.; Labrique, A.B.; Kmush, B.L. Epidemiology of Genotype 1 and 2 Hepatitis E Virus Infections. Cold Spring Harb. Perspect. Med. 2019, 9, a031732. [Google Scholar] [CrossRef]
- Doceul, V.; Bagdassarian, E.; Demange, A.; Pavio, N. Zoonotic Hepatitis E Virus: Classification, Animal Reservoirs and Transmission Routes. Viruses 2016, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Van Der Poel, W.H. Food and Environmental Routes of Hepatitis e Virus Transmission. Curr. Opin. Virol. 2014, 4, 91–96. [Google Scholar] [CrossRef]
- Li, P.; Liu, J.; Li, Y.; Su, J.; Ma, Z.; Bramer, W.M.; Cao, W.; de Man, R.A.; Peppelenbosch, M.P.; Pan, Q. The Global Epidemiology of Hepatitis E Virus Infection: A Systematic Review and Meta-analysis. Liver Int. 2020, 40, 1516–1528. [Google Scholar] [CrossRef]
- Lhomme, S.; Marion, O.; Abravanel, F.; Izopet, J.; Kamar, N. Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections. J. Clin. Med. 2020, 9, 331. [Google Scholar] [CrossRef]
- Kamar, N.; Selves, J.; Mansuy, J.-M.; Ouezzani, L.; Péron, J.-M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E Virus and Chronic Hepatitis in Organ-Transplant Recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Tedder, R.S.; Ijaz, S. Persistent Carriage of Hepatitis E Virus in Patients with HIV Infection. N. Engl. J. Med. 2009, 361, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Haboubi, H.N.Y.; Diyar, R.; Benton, A.; Ch’ng, C.L. A Case of Acute Hepatitis E Infection in a Patient with Non-Hodgkin Lymphoma Treated Successfully with Ribavirin. Case Rep. Gastrointest. Med. 2017, 2017, 8941218. [Google Scholar] [CrossRef]
- Kamar, N.; Rostaing, L.; Legrand-Abravanel, F.; Izopet, J. How Should Hepatitis E Virus Infection Be Defined in Organ-Transplant Recipients? Am. J. Transplant. 2013, 13, 1935–1936. [Google Scholar] [CrossRef]
- Kamar, N.; Izopet, J.; Dalton, H.R. Chronic Hepatitis E Virus Infection and Treatment. J. Clin. Exp. Hepatol. 2013, 3, 134–140. [Google Scholar] [CrossRef]
- Chaudhry, S.A.; Verma, N.; Koren, G. Hepatitis E Infection during Pregnancy. Can. Fam. Physician 2015, 61, 607–608. [Google Scholar]
- Kenney, S.P.; Meng, X.J. Hepatitis E Virus Genome Structure and Replication Strategy. Cold Spring Harb. Perspect. Med. 2019, 9, a031724. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.A.; Burgess, W.H.; Emerson, S.U.; Leibowitz, R.S.; Sosnovtseva, S.A.; Tsarev, S.; Purcell, R.H. Structural Characterization of Recombinant Hepatitis E Virus ORF2 Proteins in Baculovirus-Infected Insect Cells. Protein Expr. Purif. 1998, 12, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Nagashima, S.; Tanaka, T.; Okamato, H. ORF3 Protein of Hepatitis E Virus Is Essential for Virion Release from Infected Cells. J. Gen. Virol. 2009, 90, 1880–1891. [Google Scholar] [CrossRef]
- Zafrullah, M.; Ozdener, M.H.; Panda, S.K.; Jameel, S. The ORF3 Protein of Hepatitis E Virus Is a Phosphoprotein That Associates with the Cytoskeleton. J. Virol. 1997, 71, 9045–9053. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Heller, B.; Capuccino, J.M.V.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E Virus ORF3 Is a Functional Ion Channel Required for Release of Infectious Particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef]
- Sari, G.; Zhu, J.; Ambardekar, C.; Yin, X.; Boonstra, A.; Feng, Z.; Vanwolleghem, T. The Viral ORF3 Protein Is Required for Hepatitis E Virus Apical Release and Efficient Growth in Polarized Hepatocytes and Humanized Mice. J. Virol. 2021, 95, 0058521. [Google Scholar] [CrossRef]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar; Nayak, B.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, 1005521. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Selves, J.; Garrouste, C.; Esposito, L.; Lavayssière, L.; Cointault, O.; Ribes, D.; Cardeau, I.; Nogier, M.B.; et al. Influence of Immunosuppressive Therapy on the Natural History of Genotype 3 Hepatitis-E Virus Infection after Organ Transplantation. Transplantation 2010, 89, 353–360. [Google Scholar] [CrossRef]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Esposito, L.; Cardeau-Desangles, I.; Mansuy, J.M.; Selves, J.; Peron, J.M.; Otal, P.; et al. Pegylated Interferon-α for Treating Chronic Hepatitis E Virus Infection after Liver Transplantation. Clin. Infect. Dis. 2010, 50, e30–e33. [Google Scholar] [CrossRef]
- Alric, L.; Bonnet, D.; Laurent, G.; Kamar, N.; Izopet, J. Chronic Hepatitis E Virus Infection: Successful Virologic Response to Pegylated Interferon-Alpha Therapy. Ann. Intern. Med. 2010, 153, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Tripon, S.; Bismuth, M.; Hillaire, S.; Dumortier, J.; Radenne, S.; Coilly, A.; Garrigue, V.; D’Alteroche, L.; et al. Ribavirin for Chronic Hepatitis E Virus Infection in Transplant Recipients. N. Engl. J. Med. 2014, 370, 1111–1120. [Google Scholar] [CrossRef]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Lhomme, S.; Esposito, L.; Basse, G.; Cointault, O.; Ribes, D.; Nogier, M.B.; et al. Ribavirin Therapy Inhibits Viral Replication on Patients with Chronic Hepatitis E Virus Infection. Gastroenterology 2010, 139, 1612–1618. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Behrendt, P.; Hofmann, J.; Pageaux, G.P.; Barbet, C.; Moal, V.; Couzi, L.; Horvatits, T.; Man, R.A.; et al. Ribavirin for Hepatitis E Virus Infection After Organ Transplantation: A Large European Retrospective Multicenter Study. Clin. Infect. Dis. 2020, 71, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Gisa, A.; Radonic, A.; Nitsche, A.; Behrendt, P.; Suneetha, P.V.; Pischke, S.; Bremer, B.; Brown, R.J.P.; Manns, M.P.; et al. In Vivo Evidence for Ribavirin-Induced Mutagenesis of the Hepatitis E Virus Genome. Gut 2016, 65, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Ramière, C.; Dallmeier, K.; Piorkowski, G.; Trabaud, M.-A.; Lebossé, F.; Scholtès, C.; Roche, M.; Legras-Lachuer, C.; Lamballerie, X.; et al. Hepatitis E Virus Mutations Associated with Ribavirin Treatment Failure Result in Altered Viral Fitness and Ribavirin Sensitivity. J. Hepatol. 2016, 65, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A Mutation in the Hepatitis E Virus RNA Polymerase Promotes Its Replication and Associates with Ribavirin Treatment Failure in Organ Transplant Recipients. Gastroenterology 2014, 147, 1008–1011. [Google Scholar] [CrossRef]
- Gorris, M.; van der Lecq, B.M.; van Erpecum, K.J.; de Bruijne, J. Treatment for Chronic Hepatitis E Virus Infection: A Systematic Review and Meta-Analysis. J. Viral Hepat. 2021, 28, 454–463. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic siRNA: State of the Art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Kurreck, J. RNA Interference: From Basic Research to Therapeutic Applications. Angew. Chem. Int. Ed Engl. 2009, 48, 1378–1398. [Google Scholar] [CrossRef]
- Huang, F.; Hua, X.; Yang, S.; Yuan, C.; Zhang, W. Effective Inhibition of Hepatitis E Virus Replication in A549 Cells and Piglets by RNA Interference (RNAi) Targeting RNA-Dependent RNA Polymerase. Antivir. Res. 2009, 83, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Panda, S.K.; Durgapal, H.; Acharya, S.K.; Rehman, S.; Kar, U.K. Inhibition of Hepatitis E Virus Replication Using Short Hairpin RNA (shRNA). Antivir. Res. 2010, 85, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhou, J.; Yang, Z.; Cui, L.; Zhang, W.; Yuan, C.; Yang, S.; Zhu, J.; Hua, X. RNA Interference Inhibits Hepatitis E Virus mRNA Accumulation and Protein Synthesis in Vitro. Vet. Microbiol. 2010, 142, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lei, M.; Jiao, H.; Du, L.; Cheng, Y.; Zhang, D.; Hao, Y.; Man, C.; Wang, F. RNA Interference Induces Effective Inhibition of mRNA Accumulation and Protein Expression of SHEV ORF3 Gene in Vitro. Curr. Microbiol. 2011, 62, 1355–1362. [Google Scholar] [CrossRef]
- Zhang, C.; Freistaedter, A.; Schmelas, C.; Gunkel, M.; Dao Thi, V.L.; Grimm, D. An RNA Interference/Adeno-Associated Virus Vector–Based Combinatorial Gene Therapy Approach Against Hepatitis E Virus. Hepatol. Commun. 2022, 6, 878. [Google Scholar] [CrossRef]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human Immunodeficiency Virus Type 1 Escape from RNA Interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef]
- Wilson, J.A.; Richardson, C.D. Hepatitis C Virus Replicons Escape RNA Interference Induced by a Short Interfering RNA Directed against the NS5b Coding Region. J. Virol. 2005, 79, 7050–7058. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA Helicase RIG-I Has an Essential Function in Double-Stranded RNA-Induced Innate Antiviral Responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′ Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis E Sousa, C. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5′-Phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.-J. Hepatitis E Virus Inhibits Type I Interferon Induction by ORF1 Products. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [CrossRef]
- Bagdassarian, E.; Doceul, V.; Pellerin, M.; Demange, A.; Meyer, L.; Jouvenet, N.; Pavio, N. The Amino-Terminal Region of Hepatitis E Virus ORF1 Containing a Methyltransferase (Met) and a Papain-Like Cysteine Protease (PCP) Domain Counteracts Type I Interferon Response. Viruses 2018, 10, 726. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, Y.; Nan, Y.; Ma, Z.; Yang, L.; Zhang, Y.-J. The Capsid Protein of Hepatitis E Virus Inhibits Interferon Induction via Its N-Terminal Arginine-Rich Motif. Viruses 2019, 11, 1050. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, W.; Li, Y.; Zhou, X.; Yin, Y.; Wang, Y.; de Man, R.A.; van der Laan, L.J.W.; Huang, F.; Kamar, N.; et al. RIG-I Is a Key Antiviral Interferon-Stimulated Gene against Hepatitis E Virus Regardless of Interferon Production. Hepatology 2017, 65, 1823–1839. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Liu, Q.; Berube, N.; Detmer, S.; Zhou, Y. 5’-Triphosphate-Short Interfering RNA: Potent Inhibition of Influenza A Virus Infection by Gene Silencing and RIG-I Activation. J. Virol. 2012, 86, 10359–10369. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, Y.; Yan, F.; Tu, J.; Yang, X.; Xing, Y.; Chen, Z. 5’-Triphosphate-siRNA Activates RIG-I-Dependent Type I Interferon Production and Enhances Inhibition of Hepatitis B Virus Replication in HepG2.2.15 Cells. Eur. J. Pharmacol. 2013, 721, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Zhang, C.; Zhang, J.; Tian, Z. Reversal of Hepatitis B Virus-Induced Immune Tolerance by an Immunostimulatory 3p-HBx-siRNAs in a Retinoic Acid Inducible Gene I-Dependent Manner. Hepatology 2011, 54, 1179–1189. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.J.; et al. Proposed Reference Sequences for Hepatitis E Virus Subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef]
- Lu, Z.J.; Mathews, D.H. OligoWalk: An Online siRNA Design Tool Utilizing Hybridization Thermodynamics. Nucleic Acids Res. 2008, 36, W104–W108. [Google Scholar] [CrossRef]
- Naito, Y.; Yoshimura, J.; Morishita, S.; Ui-Tei, K. siDirect 2.0: Updated Software for Designing Functional siRNA with Reduced Seed-Dependent off-Target Effect. BMC Bioinform. 2009, 10, 392. [Google Scholar] [CrossRef]
- Kurreck, J. siRNA Efficiency: Structure or Sequence-That Is the Question. J. Biomed. Biotechnol. 2006, 2006, 83757. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, A.; Grössl, T.; Geisler, A.; Wang, X.; Pinkert, S.; Pozzuto, T.; Schwer, C.; Kurreck, J.; Weger, S.; Vetter, R.; et al. Inhibition of Adenovirus Infections by siRNA-Mediated Silencing of Early and Late Adenoviral Gene Functions. Antivir. Res. 2010, 88, 86–94. [Google Scholar] [CrossRef]
- Schemmerer, M.; Apelt, S.; Trojnar, E.; Ulrich, R.G.; Wenzel, J.J.; Johne, R. Enhanced Replication of Hepatitis E Virus Strain 47832c in an A549-Derived Subclonal Cell Line. Viruses 2016, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenröder, J.; Nickel, P.; Hofmann, J. An ORF1-Rearranged Hepatitis E Virus Derived from a Chronically Infected Patient Efficiently Replicates in Cell Culture. J. Viral Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef]
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Brüggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust Hepatitis E Virus Infection and Transcriptional Response in Human Hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731–1741. [Google Scholar] [CrossRef]
- Johne, R.; Trojnar, E.; Filter, M.; Hofmann, J. Thermal Stability of Hepatitis E Virus as Estimated by a Cell Culture Method. Appl. Environ. Microbiol. 2016, 82, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Jothikumar, N.; Cromeans, T.L.; Robertson, B.H.; Meng, X.J.; Hill, V.R. A Broadly Reactive One-Step Real-Time RT-PCR Assay for Rapid and Sensitive Detection of Hepatitis E Virus. J. Virol. Methods 2006, 131, 65–71. [Google Scholar] [CrossRef]
- Wang, B.; Harms, D.; Papp, C.P.; Niendorf, S.; Jacobsen, S.; Lütgehetmann, M.; Pischke, S.; Wedermeyer, H.; Hofmann, J.; Bock, C.-T. Comprehensive Molecular Approach for Characterization of Hepatitis E Virus Genotype 3 Variants. J. Clin. Microbiol. 2018, 56, e01686-17. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-Strand Cleavage Facilitates Assembly of siRNA into Ago2-Containing RNAi Enzyme Complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef]
- Zhou, Y.; Guo, M.; Wang, X.; Li, J.; Wang, Y.; Ye, L.; Dai, M.; Zhou, L.; Persidsky, Y.; Ho, W. TLR3 Activation Efficiency by High or Low Molecular Mass Poly I:C. Innate Immun. 2013, 19, 184–192. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, G.; Mahato, R.I. RNAi for Treating Hepatitis B Viral Infection. Pharm. Res. 2008, 25, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.B.; Brideau-Andersen, A.; Chisari, F.V. Interference of Hepatitis C Virus RNA Replication by Short Interfering RNAs. Proc. Natl. Acad. Sci. USA 2003, 100, 2014–2018. [Google Scholar] [CrossRef]
- Stein, E.A.; Pinkert, S.; Becher, P.M.; Geisler, A.; Zeichhardt, H.; Klopfleisch, R.; Poller, W.; Tschöpe, C.; Lassner, D.; Fechner, H.; et al. Combination of RNA Interference and Virus Receptor Trap Exerts Additive Antiviral Activity in Coxsackievirus B3-Induced Myocarditis in Mice. J. Infect. Dis. 2015, 211, 613–622. [Google Scholar] [CrossRef]
- Suzuki, H.; Saitoh, H.; Suzuki, T.; Takaku, H. Baculovirus-Mediated Bispecific Short-Hairpin Small-Interfering RNAs Have Remarkable Ability to Cope With Both Influenza Viruses A and B. Oligonucleotides 2009, 19, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Geisler, A.; Dieringer, B.; Elsner, L.; Klingel, K.; Klopfleisch, R.; Vornlocher, H.-P.; Kurreck, J.; Fechner, H. Lipid Nanoparticle-Encapsulated, Chemically Modified Anti-Adenoviral siRNAs Inhibit Hepatic Adenovirus Infection in Immunosuppressed Syrian Hamsters. Mol. Ther. Nucleic Acids 2023, 32, 923–936. [Google Scholar] [CrossRef]
- Saayman, S.; Barichievy, S.; Capovilla, A.; Morris, K.V.; Arbuthnot, P.; Weinberg, M.S. The Efficacy of Generating Three Independent Anti-HIV-1 siRNAs from a Single U6 RNA Pol III-Expressed Long Hairpin RNA. PLoS ONE 2008, 3, 2602. [Google Scholar] [CrossRef]
- Berkhout, B.; ter Brake, O. Towards a Durable RNAi Gene Therapy for HIV-AIDS. Expert Opin. Biol. Ther. 2009, 9, 161–170. [Google Scholar] [CrossRef]
- Liu, Y.P.; von Eije, K.J.; Schopman, N.C.T.; Westerink, J.-T.; ter Brake, O.; Haasnoot, J.; Berkhout, B. Combinatorial RNAi against HIV-1 Using Extended Short Hairpin RNAs. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Tolksdorf, B.; Nie, C.; Niemeyer, D.; Röhrs, V.; Berg, J.; Lauster, D.; Adler, J.M.; Haag, R.; Trimpert, J.; Kaufer, B.; et al. Inhibition of SARS-CoV-2 Replication by a Small Interfering RNA Targeting the Leader Sequence. Viruses 2021, 13, 2030. [Google Scholar] [CrossRef]
- Tolksdorf, B.; Heinze, J.; Niemeyer, D.; Röhrs, V.; Berg, J.; Drosten, C.; Kurreck, J. Development of a Highly Stable, Active Small Interfering RNA with Broad Activity against SARS-CoV Viruses. Antivir. Res. 2024, 226, 105879. [Google Scholar] [CrossRef]
- Ju, X.; Xiang, G.; Gong, M.; Yang, R.; Qin, J.; Li, Y.; Nan, Y.; Yang, Y.; Zhang, Q.C.; Ding, Q. Identification of Functional Cis-Acting RNA Elements in the Hepatitis E Virus Genome Required for Viral Replication. PLoS Pathog. 2020, 16, e1008488. [Google Scholar] [CrossRef] [PubMed]
- van Tong, H.; Hoan, N.X.; Wang, B.; Wedemeyer, H.; Bock, C.-T.; Velavan, T.P. Hepatitis E Virus Mutations: Functional and Clinical Relevance. EBioMedicine 2016, 11, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Davis, M.E. Insights into the Kinetics of siRNA-Mediated Gene Silencing from Live-Cell and Live-Animal Bioluminescent Imaging. Nucleic Acids Res. 2006, 34, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Nagai, C.; Hatakeyama, H.; Minakawa, N.; Harashima, H.; Matsuda, A. Intracellular Stability of 2′-OMe-4′-Thioribonucleoside Modified siRNA Leads to Long-Term RNAi Effect. Nucleic Acids Res. 2012, 40, 5787–5793. [Google Scholar] [CrossRef]
- Choi, M.; Hofmann, J.; Köhler, A.; Wang, B.; Bock, C.-T.; Schott, E.; Reinke, P.; Nickel, P. Prevalence and Clinical Correlates of Chronic Hepatitis E Infection in German Renal Transplant Recipients With Elevated Liver Enzymes. Transplant. Direct 2018, 4, e341. [Google Scholar] [CrossRef]
- Harms, D.; Choi, M.; Allers, K.; Wang, B.; Pietsch, H.; Papp, C.-P.; Hanisch, L.; Kurreck, J.; Hofmann, J.; Bock, C.-T. Specific Circulating microRNAs during Hepatitis E Infection Can Serve as Indicator for Chronic Hepatitis E. Sci. Rep. 2020, 10, 5337. [Google Scholar] [CrossRef]
- Rau, F.; Elsner, C.; Meister, T.L.; Gömer, A.; Kallies, R.; Dittmer, U.; Steinmann, E.; Todt, D. Monitoring of Hepatitis E Virus in Wastewater Can Identify Clinically Relevant Variants. Liver Int. 2024, 44, 637–643. [Google Scholar] [CrossRef]
- Schemmerer, M.; Wenzel, J.J.; Stark, K.; Faber, M. Molecular Epidemiology and Genotype-Specific Disease Severity of Hepatitis E Virus Infections in Germany, 2010–2019. Emerg. Microbes Infect. 2022, 11, 1754–1763. [Google Scholar] [CrossRef]
- Haldipur, B.; Bhukya, P.L.; Arankalle, V.; Lole, K. Positive Regulation of Hepatitis E Virus Replication by MicroRNA-122. J. Virol. 2018, 92, e01999-17. [Google Scholar] [CrossRef]
- Müller, C.; Obermann, W.; Schulte, F.W.; Lange-Grünweller, K.; Oestereich, L.; Elgner, F.; Glitscher, M.; Hildt, E.; Singh, K.; Wendel, H.-G.; et al. Comparison of Broad-Spectrum Antiviral Activities of the Synthetic Rocaglate CR-31-B (−) and the eIF4A-Inhibitor Silvestrol. Antivir. Res. 2020, 175, 104706. [Google Scholar] [CrossRef]
- Nasheri, N.; Doctor, T.; Chen, A.; Harlow, J.; Gill, A. Evaluation of High-Pressure Processing in Inactivation of the Hepatitis E Virus. Front. Microbiol. 2020, 11, 461. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Monini, M.; Ammendolia, M.G.; De Sabato, L.; Ostanello, F.; Vaccari, G.; Di Bartolo, I. In Vitro Replication of Swine Hepatitis E Virus (HEV): Production of Cell-Adapted Strains. Animals 2023, 13, 276. [Google Scholar] [CrossRef]
- Primadharsini, P.P.; Takahashi, M.; Nishizawa, T.; Sato, Y.; Nagashima, S.; Murata, K.; Okamoto, H. The Full-Genome Analysis and Generation of an Infectious cDNA Clone of a Genotype 6 Hepatitis E Virus Variant Obtained from a Japanese Wild Boar: In Vitro Cultivation in Human Cell Lines. Viruses 2024, 16, 842. [Google Scholar] [CrossRef]
- Fu, R.M.; Decker, C.C.; Dao Thi, V.L. Cell Culture Models for Hepatitis E Virus. Viruses 2019, 11, 608. [Google Scholar] [CrossRef] [PubMed]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell Culture Systems for the Study of Hepatitis E Virus. Antivir. Res. 2019, 163, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, M.; Hirchaud, E.; Blanchard, Y.; Pavio, N.; Doceul, V. Characterization of a Cell Culture System of Persistent Hepatitis E Virus Infection in the Human HepaRG Hepatic Cell Line. Viruses 2021, 13, 406. [Google Scholar] [CrossRef]
- Schmidt, K.; Berg, J.; Roehrs, V.; Kurreck, J.; Al-Zeer, M.A. 3D-Bioprinted HepaRG Cultures as a Model for Testing Long Term Aflatoxin B1 Toxicity in Vitro. Toxicol. Rep. 2020, 7, 1578–1587. [Google Scholar] [CrossRef]
- Merl, S.; Michaelis, C.; Jaschke, B.; Vorpahl, M.; Seidl, S.; Wessely, R. Targeting 2A Protease by RNA Interference Attenuates Coxsackieviral Cytopathogenicity and Promotes Survival in Highly Susceptible Mice. Circulation 2005, 111, 1583–1592. [Google Scholar] [CrossRef]
- Werk, D.; Schubert, S.; Lindig, V.; Grunert, H.-P.; Zeichhardt, H.; Erdmann, V.A.; Kurreck, J. Developing an Effective RNA Interference Strategy against a Plus-Strand RNA Virus: Silencing of Coxsackievirus B3 and Its Cognate Coxsackievirus-Adenovirus Receptor. Biol. Chem. 2005, 386, 857–863. [Google Scholar] [CrossRef]
- Tang, Q.; Khvorova, A. RNAi-Based Drug Design: Considerations and Future Directions. Nat. Rev. Drug Discov. 2024, 23, 341–364. [Google Scholar] [CrossRef]
- Sneller, L.; Lin, C.; Price, A.; Kottilil, S.; Chua, J.V. RNA Interference Therapeutics for Chronic Hepatitis B: Progress, Challenges, and Future Prospects. Microorganisms 2024, 12, 599. [Google Scholar] [CrossRef] [PubMed]
- Thangamani, L.; Balasubramanian, B.; Easwaran, M.; Natarajan, J.; Pushparaj, K.; Meyyazhagan, A.; Piramanayagam, S. GalNAc-siRNA Conjugates: Prospective Tools on the Frontier of Anti-Viral Therapeutics. Pharmacol. Res. 2021, 173, 105864. [Google Scholar] [CrossRef]
- Merl, S.; Wessely, R. Anti-Coxsackieviral Efficacy of RNA Interference Is Highly Dependent on Genomic Target Selection and Emergence of Escape Mutants. Oligonucleotides 2007, 17, 44–53. [Google Scholar] [CrossRef] [PubMed]
- ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA Interference: A Multiple shRNA Approach. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef]
- Poeck, H.; Besch, R.; Maihoefer, C.; Renn, M.; Tormo, D.; Morskaya, S.S.; Kirschnek, S.; Gaffal, E.; Landsberg, J.; Hellmuth, J.; et al. 5′-Triphosphate-siRNA: Turning Gene Silencing and Rig-I Activation against Melanoma. Nat. Med. 2008, 14, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Dauletbaev, N.; Cammisano, M.; Herscovitch, K.; Lands, L.C. Stimulation of the RIG-I/MAVS Pathway by Polyinosinic:Polycytidylic Acid Upregulates IFN-β in Airway Epithelial Cells with Minimal Costimulation of IL-8. J. Immunol. 2015, 195, 2829–2841. [Google Scholar] [CrossRef]
- Devhare, P.B.; Desai, S.; Lole, K.S. Innate Immune Responses in Human Hepatocyte-Derived Cell Lines Alter Genotype 1 Hepatitis E Virus Replication Efficiencies. Sci. Rep. 2016, 6, 26827. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Organism | Target | Reference |
|---|---|---|---|
| 4 | Pig | RdRp | Huang et al. [32] |
| 1 | Human | Hel, RdRp, 3′CAE | Kumar et al. [33] |
| 4 | Human | ORF2 | Huang et al. [34] |
| 4 | Pig | ORF3 | Liu et al. [35] |
| 3 | Human | Met, Y, HVR, X, Hel, RdRp, ORF2 | Zhang et al. [36] |
| Name | Target | siRNA-Sequence |
|---|---|---|
| siORF3.1 | ORF3 | 5′-GGGCUGUUCUGUUGCUGUUTT-3′ 3′-TTCCCGACAAGACAACGACAA-5′ |
| siORF3.2 | ORF3 | 5′-GGGUUGAUUCUCAGCCCUUTT-3′ 3′-TTCCCAACUAAGAGUCGGGAA-5′ |
| siORF3.3 1 | ORF3 | 5′-CCUAUAUUCAUCCAACCAATT-3′ 3′-TTGGAUAUAAGUAGGUUGGUU-5′ |
| siHel.1 | Helicase | 5′-GGAUGUUGAUGUGGUGGUUTT-3′ 3′-TTCCUACAACUACACCACCAA-5′ |
| siHel.2 | Helicase | 5′-ACCGCAUUUGUUGCUACUATT-3′ 3′-TTUGGCGUAAACAACGAUGAU-5′ |
| siHel.3 | Helicase | 5′-ACUUUCACGGAGACUACAATT-3′ 3′-TTUGAAAGUGCCUCUGAUGUU-5′ |
| siCon | no target | 5′-ACGUGACACGUUCGGAGAATT-3′ 3′-TTUGCACUGUGCAAGCCUCUU-5′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziersch, M.; Harms, D.; Neumair, L.; Kurreck, A.; Johne, R.; Bock, C.-T.; Kurreck, J. Combining RNA Interference and RIG-I Activation to Inhibit Hepatitis E Virus Replication. Viruses 2024, 16, 1378. https://doi.org/10.3390/v16091378
Ziersch M, Harms D, Neumair L, Kurreck A, Johne R, Bock C-T, Kurreck J. Combining RNA Interference and RIG-I Activation to Inhibit Hepatitis E Virus Replication. Viruses. 2024; 16(9):1378. https://doi.org/10.3390/v16091378
Chicago/Turabian StyleZiersch, Mathias, Dominik Harms, Lena Neumair, Anke Kurreck, Reimar Johne, C.-Thomas Bock, and Jens Kurreck. 2024. "Combining RNA Interference and RIG-I Activation to Inhibit Hepatitis E Virus Replication" Viruses 16, no. 9: 1378. https://doi.org/10.3390/v16091378