
HSV-2 Manipulates Autophagy through Interferon Pathway: A Strategy for Viral Survival

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus Propagation
2.3. IFN Studies
2.4. SYBR-Green PCR
2.5. ELISA
2.6. Immunoblotting
2.7. Apoptosis Analysis
2.7.1. Quantification of Bcl-2 and Bax Gene Expression
2.7.2. TUNEL Assay for Apoptosis Detection
2.7.3. Flow Cytometric Analysis of Apoptotic Cell Populations
2.8. Statistical Analysis
3. Results
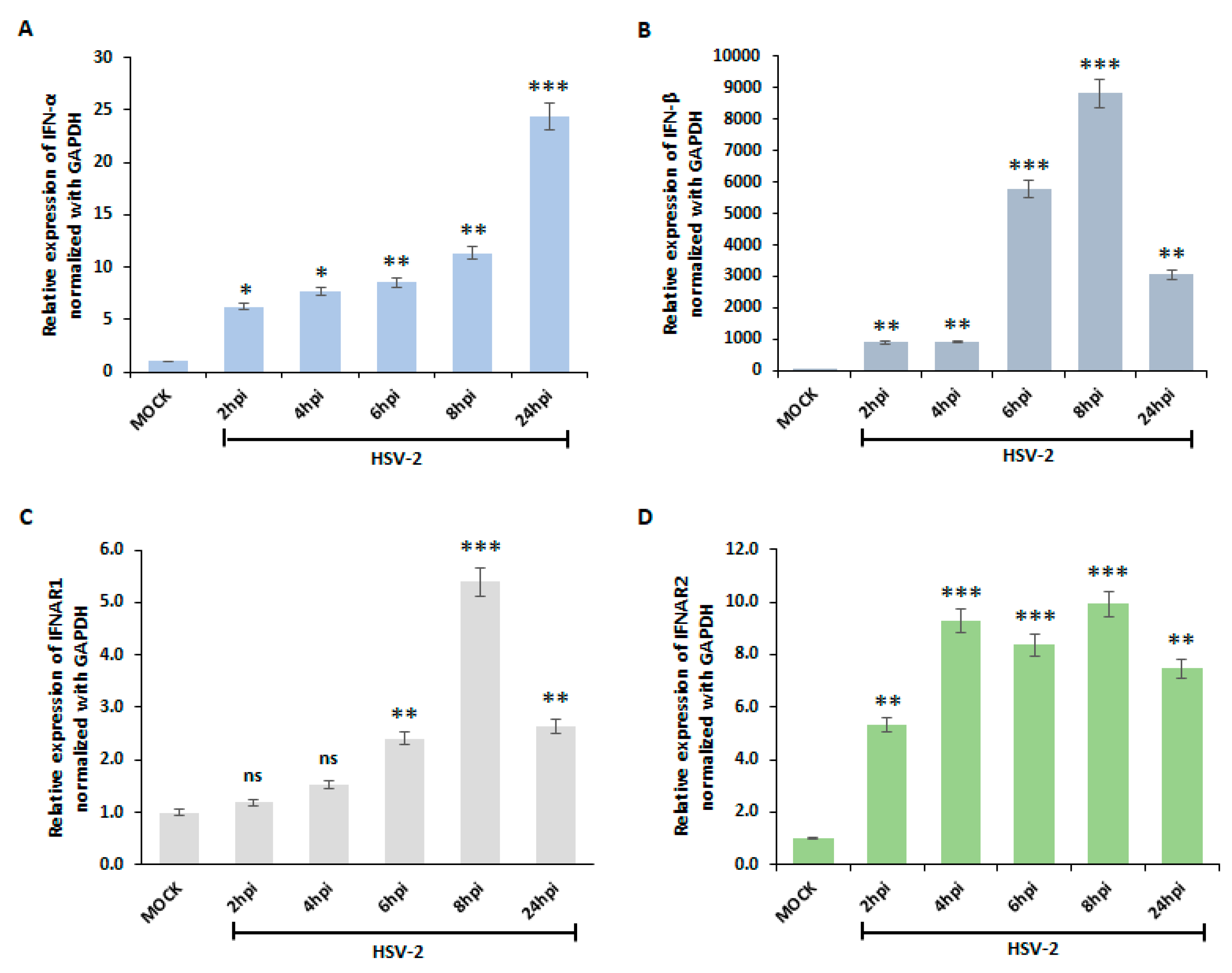
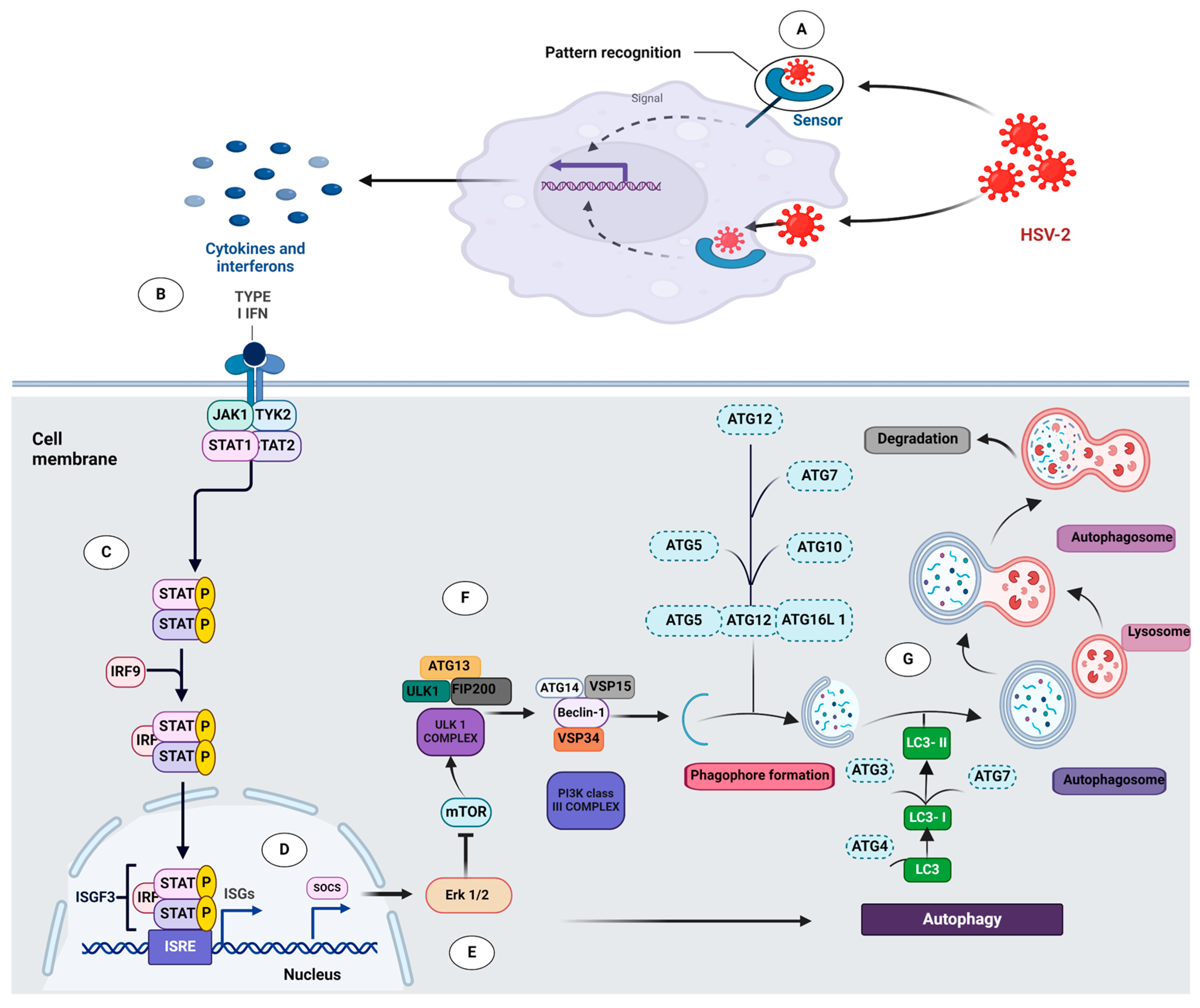
3.1. HSV-2 Activates Type I Interferon Pathway in Macrophages
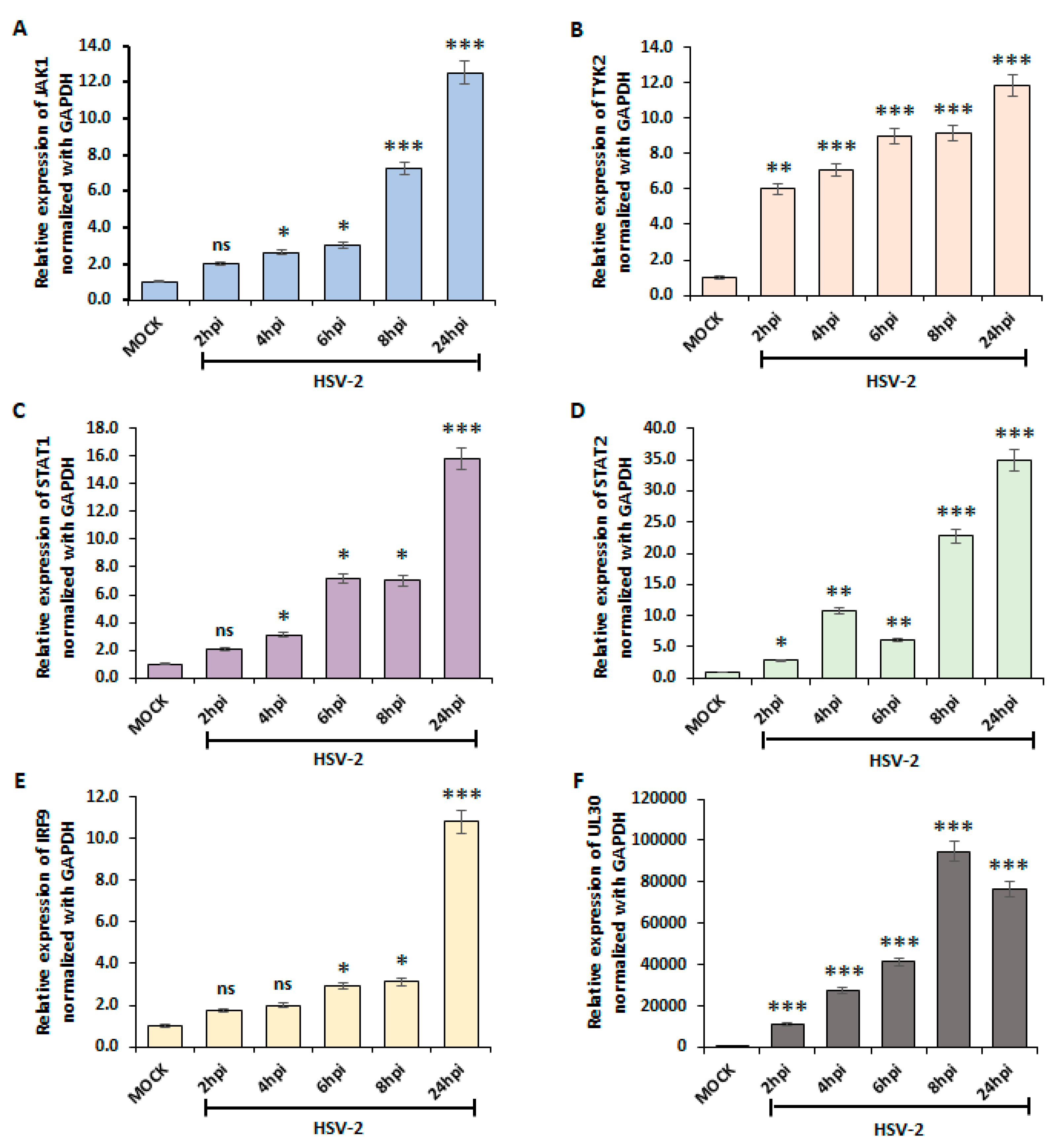
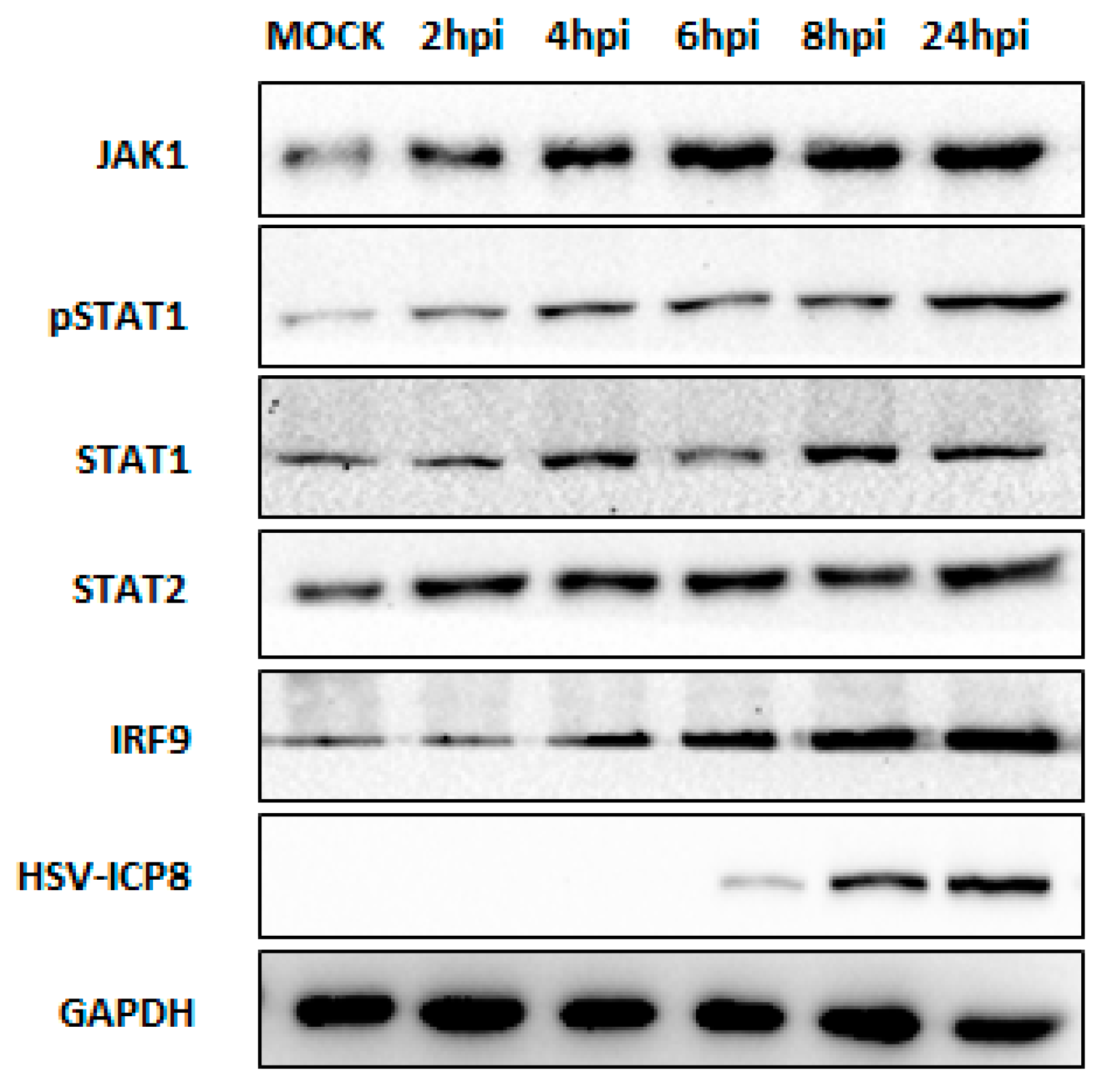
3.2. Type I IFN Pathway Mediates Downstream Signaling through the JAK-STAT Pathway upon HSV-2 Infection
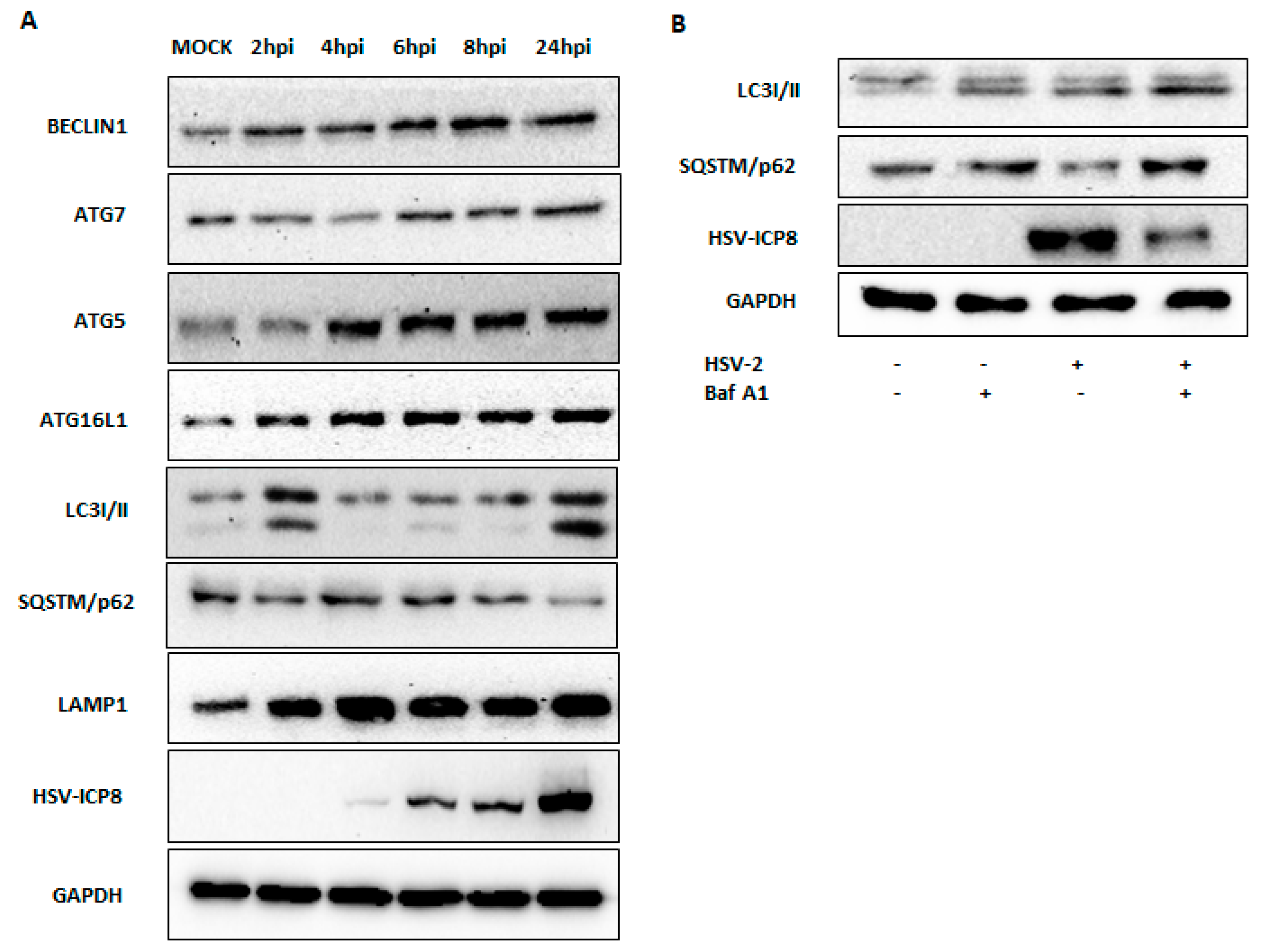
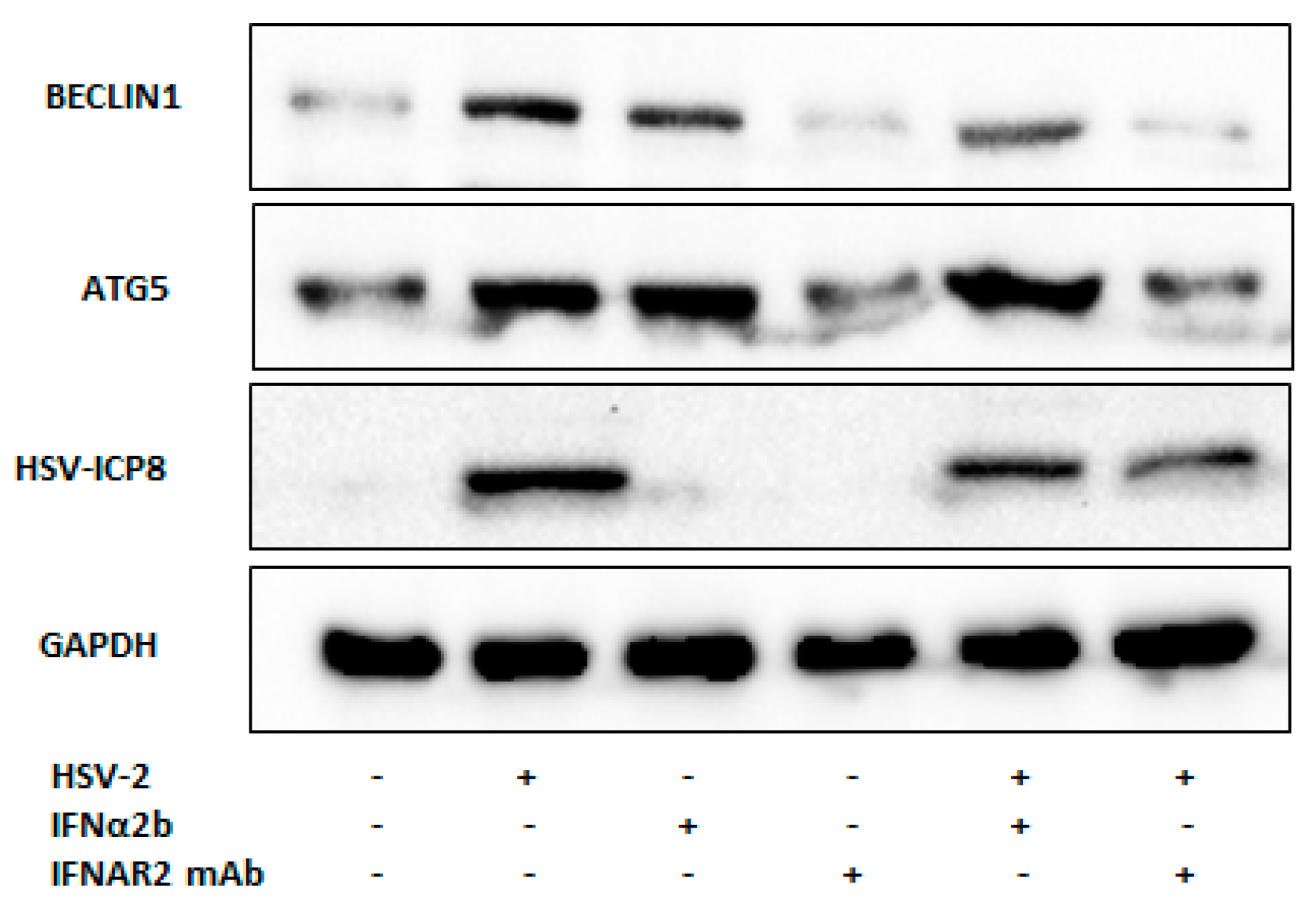
3.3. Type I IFN Induces Autophagy upon HSV-2 Infection
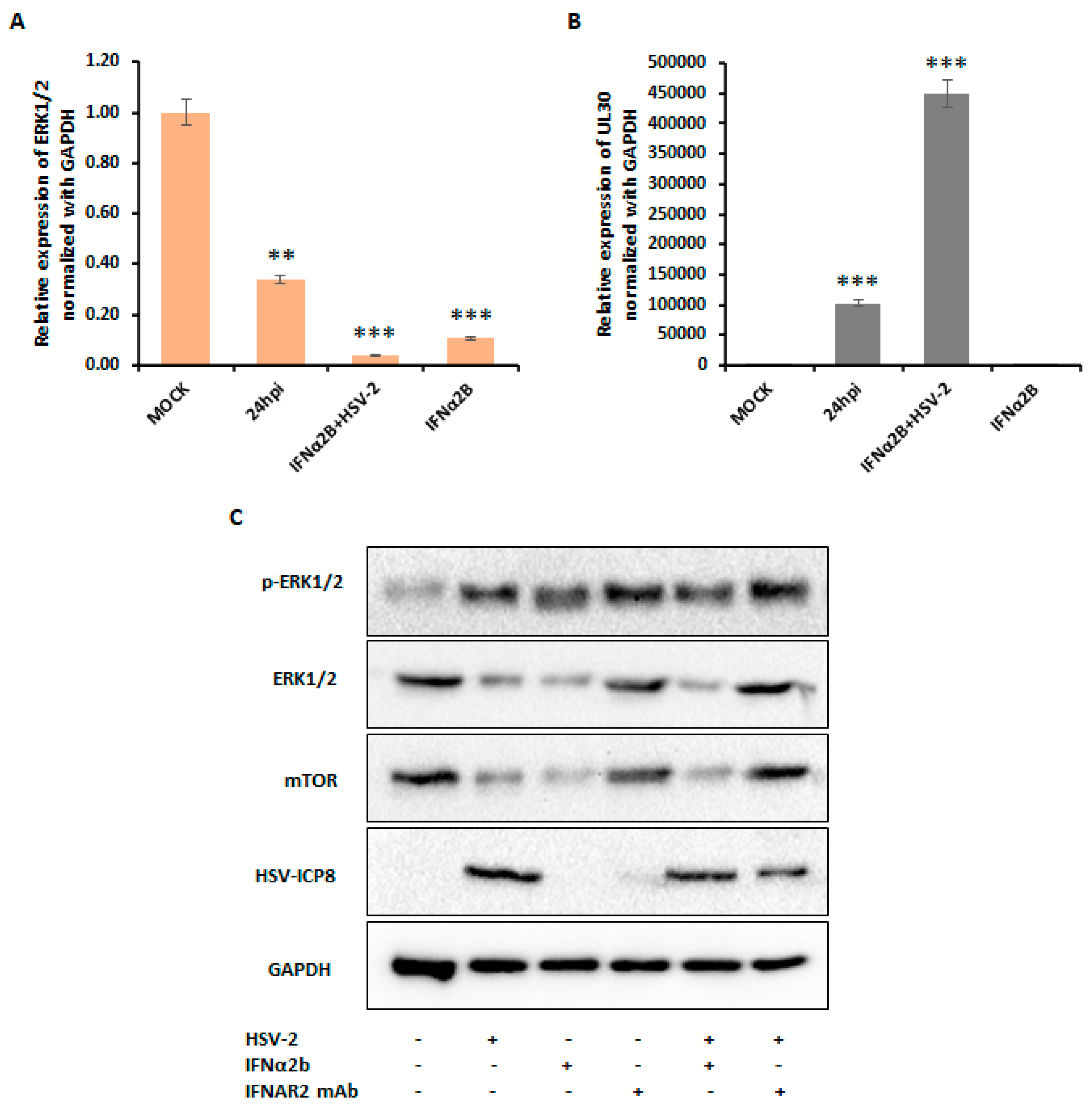
3.4. Type I IFN Induces Autophagy through Inhibition of ERK1/2 and mTOR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Looker, K.J.; Welton, N.J.; Sabin, K.M.; Dalal, S.; Vickerman, P.; Turner, K.M.E.; Boily, M.-C.; Gottlieb, S.L. Global and Regional Estimates of the Contribution of Herpes Simplex Virus Type 2 Infection to HIV Incidence: A Population Attributable Fraction Analysis Using Published Epidemiological Data. Lancet Infect. Dis. 2020, 20, 240–249. [Google Scholar] [CrossRef]
- Banerjee, A.; Mukherjee, A. Herpesviridae and microRNAs. In Current Perspectives on Viral Disease Outbreaks—Epidemiology, Detection and Control; Claborn, D., Ed.; IntechOpen: London, UK, 2022; ISBN 978-1-83881-910-1. [Google Scholar]
- Jaishankar, D.; Shukla, D. Genital Herpes: Insights into Sexually Transmitted Infectious Disease. Microb. Cell 2016, 3, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Armangue, T.; Leypoldt, F.; Málaga, I.; Raspall-Chaure, M.; Marti, I.; Nichter, C.; Pugh, J.; Vicente-Rasoamalala, M.; Lafuente-Hidalgo, M.; Macaya, A.; et al. Herpes Simplex Virus Encephalitis Is a Trigger of Brain Autoimmunity. Ann. Neurol. 2014, 75, 317–323. [Google Scholar] [CrossRef]
- Mancini, M.; Vidal, S.M. Insights into the Pathogenesis of Herpes Simplex Encephalitis from Mouse Models. Mamm. Genome 2018, 29, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Kulkarni, S.; Mukherjee, A. Herpes Simplex Virus: The Hostile Guest That Takes Over Your Home. Front. Microbiol. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Lee, H.K. Vaccines against Genital Herpes: Where Are We? Vaccines 2020, 8, 420. [Google Scholar] [CrossRef]
- Corey, L.; Wald, A.; Celum, C.L.; Quinn, T.C. The Effects of Herpes Simplex Virus-2 on HIV-1 Acquisition and Transmission: A Review of Two Overlapping Epidemics. JAIDS J. Acquir. Immune Defic. Syndr. 2004, 35, 435–445. [Google Scholar] [CrossRef]
- Kinghorn, G.R. Genital Herpes: Natural History and Treatment of Acute Episodes. J. Med. Virol. 1993, 41, 33–38. [Google Scholar] [CrossRef]
- Banerjee, A.; Dass, D.; Dhotre, K.; Wakchoure, P.; More, A.; Rana, S.; Khan, A.A.; Mukherjee, A. Combinatorial Effects of miRNAs in HSV-2 Infection of Macrophages: An In Silico and In Vitro Integration Approach. Vaccines 2023, 11, 1488. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy Balances Inflammation in Innate Immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent Roles of Autophagy in Virus Infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, Immunity, and Microbial Adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Tu, S.; Ding, L.; Jin, M.; Chen, H.; Zhou, H. The Role of Autophagy in Viral Infections. J. Biomed. Sci. 2023, 30, 5. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippé, R.; et al. Autophagy Enhances the Presentation of Endogenous Viral Antigens on MHC Class I Molecules during HSV-1 Infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- Dong, X.; Levine, B. Autophagy and Viruses: Adversaries or Allies? J. Innate Immun. 2013, 5, 480–493. [Google Scholar] [CrossRef]
- Mukhopadhyay, U.; Chanda, S.; Patra, U.; Mukherjee, A.; Rana, S.; Mukherjee, A.; Chawla-Sarkar, M. Synchronized Orchestration of miR-99b and Let-7g Positively Regulates Rotavirus Infection by Modulating Autophagy. Sci. Rep. 2019, 9, 1318. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Ge, X.; Guo, X.; Yang, H. Autophagy Promotes the Replication of Encephalomyocarditis Virus in Host Cells. Autophagy 2011, 7, 613–628. [Google Scholar] [CrossRef]
- Bekisz, J.; Schmeisser, H.; Hernandez, J.; Goldman, N.D.; Zoon, K.C. Mini ReviewHuman Interferons Alpha, Beta and Omega. Growth Factors 2004, 22, 243–251. [Google Scholar] [CrossRef]
- Schmeisser, H.; Fey, S.B.; Horowitz, J.; Fischer, E.R.; Balinsky, C.A.; Miyake, K.; Bekisz, J.; Snow, A.L.; Zoon, K.C. Type I Interferons Induce Autophagy in Certain Human Cancer Cell Lines. Autophagy 2013, 9, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.S.; Levy, D.E.; Darnell, J.E. Two Interferon-Induced Nuclear Factors Bind a Single Promoter Element in Interferon-Stimulated Genes. Proc. Natl. Acad. Sci. USA 1988, 85, 8521–8525. [Google Scholar] [CrossRef]
- Platanias, L.C. The P38 Mitogen-Activated Protein Kinase Pathway and Its Role in Interferon Signaling. Pharmacol. Ther. 2003, 98, 129–142. [Google Scholar] [CrossRef]
- Kaur, S.; Katsoulidis, E.; Platanias, L.C. Akt and mRNA Translation by Interferons. Cell Cycle 2008, 7, 2112–2116. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Sassano, A.; Dolniak, B.; Joshi, S.; Majchrzak-Kita, B.; Baker, D.P.; Hay, N.; Fish, E.N.; Platanias, L.C. Role of the Akt Pathway in mRNA Translation of Interferon-Stimulated Genes. Proc. Natl. Acad. Sci. USA 2008, 105, 4808–4813. [Google Scholar] [CrossRef] [PubMed]
- Huleihel, M.; Ishanu, V.; Tal, J.; Arad, S. (Malis) Antiviral Effect of Red Microalgal Polysaccharides on Herpes Simplex and Varicella Zoster Viruses. J. Appl. Phycol. 2001, 13, 127–134. [Google Scholar] [CrossRef]
- Yakoub, A.M.; Shukla, D. Basal Autophagy Is Required for Herpes Simplex Virus-2 Infection. Sci. Rep. 2015, 5, 12985. [Google Scholar] [CrossRef]
- Petrovski, G.; Pásztor, K.; Orosz, L.; Albert, R.; Mencel, E.; Moe, M.C.; Kaarniranta, K.; Facskó, A.; Megyeri, K. Herpes Simplex Virus Types 1 and 2 Modulate Autophagy in SIRC Corneal Cells. J. Biosci. 2014, 39, 683–692. [Google Scholar] [CrossRef]
- Zhu, S.; Cao, L.; Yu, Y.; Yang, L.; Yang, M.; Liu, K.; Huang, J.; Kang, R.; Livesey, K.M.; Tang, D. Inhibiting Autophagy Potentiates the Anticancer Activity of IFN1@/IFN-α in Chronic Myeloid Leukemia Cells. Autophagy 2013, 9, 317–327. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Zhang, Z.; Singh, R.; Aschner, M. Methods for the Detection of Autophagy in Mammalian Cells. Curr. Protoc. Toxicol. 2016, 69, 20.12.1–20.12.26. [Google Scholar] [CrossRef]
- Li, Z.; Huang, Z.; Zhang, H.; Lu, J.; Wei, Y.; Yang, Y.; Bai, L. IRE1-mTOR-PERK Axis Coordinates Autophagy and ER Stress-Apoptosis Induced by P2X7-Mediated Ca2+ Influx in Osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 695041. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Lin, E.; He, L.; Yu, J.; Tan, P.; Zhou, Y. Autophagy and Viral Infection. Adv. Exp. Med. Biol. 2019, 1209, 55–78. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Wilson, C.A.; Lee, S.J.; Benveniste, E.N. IFN-Beta-Induced SOCS-1 Negatively Regulates CD40 Gene Expression in Macrophages and Microglia. FASEB J. 2006, 20, 985–987. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR Pathways: Cross-Talk and Compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Ambjørn, M.; Ejlerskov, P.; Liu, Y.; Lees, M.; Jäättelä, M.; Issazadeh-Navikas, S. IFNB1/Interferon-β-Induced Autophagy in MCF-7 Breast Cancer Cells Counteracts Its Proapoptotic Function. Autophagy 2013, 9, 287–302. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, C.M.; Hwang, S.Y.; Holland, K.A.; Gulluyan, L.M.; Tavaria, M.; Weaver, B.; Reich, N.C.; Kola, I.; Hertzog, P.J. Cloning and Characterization of Soluble and Transmembrane Isoforms of a Novel Component of the Murine Type I Interferon Receptor, IFNAR 2. J. Biol. Chem. 1997, 272, 23865–23870. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gong, M.; Zhao, F.; Shao, J.; Xie, Y.; Zhang, Y.; Chang, H. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate Immune Recognition of Viral Infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 Complex-at the Crossroads of Autophagy and Beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef]
- Fraiberg, M.; Elazar, Z. Genetic Defects of Autophagy Linked to Disease. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2020; Volume 172, pp. 293–323. ISBN 978-0-12-822021-4. [Google Scholar]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. LC3- and P62-Based Biochemical Methods for the Analysis of Autophagy Progression in Mammalian Cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Hassan, A.; Lee, C.-H.; Xie, X.-S.; Li, X. Molecular Basis of V-ATPase Inhibition by Bafilomycin A1. Nat. Commun. 2021, 12, 1782. [Google Scholar] [CrossRef] [PubMed]
- Mastino, A.; Sciortino, M.T.; Medici, M.A.; Perri, D.; Ammendolia, M.G.; Grelli, S.; Amici, C.; Pernice, A.; Guglielmino, S. Herpes Simplex Virus 2 Causes Apoptotic Infection in Monocytoid Cells. Cell Death Differ. 1997, 4, 629–638. [Google Scholar] [CrossRef]
- Sasi, W.; Sharma, A.K.; Mokbel, K. The Role of Suppressors of Cytokine Signalling in Human Neoplasms. Mol. Biol. Int. 2014, 2014, 630797. [Google Scholar] [CrossRef]
- Carrière, A.; Cargnello, M.; Julien, L.-A.; Gao, H.; Bonneil, É.; Thibault, P.; Roux, P.P. Oncogenic MAPK Signaling Stimulates mTORC1 Activity by Promoting RSK-Mediated Raptor Phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk Implications for Tuberous Sclerosis and Cancer Pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef]
- Yao, R.; Cooper, G.M. Requirement for Phosphatidylinositol-3 Kinase in the Prevention of Apoptosis by Nerve Growth Factor. Science 1995, 267, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, C.L.; Choudhury, B.; Davies, H.; Edkins, S.; Greenman, C.; Haaften, G.V.; Mironenko, T.; Santarius, T.; Stevens, C.; Stratton, M.R.; et al. LKB1/KRAS Mutant Lung Cancers Constitute a Genetic Subset of NSCLC with Increased Sensitivity to MAPK and mTOR Signalling Inhibition. Br. J. Cancer 2009, 100, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR Inhibits Autophagy by Controlling ULK1 Ubiquitylation, Self-Association and Function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR Inhibition Activates Overall Protein Degradation by the Ubiquitin Proteasome System as Well as by Autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the Lysosome: A Control Centre for Cellular Clearance and Energy Metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 Functions as a Transcriptional Regulator of Autophagy by Preventing Nuclear Transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequences | Reverse Primer Sequences |
|---|---|---|
| UL30 | 5′-CGCTCAACACGGACTATTACTT-3′ | 5′-CTCGGTGATCTTGGCGTTATT-3′ |
| IFN-α | 5′-AATTCTGCACCGAACTCTACC-3′ | 5′-ATGGAGTCCGCATTCATCAG-3′ |
| IFN-β | 5′-GCTTCTCCACTACAGCTCTTTC-3′ | 5′-CAGTATTCAAGCCTCCCATTCA-3′ |
| IFN-αβR1 | 5′-TTAGTGACGCTGTATGTGAGAAA-3′ | 5′-TGACAAACGGGAGAGCAAATA-3′ |
| IFN-αβR2 | 5′-GTGAGGAGGGAACACCTTATTT-3′ | 5′-ATGCACTGAGAAGGCAGATAAA-3′ |
| JAK1 | 5′-GGATTACAAGGATGACGAAGGA-3′ | 5′-CGAAGAAGGCCAGGGAAATA-3′ |
| TYK2 | 5′-CTCAAAGCTGCATCCCTTCT-3′ | 5′-GTGACTTCCACAAGGACCTAAA-3′ |
| STAT1 | 5′-CACCTACGAACATGACCCTATC-3′ | 5′-GCTGTCTTTCCACCACAAAC-3′ |
| STAT2 | 5′-CAGGCTCATTGTGGTCTCTAAT-3′ | 5′-GCCCTAGTTCCAGCTCTAATG-3′ |
| IRF9 | 5′-TCAGAGGTCCCTGGAGTTT-3′ | 5′-CCCGTTGTAGATGAAGGTGAG-3′ |
| ERK1/2 | 5′-AGAGAACCCTGAGGGAGATAAA-3′ | 5′-CGATGGTTGGTGCTCGAATA-3′ |
| BCL2 | 5′-ATCGCCCTGTGGATGACTGAGT-3′ | 5′-GCCAGGAGAAATCAAACAGAGGC-3′ |
| BAX | 5′-TCAGGATGCGTCCACCAAGAAG-3′ | 5′-TGTGTCCACGGCGGCAATCATC-3′ |
| GAPDH | 5′-CAAGAGCACAAGAGGAAGAGAG-3′ | 5′-CTACATGGCAACTGTGAGGAG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dass, D.; Banerjee, A.; Dhotre, K.; Sonawane, V.; More, A.; Mukherjee, A. HSV-2 Manipulates Autophagy through Interferon Pathway: A Strategy for Viral Survival. Viruses 2024, 16, 1383. https://doi.org/10.3390/v16091383
Dass D, Banerjee A, Dhotre K, Sonawane V, More A, Mukherjee A. HSV-2 Manipulates Autophagy through Interferon Pathway: A Strategy for Viral Survival. Viruses. 2024; 16(9):1383. https://doi.org/10.3390/v16091383
Chicago/Turabian StyleDass, Debashree, Anwesha Banerjee, Kishore Dhotre, Vaishnavi Sonawane, Ashwini More, and Anupam Mukherjee. 2024. "HSV-2 Manipulates Autophagy through Interferon Pathway: A Strategy for Viral Survival" Viruses 16, no. 9: 1383. https://doi.org/10.3390/v16091383
APA StyleDass, D., Banerjee, A., Dhotre, K., Sonawane, V., More, A., & Mukherjee, A. (2024). HSV-2 Manipulates Autophagy through Interferon Pathway: A Strategy for Viral Survival. Viruses, 16(9), 1383. https://doi.org/10.3390/v16091383