

Complex Evolutionary Dynamics of H5N8 Influenza A Viruses Revealed by Comprehensive Reassortment Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
Sequence Selection for Analysis
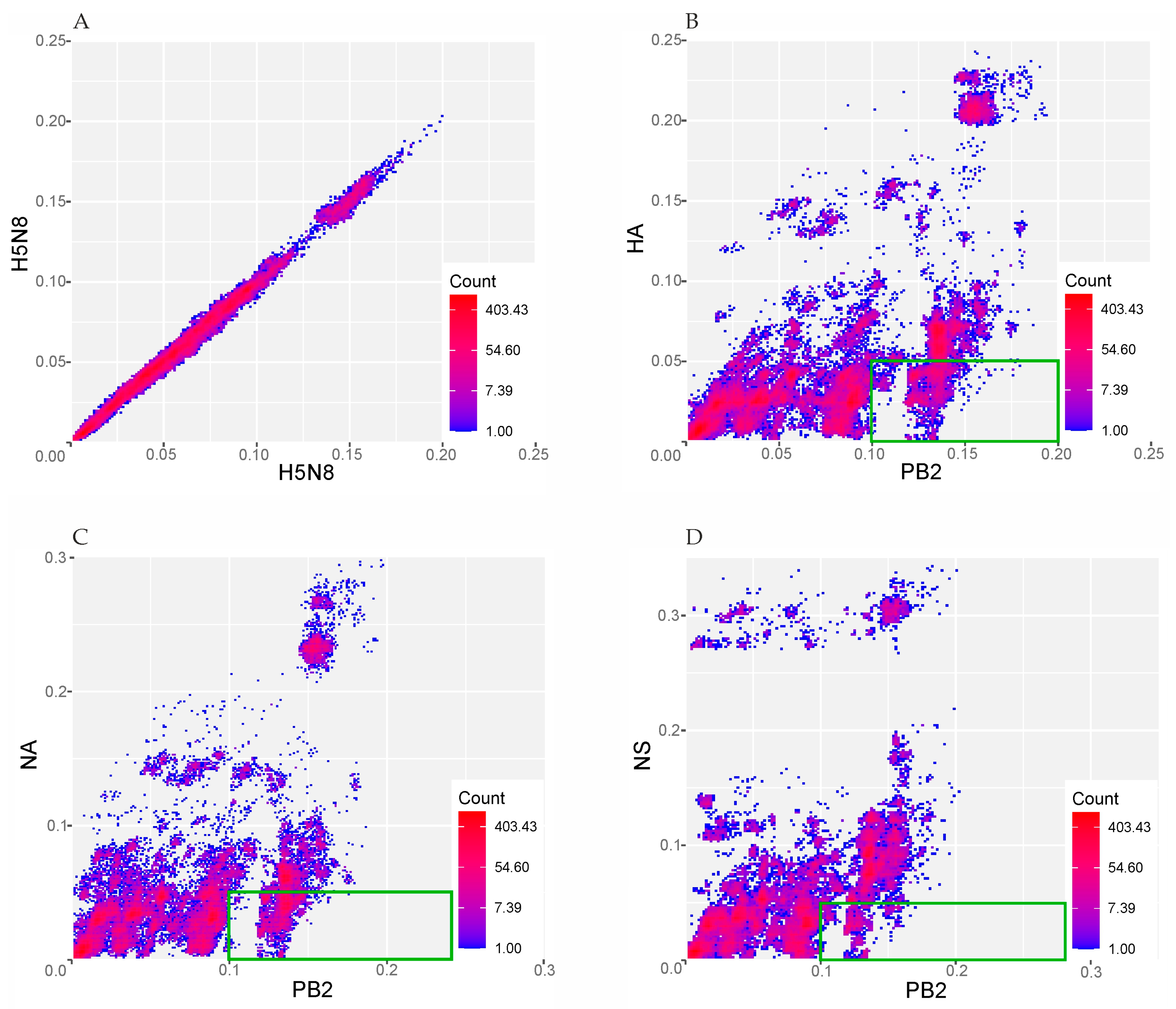
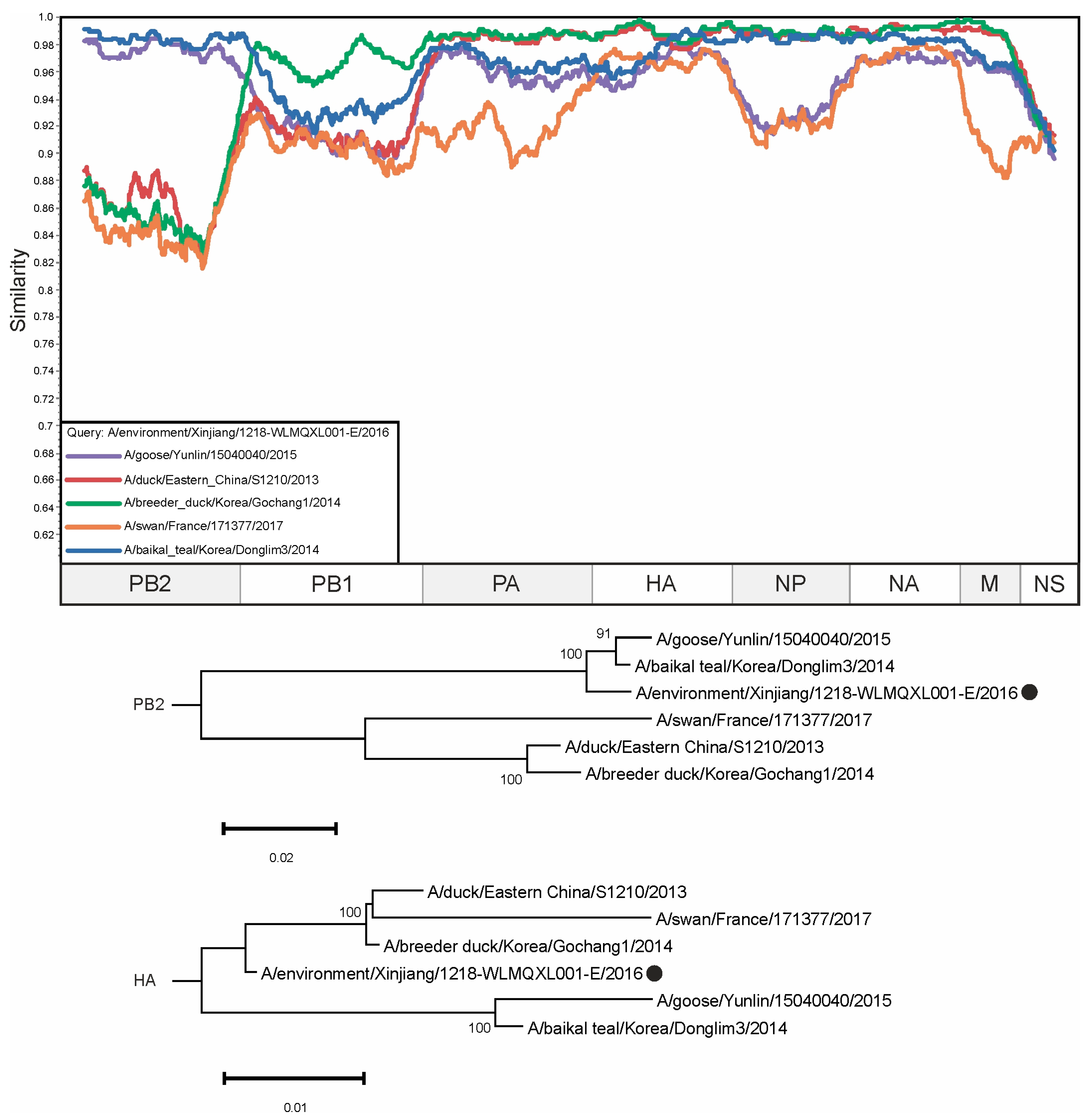
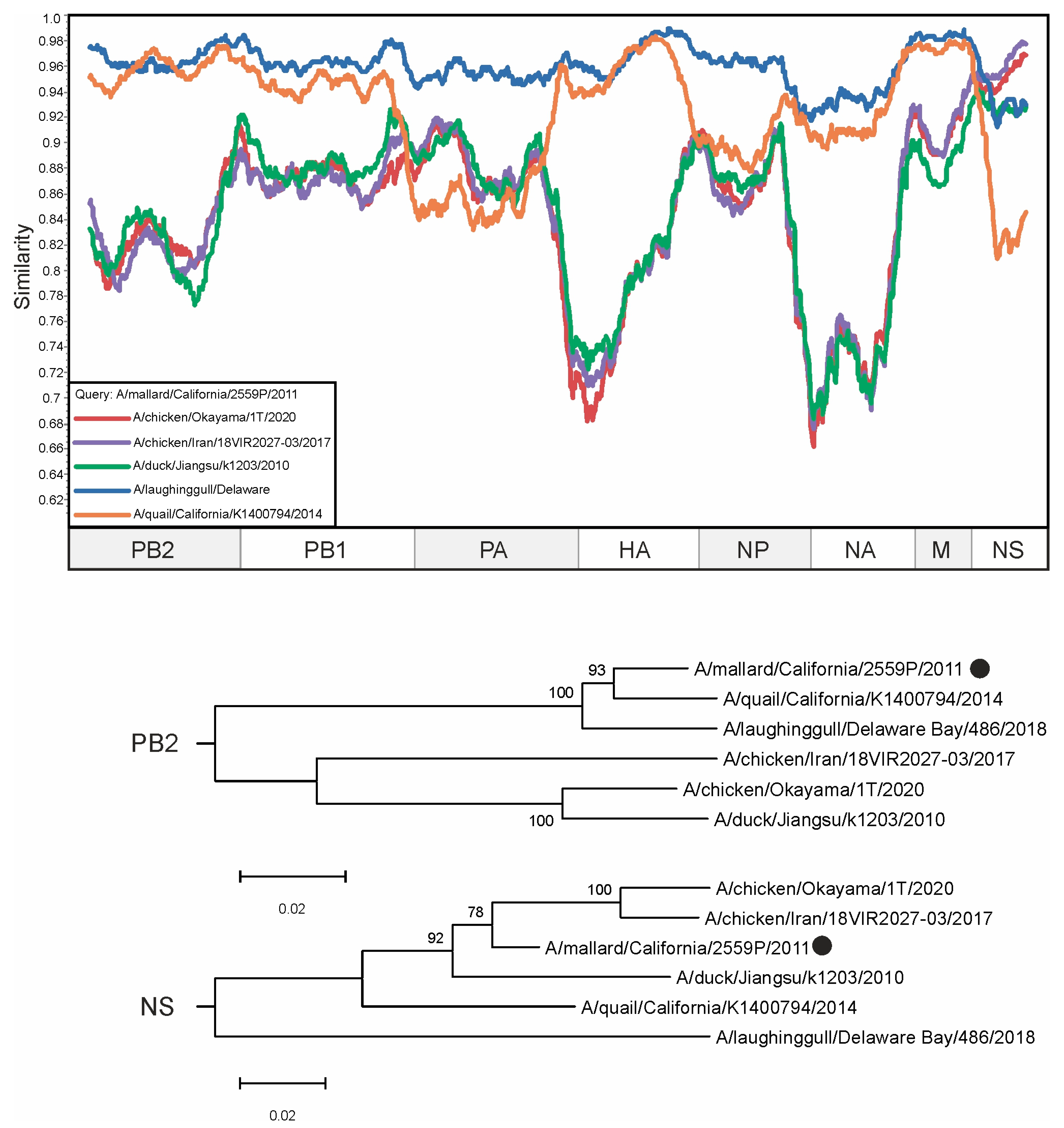
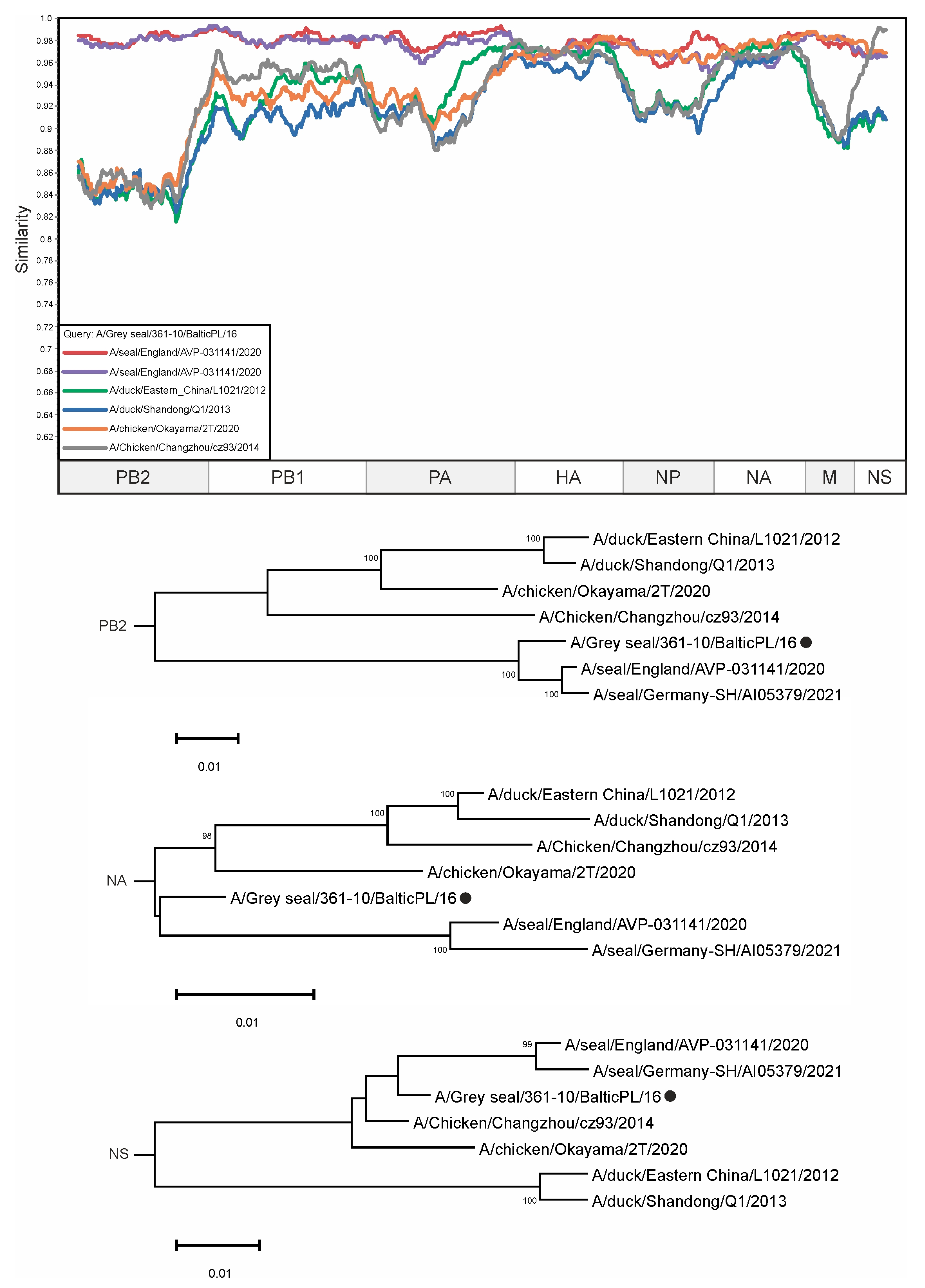
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.-T.; Linster, M.; Mendenhall, I.H.; Su, Y.C.F.; Smith, G.J.D. Avian Influenza Viruses in Humans: Lessons from Past Outbreaks. Br. Med. Bull. 2019, 132, 81–95. [Google Scholar] [CrossRef]
- Crisci, E.; Mussá, T.; Fraile, L.; Montoya, M. Review: Influenza Virus in Pigs. Mol. Immunol. 2013, 55, 200–211. [Google Scholar] [CrossRef]
- Campos, A.C.A.; Góes, L.G.B.; Moreira-Soto, A.; de Carvalho, C.; Ambar, G.; Sander, A.-L.; Fischer, C.; Ruckert da Rosa, A.; Cardoso de Oliveira, D.; Kataoka, A.P.G.; et al. Bat Influenza A(HL18NL11) Virus in Fruit Bats, Brazil. Emerg. Infect. Dis. 2019, 25, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Borland, S.; Gracieux, P.; Jones, M.; Mallet, F.; Yugueros-Marcos, J. Influenza A Virus Infection in Cats and Dogs: A Literature Review in the Light of the “One Health” Concept. Front. Public Health 2020, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Slavinski, S.; Schiff, C.; Merlino, M.; Daskalakis, D.; Liu, D.; Rakeman, J.L.; Misener, M.; Thompson, C.; Leung, Y.L.; et al. Outbreak of Influenza A(H7N2) Among Cats in an Animal Shelter With Cat-to-Human Transmission—New York City, 2016. Clin. Infect. Dis. 2017, 65, 1927–1929. [Google Scholar] [CrossRef] [PubMed]
- Runstadler, J.A.; Puryear, W. A Brief Introduction to Influenza A Virus in Marine Mammals. Anim. Influenza Virus Methods Protoc. 2020, 429–450. [Google Scholar]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- Martini, M.; Gazzaniga, V.; Bragazzi, N.L.; Barberis, I. The Spanish Influenza Pandemic: A Lesson from History 100 Years after 1918. J. Prev. Med. Hyg. 2019, 60, E64–E67. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The Biology of Influenza Viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef]
- Available online: https://ictv.global/taxonomy (accessed on 1 December 2023).
- Suarez, D.L. Influenza A Virus. In Animal Influenza; Wiley: Hoboken, NJ, USA, 2016; pp. 1–30. [Google Scholar]
- Hutchinson, E.C. Influenza Virus. Trends Microbiol. 2018, 26, 809–810. [Google Scholar] [CrossRef]
- Fereidouni, S.; Starick, E.; Karamendin, K.; Di Genova, C.; Scott, S.D.; Khan, Y.; Harder, T.; Kydyrmanov, A. Genetic Characterization of a New Candidate Hemagglutinin Subtype of Influenza A Viruses. Emerg. Microbes Infect. 2023, 12, 2225645. [Google Scholar] [CrossRef] [PubMed]
- Ciminski, K.; Ran, W.; Gorka, M.; Lee, J.; Malmlov, A.; Schinköthe, J.; Eckley, M.; Murrieta, R.A.; Aboellail, T.A.; Campbell, C.L.; et al. Bat Influenza Viruses Transmit among Bats but Are Poorly Adapted to Non-Bat Species. Nat. Microbiol. 2019, 4, 2298–2309. [Google Scholar] [CrossRef]
- Kilbourne, E.D. Influenza Pandemics of the 20th Century. Emerg. Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Han, G.-Z.; Rambaut, A. Genesis and Pathogenesis of the 1918 Pandemic H1N1 Influenza A Virus. Proc. Natl. Acad. Sci. USA 2014, 111, 8107–8112. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.; Lowen, A.C. Influenza A Virus Reassortment. In Influenza Pathogenesis and Control—Volume I; Springer: Cham, Switzerland, 2014; pp. 377–401. [Google Scholar]
- Shoham, D. Review: Molecular Evolution and the Feasibility of an Avian Influenza Virus Becoming a Pandemic Strain––a Conceptual Shift. Virus Genes 2006, 33, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, A.J.; Armstrong, J.S.; Downie, J.C. From Where Did the 2009 “swine-Origin” Influenza A Virus (H1N1) Emerge? Virol. J. 2009, 6, 207. [Google Scholar] [CrossRef]
- Perdue, M.L.; Swayne, D.E. Public Health Risk from Avian Influenza Viruses. Avian Dis. 2005, 49, 317–327. [Google Scholar] [CrossRef]
- Yoon, H.; Moon, O.-K.; Jeong, W.; Choi, J.; Kang, Y.-M.; Ahn, H.-Y.; Kim, J.-H.; Yoo, D.-S.; Kwon, Y.-J.; Chang, W.-S.; et al. H5N8 Highly Pathogenic Avian Influenza in the Republic of Korea: Epidemiology During the First Wave, from January Through July 2014. Osong Public Health Res. Perspect. 2015, 6, 106–111. [Google Scholar] [CrossRef]
- Lee, M.-S.; Chen, L.-H.; Chen, Y.-P.; Liu, Y.-P.; Li, W.-C.; Lin, Y.-L.; Lee, F. Highly Pathogenic Avian Influenza Viruses H5N2, H5N3, and H5N8 in Taiwan in 2015. Vet. Microbiol. 2016, 187, 50–57. [Google Scholar] [CrossRef]
- Kanehira, K.; Uchida, Y.; Takemae, N.; Hikono, H.; Tsunekuni, R.; Saito, T. Characterization of an H5N8 Influenza A Virus Isolated from Chickens during an Outbreak of Severe Avian Influenza in Japan in April 2014. Arch. Virol. 2015, 160, 1629–1643. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Kang, H.-M.; Lee, E.-K.; Song, B.-M.; Jeong, J.; Kwon, Y.-K.; Kim, H.-R.; Lee, K.-J.; Hong, M.-S.; Jang, I.; et al. Novel Reassortant Influenza A(H5N8) Viruses, South Korea, 2014. Emerg. Infect. Dis. 2014, 20, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, J.H.; Herfst, S.; Fouchier, R.A.M. How a Virus Travels the World. Science 2015, 347, 616–617. [Google Scholar] [CrossRef] [PubMed]
- Role for Migratory Wild Birds in the Global Spread of Avian Influenza H5N8. Science 2016, 354, 213–217. [CrossRef] [PubMed]
- McLeod, A.; Hinrichs, J. The Economics of Animal Influenza. In Animal Influenza; Wiley: Hoboken, NJ, USA, 2016; pp. 45–73. [Google Scholar]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Terregino, C.; Aznar, I.; Guajardo, I.M.; et al. Avian Influenza Overview February–May 2021. EFSA J. 2021, 19, e06951. [Google Scholar] [CrossRef] [PubMed]
- Floyd, T.; Banyard, A.C.; Lean, F.Z.X.; Byrne, A.M.P.; Fullick, E.; Whittard, E.; Mollett, B.C.; Bexton, S.; Swinson, V.; Macrelli, M.; et al. Encephalitis and Death in Wild Mammals at a Rehabilitation Center after Infection with Highly Pathogenic Avian Infl Uenza a(H5n8) Virus, United Kingdom. Emerg. Infect. Dis. 2021, 27, 2856–2863. [Google Scholar] [CrossRef]
- Postel, A.; King, J.; Kaiser, F.K.; Kennedy, J.; Lombardo, M.S.; Reineking, W.; de le Roi, M.; Harder, T.; Pohlmann, A.; Gerlach, T.; et al. Infections with Highly Pathogenic Avian Influenza A Virus (HPAIV) H5N8 in Harbor Seals at the German North Sea Coast, 2021. Emerg. Microbes Infect. 2022, 11, 725–729. [Google Scholar] [CrossRef]
- Ivanova, A.O.; Volchkov, P.Y.; Deviatkin, A.A. Concatenation of Segmented Viral Genomes for Reassortment Analysis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Golubkova, A.A.; Platonova, T.A.; Olshvang, O.Y.; Smirnova, S.S.; Kovyazina, S.A. Measles in Yekaterinburg: The historical path from the period before vaccination to the stage of elimination of the infection. Russ. J. Infect. Immun. 2018, 8, 526. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Vakulenko, Y.; Deviatkin, A.; Drexler, J.F.; Lukashev, A. Modular Evolution of Coronavirus Genomes. Viruses 2021, 13, 1270. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- King, J.; Schulze, C.; Engelhardt, A.; Hlinak, A.; Lennermann, S.-L.; Rigbers, K.; Skuballa, J.; Staubach, C.; Mettenleiter, T.C.; Harder, T.; et al. Novel HPAIV H5N8 Reassortant (Clade 2.3.4.4b) Detected in Germany. Viruses 2020, 12, 281. [Google Scholar] [CrossRef] [PubMed]
- Mine, J.; Tsunekuni, R.; Tanikawa, T.; Uchida, Y.; Dubovitskiy, N.; Derko, A.; Sobolev, I.; Shestopalov, A.; Sharshov, K.; Saito, T. Genetics of Japanese H5N8 High Pathogenicity Avian Influenza Viruses Isolated in Winter 2020–2021 and Their Genetic Relationship with Avian Influenza Viruses in Siberia. Transbound Emerg Dis 2022, 69, e2195–e2213. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/search?cond=H5N8 (accessed on 5 December 2023).
- Shin, D.-L.; Siebert, U.; Lakemeyer, J.; Grilo, M.; Pawliczka, I.; Wu, N.-H.; Valentin-Weigand, P.; Haas, L.; Herrler, G. Highly Pathogenic Avian Influenza A(H5N8) Virus in Gray Seals, Baltic Sea. Emerg. Infect. Dis. 2019, 25, 2295–2298. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, B.; Lemey, P.; Dong, L.; Mu, T.; Wiebe, R.A.; Guo, F.; Trovão, N.S.; Park, S.W.; Lewis, N.; et al. Synchrony of Bird Migration with Global Dispersal of Avian Influenza Reveals Exposed Bird Orders. Nat. Commun. 2024, 15, 1126. [Google Scholar] [CrossRef] [PubMed]
- Hayman, D.T.S.; Adisasmito, W.B.; Almuhairi, S.; Behravesh, C.B.; Bilivogui, P.; Bukachi, S.A.; Casas, N.; Becerra, N.C.; Charron, D.F.; Chaudhary, A.; et al. Developing One Health Surveillance Systems. One Health 2023, 17, 100617. [Google Scholar] [CrossRef]
- Joseph, U.; Su, Y.C.F.; Vijaykrishna, D.; Smith, G.J.D. The Ecology and Adaptive Evolution of Influenza A Interspecies Transmission. Influenza Other Respir. Viruses 2017, 11, 74–84. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Goraya, M.; Wang, S.; Chen, J.-L. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef]
- Li, C.; Hatta, M.; Nidom, C.A.; Muramoto, Y.; Watanabe, S.; Neumann, G.; Kawaoka, Y. Reassortment between Avian H5N1 and Human H3N2 Influenza Viruses Creates Hybrid Viruses with Substantial Virulence. Proc. Natl. Acad. Sci. USA 2010, 107, 4687–4692. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degtyarev, E.; Feoktistova, S.; Volchkov, P.; Deviatkin, A. Complex Evolutionary Dynamics of H5N8 Influenza A Viruses Revealed by Comprehensive Reassortment Analysis. Viruses 2024, 16, 1405. https://doi.org/10.3390/v16091405
Degtyarev E, Feoktistova S, Volchkov P, Deviatkin A. Complex Evolutionary Dynamics of H5N8 Influenza A Viruses Revealed by Comprehensive Reassortment Analysis. Viruses. 2024; 16(9):1405. https://doi.org/10.3390/v16091405
Chicago/Turabian StyleDegtyarev, Egor, Sofia Feoktistova, Pavel Volchkov, and Andrey Deviatkin. 2024. "Complex Evolutionary Dynamics of H5N8 Influenza A Viruses Revealed by Comprehensive Reassortment Analysis" Viruses 16, no. 9: 1405. https://doi.org/10.3390/v16091405