
Comparison of Different HIV-1 Resistance Interpretation Tools for Next-Generation Sequencing in Italy

 , , , ,
, , , ,  ,
,  , ,
, ,  , , , , and
on behalf of the Italian HIV NGS network and the ARCA Cohortadd
Show full author list
, , , , and
on behalf of the Italian HIV NGS network and the ARCA Cohortadd
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. RNA Extraction
2.3. Library Preparation for Illumina iSeq100/MiSeq Platforms
2.4. Sequence Data Analysis and Coverage Evaluation
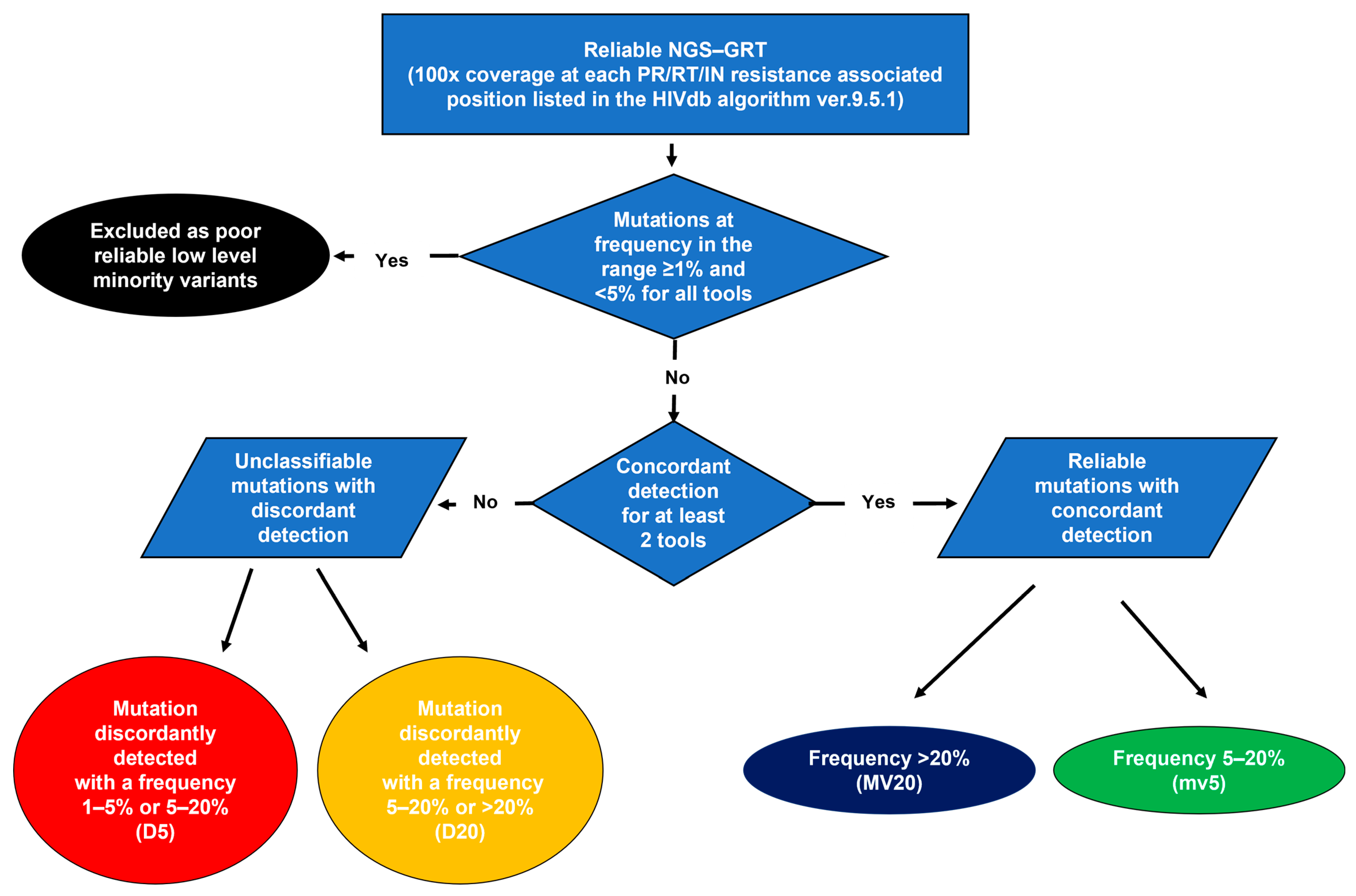
2.5. Resistance Analysis and Variant Detection Concordance
2.6. Statistical Analyses
3. Results
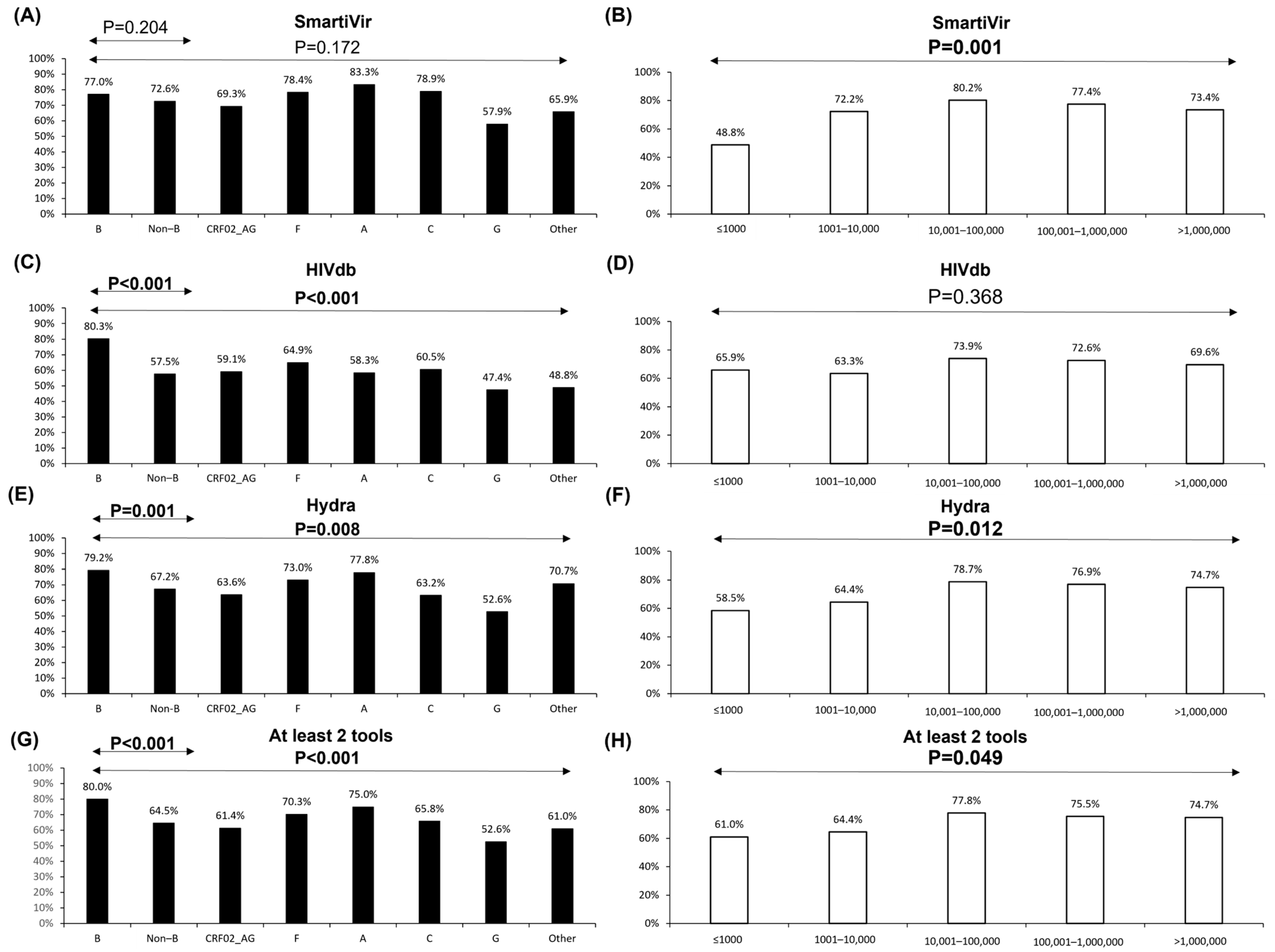
3.1. Samples’ Characteristics and Coverage Reliability Rate
3.2. Resistance Interpretation
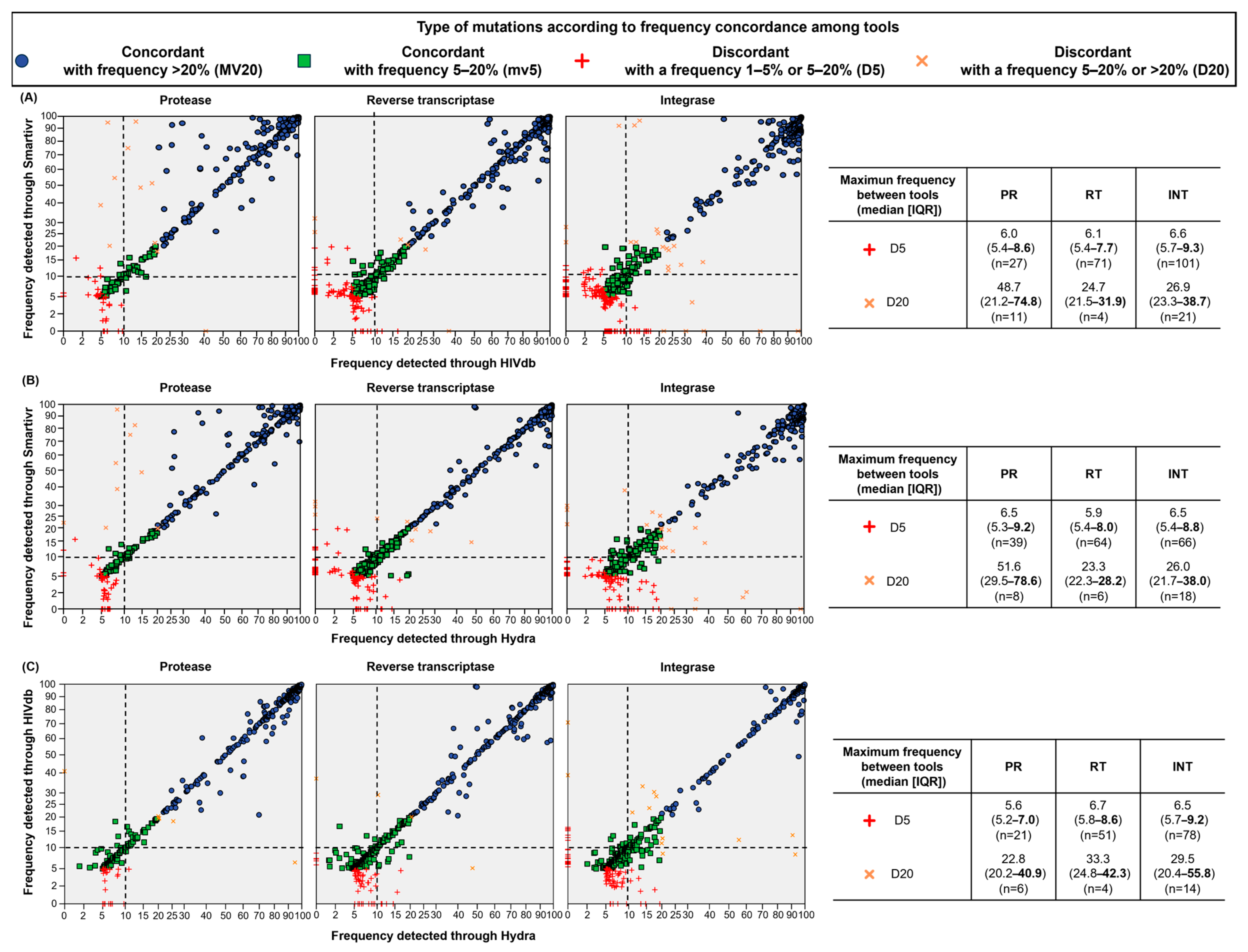
3.3. Concordance in Detecting Mutations at Resistance-Associated Positions of PR/RT/IN
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European AIDS Clinical Society. Guidelines—Version 12.0. 2023. Available online: https://www.eacsociety.org/media/guidelines-12.0.pdf (accessed on 31 July 2024).
- Panel on Antiretroviral Guidelines for Adults and Adolescents, Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV, Department of Health and Human Services. 2024. Available online: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/adult-adolescent-arv/guidelines-adult-adolescent-arv.pdf (accessed on 31 July 2024).
- Parkin, N.T.; Avila-Rios, S.; Bibby, D.F.; Brumme, C.J.; Eshleman, S.H.; Harrigan, P.R.; Howison, M.; Hunt, G.; Ji, H.; Kantor, R.; et al. Multi-Laboratory Comparison of Next-Generation to Sanger-Based Sequencing for HIV-1 Drug Resistance Genotyping. Viruses 2020, 12, 694. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.Y.; Kao, S.W.; Liu, Z.H.; Wu, T.S.; Tsai, C.L.; Lin, H.H.; Wong, W.W.; Chang, Y.Y.; Chen, S.S.; Ku, S.W.W. Shall I Trust the Report? Variable Performance of Sanger Sequencing Revealed by Deep Sequencing on HIV Drug Resistance Mutation Detection. Int. J. Infect. Dis. 2020, 93, 182–191. [Google Scholar] [CrossRef]
- Labate, L.; Bruzzone, B.; Spagnuolo, V.; Zazzi, M.; Santoro, M.; di Biagio, A.; Castagna, A. PRESTIGIO RING: “A 59-year-old HIV-1 positive, highly treatment-experienced woman failing darunavir/ritonavir plus raltegravir”. New Microbiol. 2023, 46, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Chabria, S.B.; Gupta, S.; Kozal, M.J. Deep Sequencing of HIV: Clinical and Research Applications. Annu. Rev. Genom. Hum. Genet. 2014, 15, 295–325. [Google Scholar] [CrossRef]
- Ávila-Ríos, S.; Parkin, N.; Swanstrom, R.; Paredes, R.; Shafer, R.; Ji, H.; Kantor, R. Next-Generation Sequencing for HIV Drug Resistance Testing: Laboratory, Clinical, and Implementation Considerations. Viruses 2020, 12, 617. [Google Scholar] [CrossRef] [PubMed]
- Tzou, P.L.; Ariyaratne, P.; Varghese, V.; Lee, C.; Rakhmanaliev, E.; Villy, C.; Yee, M.; Tan, K.; Michel, G.; Pinsky, B.A.; et al. Comparison of an In Vitro Diagnostic Next-Generation Sequencing Assay with Sanger Sequencing for HIV-1 Genotypic Resistance Testing. J. Clin. Microbiol. 2018, 56, e00105-18. [Google Scholar] [CrossRef] [PubMed]
- Alidjinou, E.K.; Deldalle, J.; Hallaert, C.; Robineau, O.; Ajana, F.; Choisy, P.; Hober, D.; Bocket, L. RNA and DNA Sanger Sequencing versus Next-Generation Sequencing for HIV-1 Drug Resistance Testing in Treatment-Naive Patients. J. Antimicrob. Chemother. 2017, 72, 2823–2830. [Google Scholar] [CrossRef]
- Nicot, F.; Jeanne, N.; Raymond, S.; Delfour, O.; Carcenac, R.; Lefebvre, C.; Sauné, K.; Delobel, P.; Izopet, J. Performance Comparison of Deep Sequencing Platforms for Detecting HIV-1 Variants in the Pol Gene. J. Med. Virol. 2018, 90, 1486–1492. [Google Scholar] [CrossRef]
- Dudley, D.M.; Bailey, A.L.; Mehta, S.H.; Hughes, A.L.; Kirk, G.D.; Westergaard, R.P.; O’Connor, D.H. Cross-Clade Simultaneous HIV Drug Resistance Genotyping for Reverse Transcriptase, Protease, and Integrase Inhibitor Mutations by Illumina MiSeq. Retrovirology 2014, 11, 122. [Google Scholar] [CrossRef]
- Ram, D.; Leshkowitz, D.; Gonzalez, D.; Forer, R.; Levy, I.; Chowers, M.; Lorber, M.; Hindiyeh, M.; Mendelson, E.; Mor, O. Evaluation of GS Junior and MiSeq Next-Generation Sequencing Technologies as an Alternative to Trugene Population Sequencing in the Clinical HIV Laboratory. J. Virol. Methods 2015, 212, 12–16. [Google Scholar] [CrossRef]
- Taylor, T.; Lee, E.R.; Nykoluk, M.; Enns, E.; Liang, B.; Capina, R.; Gauthier, M.K.; Van Domselaar, G.; Sandstrom, P.; Brooks, J.; et al. A MiSeq-HyDRA Platform for Enhanced HIV Drug Resistance Genotyping and Surveillance. Sci. Rep. 2019, 9, 8970. [Google Scholar] [CrossRef] [PubMed]
- Bonifacio, M.A.; Genchi, C.; Lagioia, A.; Talamo, V.; Volpe, A.; Mariggiò, M.A. Analytical Assessment of the Vela Diagnostics NGS Assay for HIV Genotyping and Resistance Testing: The Apulian Experience. Int. J. Mol. Sci. 2022, 23, 2727. [Google Scholar] [CrossRef]
- Brumme, C.J.; Poon, A.F.Y. Promises and Pitfalls of Illumina Sequencing for HIV Resistance Genotyping. Virus Res. 2017, 239, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Casadellà, M.; Paredes, R. Deep Sequencing for HIV-1 Clinical Management. Virus Res. 2017, 239, 69–81. [Google Scholar] [CrossRef]
- Lee, E.R.; Parkin, N.; Jennings, C.; Brumme, C.J.; Enns, E.; Casadellà, M.; Howison, M.; Coetzer, M.; Avila-Rios, S.; Capina, R.; et al. Performance Comparison of next Generation Sequencing Analysis Pipelines for HIV-1 Drug Resistance Testing. Sci. Rep. 2020, 10, 1634. [Google Scholar] [CrossRef]
- Noguera-Julian, M.; Lee, E.R.; Shafer, R.W.; Kantor, R.; Ji, H. Dry Panels Supporting External Quality Assessment Programs for Next Generation Sequencing-Based HIV Drug Resistance Testing. Viruses 2020, 12, 666. [Google Scholar] [CrossRef] [PubMed]
- Tzou, P.L.; Kosakovsky Pond, S.L.; Avila-Rios, S.; Holmes, S.P.; Kantor, R.; Shafer, R.W. Analysis of Unusual and Signature APOBEC-Mutations in HIV-1 Pol Next-Generation Sequences. PLoS ONE 2020, 15, e0225352. [Google Scholar] [CrossRef]
- Pyne, M.T.; Simmon, K.E.; Mallory, M.A.; Hymas, W.C.; Stevenson, J.; Barker, A.P.; Hillyard, D.R. HIV-1 Drug Resistance Assay Using Ion Torrent Next Generation Sequencing and On-Instrument End-to-End Analysis Software. J. Clin. Microbiol. 2022, 60, e00253-22. [Google Scholar] [CrossRef]
- Balakrishna, S.; Loosli, T.; Zaheri, M.; Frischknecht, P.; Huber, M.; Kusejko, K.; Yerly, S.; Leuzinger, K.; Perreau, M.; Ramette, A.; et al. Frequency Matters: Comparison of Drug Resistance Mutation Detection by Sanger and Next-Generation Sequencing in HIV-1. J. Antimicrob. Chemother. 2023, 78, 656–664. [Google Scholar] [CrossRef]
- Weber, J.; Volkova, I.; Sahoo, M.K.; Tzou, P.L.; Shafer, R.W.; Pinsky, B.A. Prospective Evaluation of the Vela Diagnostics Next-Generation Sequencing Platform for HIV-1 Genotypic Resistance Testing. J. Mol. Diagn. 2019, 21, 961. [Google Scholar] [CrossRef]
- Mohamed, S.; Boulmé, R.; Sayada, C. From Capillary Electrophoresis to Deep Sequencing: An Improved HIV-1 Drug Resistance Assessment Solution Using In Vitro Diagnostic (IVD) Assays and Software. Viruses 2023, 15, 571. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Serna, A.; Min, J.E.; Woods, C.; Chan, D.; Lima, V.D.; Montaner, J.S.; Harrigan, P.R.; Swenson, L.C. Performance of HIV-1 drug resistance testing at low-level viremia and its ability to predict future virologic outcomes and viral evolution in treatment-naive individuals. Clin. Infect. Dis. 2014, 58, 1165–1173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Santoro, M.M.; Fabeni, L.; Armenia, D.; Alteri, C.; Di Pinto, D.; Forbici, F.; Bertoli, A.; Di Carlo, D.; Gori, C.; Carta, S.; et al. Reliability and clinical relevance of the HIV-1 drug resistance test in patients with low viremia levels. Clin. Infect. Dis. 2014, 58, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Swenson, L.C.; Min, J.E.; Woods, C.K.; Cai, E.; Li, J.Z.; Montaner, J.S.; Harrigan, P.R.; Gonzalez-Serna, A. HIV drug resistance detected during low-level viraemia is associated with subsequent virologic failure. AIDS 2014, 28, 1125–1134. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Williams, A.; Menon, S.; Crowe, M.; Agarwal, N.; Biccler, J.; Bbosa, N.; Ssemwanga, D.; Adungo, F.; Moecklinghoff, C.; Macartney, M.; et al. Geographic and Population Distributions of Human Immunodeficiency Virus (HIV)-1 and HIV-2 Circulating Subtypes: A Systematic Literature Review and Meta-Analysis (2010–2021). J. Infect. Dis. 2023, 228, 1583–1591. [Google Scholar] [CrossRef]
- Fabeni, L.; Armenia, D.; Abbate, I.; Gagliardini, R.; Mazzotta, V.; Bertoli, A.; Gennari, W.; Forbici, F.; Berno, G.; Piermatteo, L.; et al. HIV-1 Transmitted Drug Resistance in Newly Diagnosed Individuals in Italy over the Period 2015–21. J. Antimicrob. Chemother. 2024, dkae189. [Google Scholar] [CrossRef]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV Subtype Diversity Worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160. [Google Scholar] [CrossRef]
- Wu, F.; Simonetti, F.R. Learning from Persistent Viremia: Mechanisms and Implications for Clinical Care and HIV-1 Cure. Curr. HIV/AIDS Rep. 2023, 20, 428–439. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Overall (n = 629) |
|---|---|
| Age, years, Median (IQR) | 44 (34–53) |
| Year of genotyping, n (%) | |
| <2020 | 28 (4.4) |
| 2020–2021 | 104 (16.5) |
| 2022–2023 | 497 (79.1) |
| Plasma HIV-1 RNA, log (copies/mL), Median (IQR) | 4.9 (4.2–5.6) |
| ≤1000 copies/mL, n (%) | 41 (6.5) |
| 1001–10,000 copies/mL, n (%) | 90 (14.3) |
| 10,001–100,000 copies/mL, n (%) | 207 (32.9) |
| 100,001–1,000,000 copies/mL, n (%) | 212 (33.7) |
| >1,000,000 copies/mL, n (%) | 79 (12.6) |
| HIV-1Subtype, n (%) 1 | |
| B | 370 (58.8) |
| CRF02_AG | 88 (14.0) |
| F | 37 (5.9) |
| C | 36 (5.7) |
| A | 38 (6.0) |
| G | 19 (3.8) |
| Others 2 | 41 (6.5) |
| Treatment status, n (%) | |
| Naive | 313 (49.8) |
| Experienced | 152 (24.2) |
| Unknown | 164 (26.1) |
| Platform used for NGS | |
| iSeq100 | 485 (77.1) |
| MiSeq | 144 (22.9) |
| Variables | Odd Ratio to Have Reliable Coverage for Resistance Interpretation (100X Coverage at All PR/RT/IN Resistance Positions) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| At Least 2 Tools | SmartVir | HIVdb | Hydra | |||||||||||||
| Crude | Adjusted 1 | Crude | Adjusted 1 | Crude | Adjusted 1 | Crude | Adjusted 1 | |||||||||
| OR (95% C.I.) | p Value | OR (95% C.I.) | p Value | OR (95% C.I.) | p Value | OR (95% C.I.) | p Value | OR (95% C.I.) | p Value | OR (95% C.I.) | p Value | OR (95% C.I.) | p Value | OR (95% C.I.) | p Value | |
| Subtype | ||||||||||||||||
| B 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| CRF02_AG | 0.4 (0.2–0.7) | <0.001 | 0.4 (0.2–0.6) | <0.001 | 0.7 (0.4–1.1) | 0.132 | 0.6 (0.3–1.0) | 0.051 | 0.4 (0.2–0.6) | <0.001 | 0.4 (0.2–0.6) | <0.001 | 0.5 (0.3–0.8) | 0.002 | 0.4 (0.2–0.7) | 0.001 |
| F | 0.6 (0.3–1.3) | 0.169 | 0.5 (0.3–1.2) | 0.11 | 1.1 (0.5–2.5) | 0.852 | 1.1 (0.5–2.5) | 0.892 | 0.5 (0.2–0.9) | 0.032 | 0.5 (0.2–1) | 0.043 | 0.7 (0.3–1.5) | 0.381 | 0.7 (0.3–1.4) | 0.293 |
| A | 0.8 (0.3–1.7) | 0.479 | 0.7 (0.3–1.6) | 0.414 | 1.5 (0.6–3.7) | 0.389 | 1.3 (0.5–3.4) | 0.541 | 0.3 (0.2–0.7) | 0.003 | 0.3 (0.2–0.7) | 0.003 | 0.9 (0.4–2.1) | 0.843 | 0.9 (0.4–2) | 0.764 |
| C | 0.5 (0.2–1) | 0.045 | 0.5 (0.2–1) | 0.052 | 1.1 (0.5–2.5) | 0.788 | 1.1 (0.5–2.7) | 0.752 | 0.4 (0.2–0.8) | 0.006 | 0.4 (0.2–0.8) | 0.013 | 0.5 (0.2–0.9) | 0.027 | 0.4 (0.2–0.9) | 0.025 |
| G | 0.3 (0.1–0.7) | 0.007 | 0.3 (0.1–0.7) | 0.005 | 0.4 (0.2–1.1) | 0.064 | 0.4 (0.1–1.0) | 0.040 | 0.2 (0.1–0.6) | 0.002 | 0.2 (0.1–0.5) | 0.001 | 0.3 (0.1–0.7) | 0.010 | 0.3 (0.1–0.7) | 0.009 |
| Others | 0.4 (0.2–0.8) | 0.007 | 0.3 (0.2–0.7) | 0.002 | 0.6 (0.3–1.1) | 0.116 | 0.5 (0.2–0.9) | 0.028 | 0.2 (0.1–0.5) | <0.001 | 0.2 (0.1–0.5) | <0.001 | 0.6 (0.3–1.3) | 0.215 | 0.5 (0.2–1.1) | 0.079 |
| Platform used | ||||||||||||||||
| Illumina Iseq100 2 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||
| Illumina Miseq | 1.5 (0.9–2.3) | 0.086 | 1.6 (1–2.5) | 0.065 | 0.9 (0.6–1.4) | 0.615 | - | - | 2.1 (1.3–3.3) | 0.002 | 2.1 (1.2–3.6) | 0.008 | 1.2 (0.8–1.9) | 0.376 | - | - |
| Viremia (copies/mL) | ||||||||||||||||
| ≤1000 | 0.5 (0.3–1) | 0.058 | 0.4 (0.2–0.7) | 0.005 | 0.3 (0.1–0.6) | <0.001 | 0.3 (0.1–0.5) | <0.001 | 0.7 (0.4–1.5) | 0.379 | - | - | 0.4 (0.2–0.9) | 0.016 | 0.3 (0.2–0.7) | 0.004 |
| 1001–10,000 | 0.6 (0.3–1) | 0.052 | 0.5 (0.3–0.9) | 0.014 | 0.8 (0.4–1.3) | 0.341 | 0.7 (0.4–1.3) | 0.279 | 0.7 (0.4–1.1) | 0.108 | - | - | 0.5 (0.3–0.9) | 0.027 | 0.5 (0.3–0.9) | 0.012 |
| 10,001–100,000 | 1.1 (0.7–1.8) | 0.577 | 1.1 (0.7–1.7) | 0.821 | 1.2 (0.7–1.9) | 0.478 | 1.2 (0.7–1.9) | 0.559 | 1.1 (0.7–1.6) | 0.769 | - | - | 1.1 (0.7–1.8) | 0.647 | 1 (0.6–1.7) | 0.901 |
| 100,001–1,000,000 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| >1,000,000 | 1 (0.5–1.7) | 0.89 | 1 (0.5–1.8) | 0.874 | 0.8 (0.4–1.5) | 0.483 | 0.8 (0.4–1.4) | 0.383 | 0.9 (0.5–1.5) | 0.611 | – | – | 0.9 (0.5–1.6) | 0.694 | 0.9 (0.5–1.6) | 0.629 |
| Treatment status | ||||||||||||||||
| cART naïve 2 | 1 | 1 | 1 | 1 | ||||||||||||
| cART experienced | 1.3 (0.8–2) | 0.249 | - | - | 1.0 (0.6–1.5) | 0.873 | 1.1 (0.7–1.9) | 0.622 | 1.7 (1.1–2.7) | 0.015 | 1.4 (0.9–2.2) | 0.192 | 1.2 (0.8–1.9) | 0.377 | - | - |
| Unknown | 1.3 (0.8–1.9) | 0.31 | - | - | 0.6 (0.4–1.0) | 0.038 | 0.7 (0.4–1.1) | 0.101 | 1.6 (1.0–2.4) | 0.040 | 0.9 (0.6–1.6) | 0.829 | 1 (0.7–1.6) | 0.885 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armenia, D.; Carioti, L.; Micheli, V.; Bon, I.; Allice, T.; Bonura, C.; Bruzzone, B.; Bracchitta, F.; Cerutti, F.; Giammanco, G.M.; et al. Comparison of Different HIV-1 Resistance Interpretation Tools for Next-Generation Sequencing in Italy. Viruses 2024, 16, 1422. https://doi.org/10.3390/v16091422
Armenia D, Carioti L, Micheli V, Bon I, Allice T, Bonura C, Bruzzone B, Bracchitta F, Cerutti F, Giammanco GM, et al. Comparison of Different HIV-1 Resistance Interpretation Tools for Next-Generation Sequencing in Italy. Viruses. 2024; 16(9):1422. https://doi.org/10.3390/v16091422
Chicago/Turabian StyleArmenia, Daniele, Luca Carioti, Valeria Micheli, Isabella Bon, Tiziano Allice, Celestino Bonura, Bianca Bruzzone, Fiorenza Bracchitta, Francesco Cerutti, Giovanni Maurizio Giammanco, and et al. 2024. "Comparison of Different HIV-1 Resistance Interpretation Tools for Next-Generation Sequencing in Italy" Viruses 16, no. 9: 1422. https://doi.org/10.3390/v16091422
APA StyleArmenia, D., Carioti, L., Micheli, V., Bon, I., Allice, T., Bonura, C., Bruzzone, B., Bracchitta, F., Cerutti, F., Giammanco, G. M., Stefanelli, F., Bonifacio, M. A., Bertoli, A., Vatteroni, M., Ibba, G., Novazzi, F., Lipsi, M. R., Cuomo, N., Vicenti, I., ... Santoro, M. M., on behalf of the Italian HIV NGS network and the ARCA Cohort. (2024). Comparison of Different HIV-1 Resistance Interpretation Tools for Next-Generation Sequencing in Italy. Viruses, 16(9), 1422. https://doi.org/10.3390/v16091422