
Bioinformatics Goes Viral: I. Databases, Phylogenetics and Phylodynamics Tools for Boosting Virus Research


Abstract
:1. Bioinformatics: From Pioneering Tools to Current, Reliable Toolbox
1.1. Database and Tool Design: Genomics Make Them Bigger and Better
1.2. From Big Data to Huge Data: Exploring the Planetary Virome in the Petabase Era
1.3. From Sequence Alignment to Evolutionary Studies and Functional Inference
2. Bio-Databases
2.1. Sequence Databases
2.1.1. Open-Source Databases
2.1.2. Registered Users Databases
2.2. Protein-Protein Interaction (PPI) Databases
2.2.1. Generic PPI Databases
BioGRID
IntAct
MINT
2.2.2. Virus–Host PPI Databases
VirusMentha
VirHostNet
HPIDB
Viruses.STRING
HCVpro
PHISTO
HVIDB
3. Phylogenetics and Phylodynamics Tools
3.1. RAxML
3.2. BEAST
3.3. IQ-TREE

3.4. Nextstrain
3.5. PhyloGeoTool
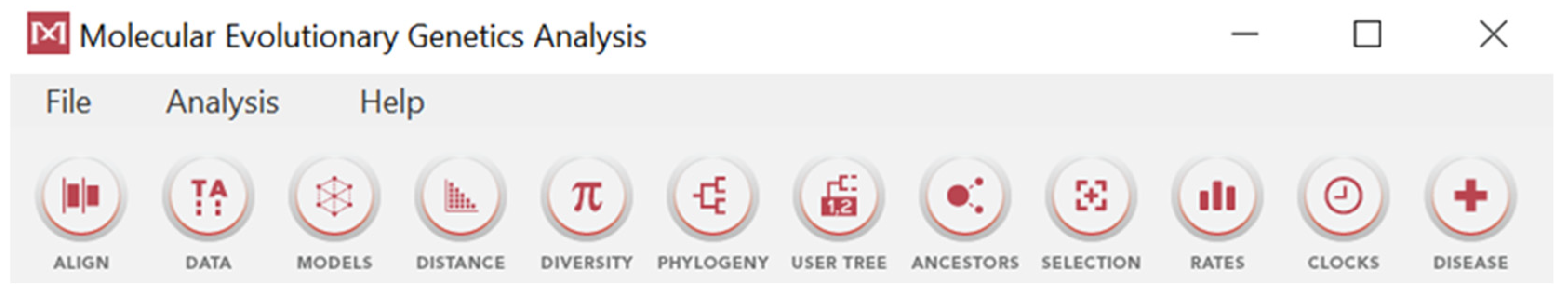
3.6. MEGA
- DATA: users can import and explore datasets of sequences in different formats through this option;
- MODELS: enables the construction of various mutation models, with possible automatic selection of the best algorithm;
- DISTANCE: allows the calculation of pairwise or overall mean distances within sequence datasets;
- DIVERSITY: enables the calculation of the diversity within an entire group or a previously defined subgroup;
- PHYLOGENY: users can build phylogenetic trees from the downloaded sequences using methods such as NJ or ML, possibly adding bootstrap analysis;
- USER TREE: allows the submission and analysis of existing tree files using different methods like ML or Parsimony;
- ANCESTORS: infers ancestral sequences from a tree file using ML or parsimony;
- SELECTION: performs analyses on SNPs, including Tajima’s test of neutrality [164];
- RATES: calculates the nucleotide substitution rates in sequence alignments, using position-by-position rates (ML) or Gamma rates [165];
- DISEASE: assesses the impact of non-synonymous mutations on health using MEGA Molecular Diagnoses, with the possibility to access pre-computed diagnoses.
3.7. Virus Pop
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hagen, J.B. The origins of bioinformatics. Nat. Rev. Genet. 2000, 1, 231–236. [Google Scholar] [CrossRef]
- Mullis, K.B.; Faloona, F.A. Specific Synthesis of DNA in Vitro via a Polymerase-Catalyzed Chain Reaction. Methods Enzymol. 1987, 155, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Vincent, A.T.; Charette, S.J.; Derome, N. A brief history of bioinformatics. Brief. Bioinform. 2019, 20, 1981–1996. [Google Scholar] [CrossRef] [PubMed]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef] [PubMed]
- Marz, M.; Beerenwinkel, N.; Drosten, C.; Fricke, M.; Frishman, D.; Hofacker, I.L.; Hoffmann, D.; Middendorf, M.; Rattei, T.; Stadler, P.F.; et al. Challenges in RNA virus bioinformatics. Bioinformatics 2014, 30, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Kapli, P.; Yang, Z.; Telford, M.J. Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 2020, 21, 428–444. [Google Scholar] [CrossRef]
- König, S.; Romoth, L.; Stanke, M. Comparative Genome Annotation. Methods Mol. Biol. 2018, 1704, 189–212. [Google Scholar] [CrossRef]
- Wang, C.; Kurgan, L. Review and comparative assessment of similarity-based methods for prediction of drug-protein interactions in the druggable human proteome. Brief. Bioinform. 2019, 20, 2066–2087. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; O’Sullivan, C.; Karsch-Mizrachi, I. Using GenBank and SRA. Methods Mol. Biol. 2022, 2443, 1–25. [Google Scholar] [CrossRef]
- GenBank and WGS Statistics. Available online: https://www.ncbi.nlm.nih.gov/genbank/statistics/ (accessed on 28 January 2024).
- Thakur, M.; Buniello, A.; Brooksbank, C.; Gurwitz, K.T.; Hall, M.; Hartley, M.; Hulcoop, D.G.; Leach, A.R.; Marques, D.; Martin, M.; et al. EMBL’s European Bioinformatics Institute (EMBL-EBI) in 2023. Nucleic Acids Res. 2024, 52, D10–D17. [Google Scholar] [CrossRef]
- Edgar, R.C.; Taylor, B.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Serratus. Available online: https://serratus.io (accessed on 19 August 2024).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Ladunga, I. Finding Homologs in Amino Acid Sequences Using Network BLAST Searches. Curr. Protoc. Bioinform. 2017, 59, 3.4.1–3.4.24. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. BLAST and FASTA similarity searching for multiple sequence alignment. Methods Mol. Biol. 2014, 1079, 75–101. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Kim, Y.; Koyutürk, M.; Topkara, U.; Grama, A.; Subramaniam, S. Inferring functional information from domain co-evolution. Bioinformatics 2006, 22, 40–49. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Bernasconi, A.; Canakoglu, A.; Masseroli, M.; Pinoli, P.; Ceri, S. A review on viral data sources and search systems for perspective mitigation of COVID-19. Brief. Bioinform. 2021, 22, 664–675. [Google Scholar] [CrossRef]
- Jangra, R.K.; Llabrés, M.; Guardado-Calvo, P.; Mittler, E.; Lasso, G. Editorial: Influence of Protein-Protein Interactions (PPIs) on the Outcome of Viral Infections. Front. Microbiol. 2022, 13, 943379. [Google Scholar] [CrossRef]
- INSDC. Available online: https://www.insdc.org/ (accessed on 28 May 2024).
- Tanizawa, Y.; Fujisawa, T.; Kodama, Y.; Kosuge, T.; Mashima, J.; Tanjo, T.; Nakamura, Y. DNA Data Bank of Japan (DDBJ) update report 2022. Nucleic Acids Res. 2023, 51, D101–D105. [Google Scholar] [CrossRef]
- Ara, T.; Kodama, Y.; Tokimatsu, T.; Fukuda, A.; Kosuge, T.; Mashima, J.; Tanizawa, Y.; Tanjo, T.; Ogasawara, O.; Fujisawa, T.; et al. DDBJ update in 2023: The MetaboBank for metabolomics data and associated metadata. Nucleic Acids Res. 2024, 52, D67–D71. [Google Scholar] [CrossRef] [PubMed]
- European Nucleotide Archive. Available online: https://www.ebi.ac.uk/ena (accessed on 29 May 2024).
- Yuan, D.; Ahamed, A.; Burgin, J.; Cummins, C.; Devraj, R.; Gueye, K.; Gupta, D.; Gupta, V.; Haseeb, M.; Ihsan, M.; et al. The European Nucleotide Archive in 2023. Nucleic Acids Res. 2024, 52, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Pathogens. Available online: https://www.ebi.ac.uk/ena/pathogens/ (accessed on 29 May 2024).
- UniProt. Available online: https://www.uniprot.org/ (accessed on 29 May 2024).
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H. Protein sequence databases. Curr. Opin. Chem. Biol. 2004, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Brister, J.R.; Chan, J.; Comeau, D.C.; Connor, R.; DiCuccio, M.; Farrell, C.M.; Feldgarden, M.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2024, 52, D33–D43. [Google Scholar] [CrossRef] [PubMed]
- NCBI-NLM-NIH. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 29 May 2024).
- Wang, Z.; Lachmann, A.; Ma’ayan, A. Mining data and metadata from the gene expression omnibus. Biophys. Rev. 2019, 1, 103–110. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource—Improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- NCBI Virus. Available online: https://www.ncbi.nlm.nih.gov/labs/virus (accessed on 30 May 2024).
- Influenza Virus Resource. Available online: https://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi?go=database (accessed on 30 May 2024).
- Zhao, M.; Chen, J.; Wang, Q.; Lu, Z.; Jia, Z. A Landscape Analysis on Virus: Based on NCBI Database. China CDC Wkly. 2022, 4, 120–125. [Google Scholar] [CrossRef]
- CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res. 2023, 51, D18–D28. [Google Scholar] [CrossRef]
- Genome Sequence Archive (GSA). Available online: https://ngdc.cncb.ac.cn/gsa/ (accessed on 8 July 2024).
- MpoxVR. Available online: https://ngdc.cncb.ac.cn/gwh/poxvirus/?lang=en (accessed on 6 July 2024).
- ViMIC. Available online: http://bmtongji.cn/ViMIC/index.php (accessed on 7 July 2024).
- Wang, Y.; Tong, Y.; Zhang, Z.; Zheng, R.; Huang, D.; Yang, J.; Zong, H.; Tan, F.; Xie, Y.; Huang, H.; et al. ViMIC: A database of human disease-related virus mutations, integration sites and cis-effects. Nucleic Acids Res. 2022, 50, D918–D927. [Google Scholar] [CrossRef]
- VirusDB. Available online: http://yaulab.math.tsinghua.edu.cn/VirusDB/ (accessed on 8 July 2024).
- Dong, R.; Zheng, H.; Tian, K.; Yau, S.C.; Mao, W.; Yu, W.; Yin, C.; Yu, C.; He, R.L.; Yang, J.; et al. Virus Database and Online Inquiry System Based on Natural Vectors. Evol. Bioinform. Online 2017, 13, 1176934317746667. [Google Scholar] [CrossRef]
- IVDB. Available online: https://ngdc.cncb.ac.cn/databasecommons/database/id/1557 (accessed on 8 July 2024).
- Chang, S.; Zhang, J.; Liao, X.; Zhu, X.; Wang, D.; Zhu, J.; Feng, T.; Zhu, B.; Gao, G.F.; Wang, J.; et al. Influenza Virus Database (IVDB): An integrated information resource and analysis platform for influenza virus research. Nucleic Acids Res. 2007, 35, D376–D380. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Dong, R.; He, R.L.; Yau, S.S. A novel alignment-free method for HIV-1 subtype classification. Infect. Genet. Evol. 2020, 77, 104080. [Google Scholar] [CrossRef] [PubMed]
- HIV Sequence Database. Available online: https://www.hiv.lanl.gov/content/index (accessed on 8 July 2024).
- De Castro, E.; Hulo, C.; Masson, P.; Auchincloss, A.; Bridge, A.; Le Mercier, P. ViralZone 2024 provides higher-resolution images and advanced virus-specific resources. Nucleic Acids Res. 2024, 52, D817–D821. [Google Scholar] [CrossRef]
- ViralZone. Available online: https://viralzone.expasy.org/ (accessed on 8 July 2024).
- GISAID. Available online: https://gisaid.org/ (accessed on 28 May 2024).
- Bogner, P.; Capua, I.; Lipman, D.J. A global initiative on sharing avian flu data. Nature 2006, 442, 981. [Google Scholar] [CrossRef]
- Lenharo, M. GISAID in crisis: Can the controversial COVID genome database survive? Nature 2023, 617, 455–457. [Google Scholar] [CrossRef] [PubMed]
- NextStrain. Available online: https://nextstrain.org/ncov (accessed on 28 May 2024).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Microreact. Available online: https://microreact.org/project/COVID-19 (accessed on 28 May 2024).
- Argimón, S.; Abudahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genom. 2016, 2, e000093. [Google Scholar] [CrossRef]
- CoVsurver. Available online: https://corona.bii.a-star.edu.sg/ (accessed on 28 May 2024).
- FluSurver. Available online: http://flusurver.bii.a-star.edu.sg/ (accessed on 28 May 2024).
- COVID-19 Genome Tracker. Available online: http://cov.genometracker.org/ (accessed on 28 May 2024).
- Akther, S.; Bezrucenkovas, E.; Sulkow, B.; Panlasigui, C.; Li, L.; Qiu, W.; Lia, D. CoV Genome Tracker: Tracing genomic footprints of COVID-19 pandemic. bioRxiv 2020. [Google Scholar] [CrossRef]
- BV-BCR. Available online: https://www.bv-brc.org/ (accessed on 8 July 2024).
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Yang, X.; Yang, S.; Zhang, Z. Current status and future perspectives of computational studies on human-virus protein-protein interactions. Brief. Bioinform. 2021, 22, bbab029. [Google Scholar] [CrossRef] [PubMed]
- Righetto, I.; Milani, A.; Cattoli, G.; Filippini, F. Comparative structural analysis of haemagglutinin proteins from type A influenza viruses: Conserved and variable features. BMC Bioinform. 2014, 15, 363. [Google Scholar] [CrossRef] [PubMed]
- Heidari, A.; Righetto, I.; Filippini, F. Electrostatic Variation of Haemagglutinin as a Hallmark of the Evolution of Avian Influenza Viruses. Sci. Rep. 2018, 8, 1929. [Google Scholar] [CrossRef]
- Righetto, I.; Filippini, F. Normal modes analysis and surface electrostatics of haemagglutinin proteins as fingerprints for high pathogenic type A influenza viruses. BMC Bioinform. 2020, 21 (Suppl. S10), 354. [Google Scholar] [CrossRef]
- Valiente, G. The Landscape of Virus-Host Protein-Protein Interaction Databases. Front. Microbiol. 2022, 13, 827742. [Google Scholar] [CrossRef]
- BioGRID. Available online: https://www.thebiogrid.org/ (accessed on 30 May 2024).
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Cytoscape. Available online: https://cytoscape.org/ (accessed on 30 May 2024).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- NDEx. Available online: https://ndexbio.org/ (accessed on 31 May 2024).
- Pillich, R.T.; Chen, J.; Rynkov, V.; Welker, D.; Pratt, D. NDEx: A Community Resource for Sharing and Publishing of Biological Networks. Methods Mol. Biol. 2017, 1558, 271–301. [Google Scholar] [CrossRef]
- esyN. Available online: https://www.esyn.org/ (accessed on 31 May 2024).
- Bean, D.M.; Heimbach, J.; Ficorella, L.; Micklem, G.; Oliver, S.G.; Favrin, G. esyN: Network building, sharing and publishing. PLoS ONE 2014, 9, e106035. [Google Scholar] [CrossRef]
- IntAct. Available online: https://www.ebi.ac.uk/intact/home (accessed on 31 May 2024).
- Orchard, S.; Salwinski, L.; Kerrien, S.; Montecchi-Palazzi, L.; Oesterheld, M.; Stümpflen, V.; Ceol, A.; Chatr-aryamontri, A.; Armstrong, J.; Woollard, P.; et al. The minimum information required for reporting a molecular interaction experiment (MIMIx). Nat. Biotechnol. 2007, 25, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Del Toro, N.; Shrivastava, A.; Ragueneau, E.; Meldal, B.; Combe, C.; Barrera, E.; Perfetto, L.; How, K.; Ratan, P.; Shirodkar, G.; et al. The IntAct database: Efficient access to fine-grained molecular interaction data. Nucleic Acids Res. 2022, 50, D648–D653. [Google Scholar] [CrossRef] [PubMed]
- Meldal, B.H.M.; Perfetto, L.; Combe, C.; Lubiana, T.; Ferreira Cavalcante, J.V.; Bye-A-Jee, H.; Waagmeester, A.; Del-Toro, N.; Shrivastava, A.; Barrera, E.; et al. Complex Portal 2022: New curation frontiers. Nucleic Acids Res. 2022, 50, D578–D586. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Y.; Gupta, S.; Paramo, M.I.; Hou, Y.; Mao, C.; Luo, Y.; Judd, J.; Wierbowski, S.; Bertolotti, M.; et al. A comprehensive SARS-CoV-2-human protein-protein interactome reveals COVID-19 pathobiology and potential host therapeutic targets. Nat. Biotechnol. 2023, 41, 128–139. [Google Scholar] [CrossRef]
- MINT. Available online: https://mint.bio.uniroma2.it/ (accessed on 31 May 2024).
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef]
- Chatr-aryamontri, A.; Ceol, A.; Peluso, D.; Nardozza, A.; Panni, S.; Sacco, F.; Tinti, M.; Smolyar, A.; Castagnoli, L.; Vidal, M.; et al. VirusMINT: A viral protein interaction database. Nucleic Acids Res. 2009, 37, D669–D673. [Google Scholar] [CrossRef]
- VirusMentha. Available online: https://virusmentha.uniroma2.it/ (accessed on 31 May 2024).
- Calderone, A.; Licata, L.; Cesareni, G. VirusMentha: A new resource for virus-host protein interactions. Nucleic Acids Res. 2015, 43, D588–D592. [Google Scholar] [CrossRef]
- HIV Human Interaction Database. Available online: http://www.ncbi.nlm.nih.gov/projects/RefSeq/HIVInteractions/ (accessed on 31 May 2024).
- Pinney, J.W.; Dickerson, J.E.; Fu, W.; Sanders-Beer, B.E.; Ptak, R.G.; Robertson, D.L. HIV-host interactions: A map of viral perturbation of the host system. AIDS 2009, 23, 549–554. [Google Scholar] [CrossRef]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef]
- Chautard, E.; Fatoux-Ardore, M.; Ballut, L.; Thierry-Mieg, N.; Ricard-Blum, S. MatrixDB, the extracellular matrix interaction database. Nucleic Acids Res. 2011, 39, D235–D240. [Google Scholar] [CrossRef] [PubMed]
- Nathan, K.G.; Lal, S.K. The Multifarious Role of 14-3-3 Family of Proteins in Viral Replication. Viruses 2020, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Guirimand, T.; Delmotte, S.; Navratil, V. VirHostNet 2.0: Surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015, 43, D583–D587. [Google Scholar] [CrossRef]
- VirHostNet 3.0. Available online: https://virhostnet.prabi.fr/ (accessed on 31 May 2024).
- Ashraf, U.; Benoit-Pilven, C.; Navratil, V.; Ligneau, C.; Fournier, G.; Munier, S.; Sismeiro, O.; Coppée, J.Y.; Lacroix, V.; Naffakh, N. Influenza virus infection induces widespread alterations of host cell splicing. NAR Genom. Bioinform. 2020, 2, lqaa095. [Google Scholar] [CrossRef]
- HPIDB. Available online: https://hpidb.igbb.msstate.edu/index.html (accessed on 31 May 2024).
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. HPIDB 2.0: A curated database for host-pathogen interactions. Database 2016, 2016, baw103. [Google Scholar] [CrossRef]
- Krishnan, D.; Babu, S.; Raju, R.; Veettil, M.V.; Prasad, T.S.K.; Abhinand, C.S. Epstein-Barr Virus: Human Interactome Reveals New Molecular Insights into Viral Pathogenesis for Potential Therapeutics and Antiviral Drug Discovery. OMICS 2024, 28, 32–44. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- STRING Viruses. Available online: http://viruses.string-db.org/ (accessed on 31 May 2024).
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Cook, H.V.; Doncheva, N.T.; Szklarczyk, D.; von Mering, C.; Jensen, L.J. Viruses.STRING: A Virus-Host Protein-Protein Interaction Database. Viruses 2018, 10, 519. [Google Scholar] [CrossRef]
- Rouka, E.; Hatzoglou, C.; Gourgoulianis, K.I.; Zarogiannis, S.G. Interactome networks between the human respiratory syncytial virus (HRSV), the human metapneumovirus (HMPV), and their host: In silico investigation and comparative functional enrichment analysis. Microb. Pathog. 2020, 141, 104000. [Google Scholar] [CrossRef] [PubMed]
- Kwofie, S.K.; Schaefer, U.; Sundararajan, V.S.; Bajic, V.B.; Christoffels, A. HCVpro: Hepatitis C virus protein interaction database. Infect. Genet. Evol. 2011, 11, 1971–1977. [Google Scholar] [CrossRef]
- Durmuş Tekir, S.; Çakır, T.; Ardiç, E.; Sayılırbaş, A.S.; Konuk, G.; Konuk, M.; Sarıyer, H.; Uğurlu, A.; Karadeniz, İ.; Özgür, A.; et al. PHISTO: Pathogen-host interaction search tool. Bioinformatics 2013, 29, 1357–1358. [Google Scholar] [CrossRef] [PubMed]
- PHISTO. Available online: https://phisto.org/ (accessed on 31 May 2024).
- Durmuş Tekir, S.; Cakir, T.; Ulgen, K.Ö. Infection Strategies of Bacterial and Viral Pathogens through Pathogen-Human Protein-Protein Interactions. Front. Microbiol. 2012, 3, 46. [Google Scholar] [CrossRef]
- HVIDB. Available online: http://zzdlab.com/hvidb/ (accessed on 31 May 2024).
- CORUM. Available online: http://mips.helmholtz-muenchen.de/corum/ (accessed on 31 May 2024).
- Tsitsiridis, G.; Steinkamp, R.; Giurgiu, M.; Brauner, B.; Fobo, G.; Frishman, G.; Montrone, C.; Ruepp, A. CORUM: The comprehensive resource of mammalian protein complexes—2022. Nucleic Acids Res. 2023, 51, D539–D545. [Google Scholar] [CrossRef] [PubMed]
- Hu.MAP. Available online: http://hu.proteincomplexes.org/ (accessed on 31 May 2024).
- Drew, K.; Wallingford, J.B.; Marcotte, E.M. hu.MAP 2.0: Integration of over 15,000 proteomic experiments builds a global compendium of human multiprotein assemblies. Mol. Syst. Biol. 2021, 17, e10016. [Google Scholar] [CrossRef]
- Yang, X.; Lian, X.; Fu, C.; Wuchty, S.; Yang, S.; Zhang, Z. HVIDB: A comprehensive database for human-virus protein-protein interactions. Brief. Bioinform. 2021, 22, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Abdurakhmonov, I.Y. (Ed.) Bioinformatics: Basics, Development, and Future, Bioinformatics. In Bioinformatics—Updated Features and Applications; Intech Open: London, UK, 2016. [Google Scholar] [CrossRef]
- Nicoll, C.R.; Bailleul, G.; Fiorentini, F.; Mascotti, M.L.; Fraaije, M.W.; Mattevi, A. Ancestral-sequence reconstruction unveils the structural basis of function in mammalian FMOs. Nat. Struct. Mol. Biol. 2020, 27, 14–24. [Google Scholar] [CrossRef]
- Theys, K.; Lemey, P.; Vandamme, A.M.; Baele, G. Advances in Visualization Tools for Phylogenomic and Phylodynamic Studies of Viral Diseases. Front. Public Health 2019, 7, 208. [Google Scholar] [CrossRef]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Lemey, P. Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 2011, 1, 423–429. [Google Scholar] [CrossRef]
- Minin, V.N.; Suchard, M.A. Fast, accurate and simulation-free stochastic mapping. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3985–3995. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.P.; Ludwig, T.; Meier, H.; Wolf, M.J. AxML: A fast program for sequential and parallel phylogenetic tree calculations based on the maximum likelihood method. In Proceedings of the Proceedings. IEEE Computer Society Bioinformatics Conference, Stanford, CA, USA, 16 August 2002; Volume 1, pp. 21–28. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.P. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- RAxML-NG. Available online: https://github.com/amkozlov/raxml-ng (accessed on 31 May 2024).
- Legason, I.D.; Burns, A.; Ridout, K.; Jennings, D.; Ogwang, M.D.; Otim, I.; Nabalende, H.; Achola, C.; Ayitewala, A.; Sseruyange, J.; et al. Genomic Landscape of Epstein-Barr Virus in Endemic Burkitt Lymphoma. Blood 2023, 142 (Suppl. S1), 2985. [Google Scholar] [CrossRef]
- Salichos, L.; Minosse, C.; Visco-Comandini, U.; Taibi, C.; Zulian, V.; D’Offizi, G.; Pallothu, N.; McPhee, F.; Garbuglia, A.R. Phylogenetic and Phylodynamic Analysis of Delta Strains Circulating in Italy. Viruses 2023, 15, 1791. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Hastings, W.K. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Gass, J.D., Jr.; Hill, N.J.; Damodaran, L.; Naumova, E.N.; Nutter, F.B.; Runstadler, J.A. Ecogeographic Drivers of the Spatial Spread of Highly Pathogenic Avian Influenza Outbreaks in Europe and the United States, 2016–Early 2022. Int. J. Environ. Res. Public Health 2023, 20, 6030. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, F.; Brierley, L.; Robertson, G.; Chase-Topping, M.; Lycett, S.; Woolhouse, M. Temporal Dynamics, Discovery, and Emergence of Human-Transmissible RNA Viruses. Mol. Biol. Evol. 2024, 41, msad272. [Google Scholar] [CrossRef]
- BEAST. Available online: https://beast.community/index.html (accessed on 31 May 2024).
- Ayres, D.L.; Cummings, M.P.; Baele, G.; Darling, A.E.; Lewis, P.O.; Swofford, D.L.; Huelsenbeck, J.P.; Lemey, P.; Rambaut, A.; Suchard, M.A. BEAGLE 3: Improved Performance, Scaling, and Usability for a High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2019, 68, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- BEAGLE Library. Available online: https://github.com/beagle-dev/beagle-lib (accessed on 31 May 2024).
- Rambaut, A. FigTree—Tree Figure Drawing Tool Version v. 1.4.4; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2018. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- IQ-TREE. Available online: http://www.iqtree.org (accessed on 31 May 2024).
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- W-IQ-TREE. Available online: http://iqtree.cibiv.univie.ac.at (accessed on 31 May 2024).
- Sulaiman, L.; Shittu, I.; Fusaro, A.; Inuwa, B.; Zecchin, B.; Gado, D.; Schivo, A.; Bianco, A.; Laleye, A.; Gobbo, F.; et al. Live Bird Markets in Nigeria: A Potential Reservoir for H9N2 Avian Influenza Viruses. Viruses 2021, 13, 1445. [Google Scholar] [CrossRef]
- Mei, X.; Guo, J.; Fang, P.; Ma, J.; Li, M.; Fang, L. The Characterization and Pathogenicity of a Recombinant Porcine Epidemic Diarrhea Virus Variant ECQ1. Viruses 2023, 15, 1492. [Google Scholar] [CrossRef] [PubMed]
- Soubrier, J.; Steel, M.; Lee, M.S.; Der Sarkissian, C.; Guindon, S.; Ho, S.Y.; Cooper, A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012, 29, 3345–3358. [Google Scholar] [CrossRef]
- Lewis, P.O. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 2001, 50, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gil, M.; Dufayard, J.F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Xie, R.; Edwards, K.M.; Wille, M.; Wei, X.; Wong, S.S.; Zanin, M.; El-Shesheny, R.; Ducatez, M.; Poon, L.L.M.; Kayali, G.; et al. The episodic resurgence of highly pathogenic avian influenza H5 virus. Nature 2023, 622, 810–817. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. How SARS-CoV-2 Big Data Are Challenging Viral Taxonomy Rules. Viruses 2023, 15, 715. [Google Scholar] [CrossRef]
- Libin, P.; Vanden Eynden, E.; Incardona, F.; Nowé, A.; Bezenchek, A.; EucoHIV Study Group; Sönnerborg, A.; Vandamme, A.M.; Theys, K.; Baele, G. PhyloGeoTool: Interactively exploring large phylogenies in an epidemiological context. Bioinformatics 2017, 33, 3993–3995. [Google Scholar] [CrossRef]
- PhyloGeoTool. Available online: https://github.com/rega-cev/phylogeotool (accessed on 31 May 2024).
- MEGA. Available online: https://www.megasoftware.net (accessed on 31 May 2024).
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput. Appl. Biosci. 1994, 10, 189–191. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Menon, S. Evolutionary Analysis of SARS-CoV-2 genome and protein insights the-origin of the virus, Wuhan. Int. J. Creat. Comput. 2021, 9, b696–b704. [Google Scholar]
- Aslanyan, L.; Avagyan, H.; Karalyan, Z. Whole-genome-based phylogeny of African swine fever virus. Vet. World 2020, 13, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef]
- Tajima, F. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 1993, 135, 599–607. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kende, J.; Bonomi, M.; Temmam, S.; Regnault, B.; Pérot, P.; Eloit, M.; Bigot, T. Virus Pop-Expanding Viral Databases by Protein Sequence Simulation. Viruses 2023, 15, 1227. [Google Scholar] [CrossRef] [PubMed]
- Virus Pop. Available online: https://gitlab.pasteur.fr/tbigot/viruspop (accessed on 31 May 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | Key Features | Website |
|---|---|---|---|
| Pathogens | web portal | nucleotide sequences, raw genomic data, sample metadata, scientific literature on HIV, influenza, Hepatitis B, P. falciparum and lesser-known pathogens affecting humans | [27] |
| ViMIC | database | information on virus mutation sites, viral integration sites, viral target genes, >1,000,000 sequences of eight viruses in almost 80 human diseases, cis-effects of virus–host interactions | [42] |
| VirusDB | database and tool | virus classification by closest neighbour genome search | [44] |
| IVDB | database | analysis platform for genetic, genomic, and phylogenetic studies of the influenza viruses | [46] |
| IVDT | tool | shows the worldwide geographic distribution of chosen viral genotypes and couples genomic data with epidemiological data | [47] |
| Los Alamos HIV databases | web portal | information available in eight sections: Sequence DB, Immunology DB, HXB2 Feature DB, Env Feature DB, Neutralisation DB, HCV DB, HFV DB, and COVID-19 Genome Analysis Pipeline. Online tools for alignment, genome navigation, annotation, consensus building, motif scanning, peptide mapping, heat maps | [49] |
| ViralZone | web portal | allows you to navigate in five main sections (Taxonomy, Biology, Hosts, Genomes, Re-sources) | [51] |
| GISAID | database | Sections: EpiFlu™ (influenza virus), EpiCoV™ (SARS-CoV-2), EpiPox™ (MPXV), EpiArbo™ (Arbo-viruses) | [52] |
| BV-BRC | web portal | unified interface for sequence and data mining, and a powerful suite including several command-line bioinformatic tools for bacterial and viral research | [63] |
| BioGRID | database | hosting protein, genetic and chemical interactions from different species | [70] |
| IntAct | database | general PPI database, including information on viruses | [78] |
| MINT | database and tool | general PPI database, including information on viruses, with VirusMINT search tool | [84] |
| VirusMentha | database | detects all published virus–host PPI integrating other databases | [87] |
| VirHostNet | database and tool | modelling of host/virus interactome using an undirected graph for data exploration and examination | [95] |
| HPIDB | database | contains almost 70,000 manually curated Host–Pathogen Interaction (HPI) entries with gene ontology functional information | [97] |
| Viruses.STRING | database and tool | database tailored on virus–virus and virus–host interactions, with the CytoscapeSTRING app | [101] |
| HCVpro | database | manually verified hepatitis C virus–virus and virus–human protein interactions; proteins are mapped onto Gene ontologies, canonical pathways, OMIM, and extensively cross-referenced | [105] |
| PHISTO | database and tool | integrates tools for the visualisation of HPI networks, graph–theoretical analysis of targeted human proteins, BLAST search and text mining | [107] |
| HVIDB | database | combines multiple data resources for predicting human–virus PPIs; enriched with experimentally derived PPIs and 3D structures of PPIs (from PDB or predicted by using homology modelling of protein complexes) | [109] |
| RAxML | tool | evolutionary relationships reconstruction based on a Maximum Likelihood (ML) approach | [125] |
| BEAST | tool | versatile cross-platform software suite for Bayesian analysis via Markov Chain Monte Carlo method | [132] |
| IQ-TREE | tool | incorporates sophisticated models of sequence evolution and performs an iterative optimisation process to enhance the accuracy of phylogenetic tree inference | [140] |
| Nextstrain | tool | programme for real-time monitoring of pathogen evolution | [55] |
| PhyloGeoTool | tool | facilitaties interactive exploration of large phylogenies and associated clinical data based on automatic phylogeny partitioning into clusters refined through recursive processes | [155] |
| MEGA | tool | Molecular Evolutionary Genetics Analysis software integrating various methods such as ML and Neighbour-Joining. It also incorporates powerful statistical methods | [156] |
| Virus Pop | tool | pipeline for adding new branches to a protein phylogenetic tree | [170] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vello, F.; Filippini, F.; Righetto, I. Bioinformatics Goes Viral: I. Databases, Phylogenetics and Phylodynamics Tools for Boosting Virus Research. Viruses 2024, 16, 1425. https://doi.org/10.3390/v16091425
Vello F, Filippini F, Righetto I. Bioinformatics Goes Viral: I. Databases, Phylogenetics and Phylodynamics Tools for Boosting Virus Research. Viruses. 2024; 16(9):1425. https://doi.org/10.3390/v16091425
Chicago/Turabian StyleVello, Federico, Francesco Filippini, and Irene Righetto. 2024. "Bioinformatics Goes Viral: I. Databases, Phylogenetics and Phylodynamics Tools for Boosting Virus Research" Viruses 16, no. 9: 1425. https://doi.org/10.3390/v16091425