

Coxsackievirus B3 Activates Macrophages Independently of CAR-Mediated Viral Entry

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viral Infection
2.2. Chemical Treatment
2.3. Immunofluorescence and Confocal Microscopy
2.4. Western Blot Analysis
2.5. Real-Time Quantitative RT-PCR (RT-qPCR)
2.6. Lentivirus Production
2.7. Quantification and Statistical Analysis
3. Results
3.1. CVB3 Induced Pro-Inflammatory Gene Expression in Macrophages Independent of Viral Replication
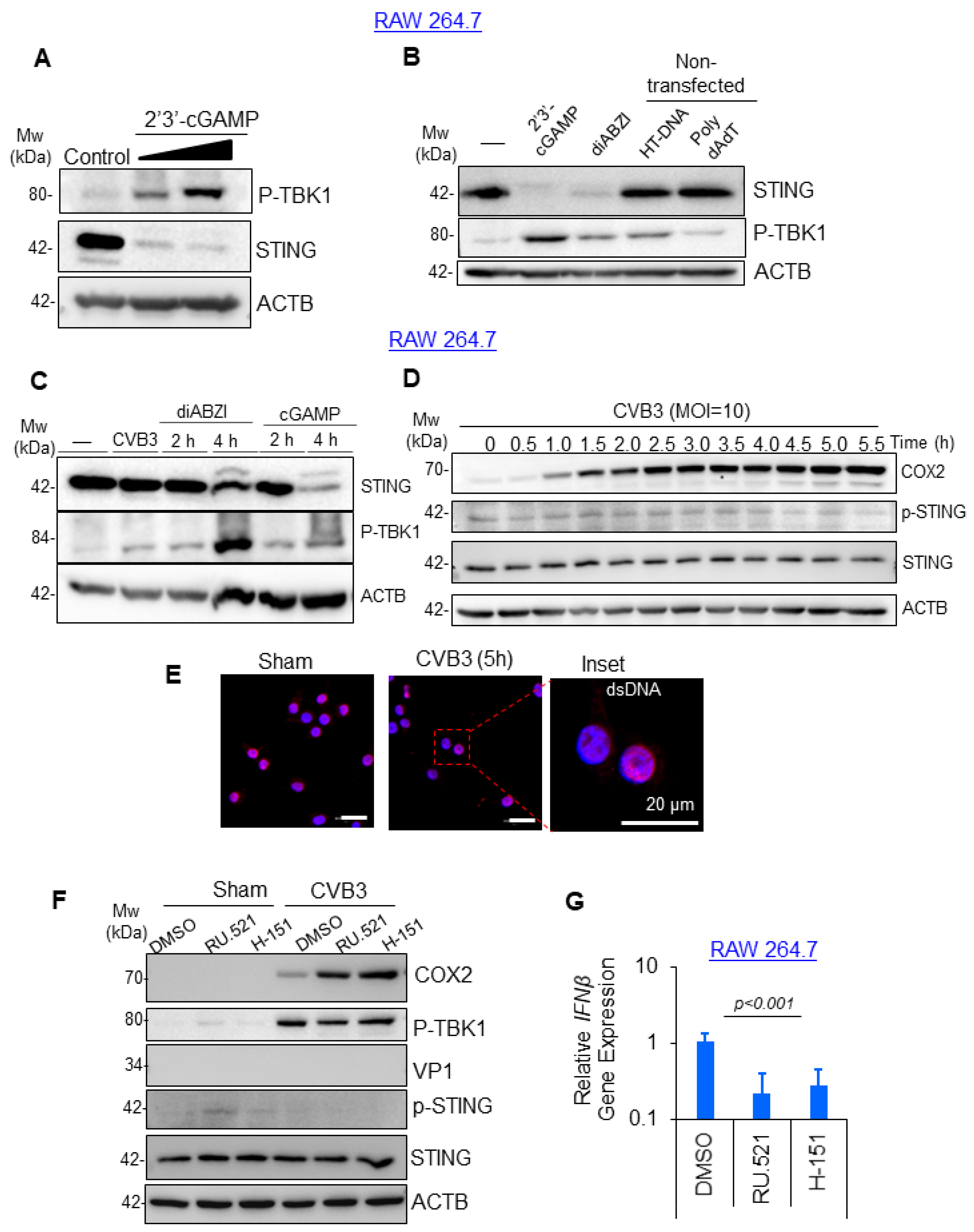
3.2. CVB3-Induced Macrophage Activation Was Independent of cGAS-STING Pathway
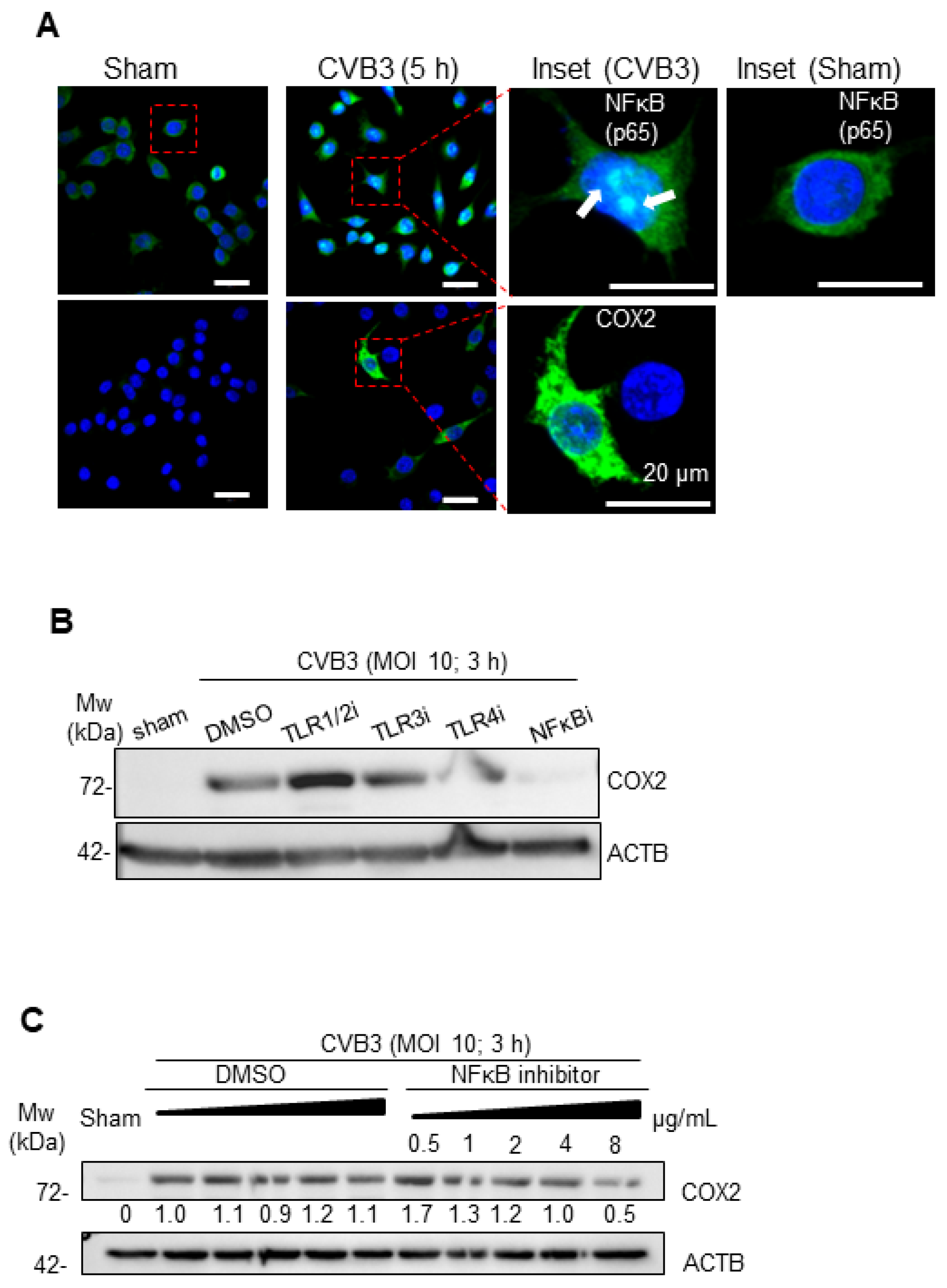
3.3. CVB3-Induced Macrophage Activation Is Dependent on NF-κB
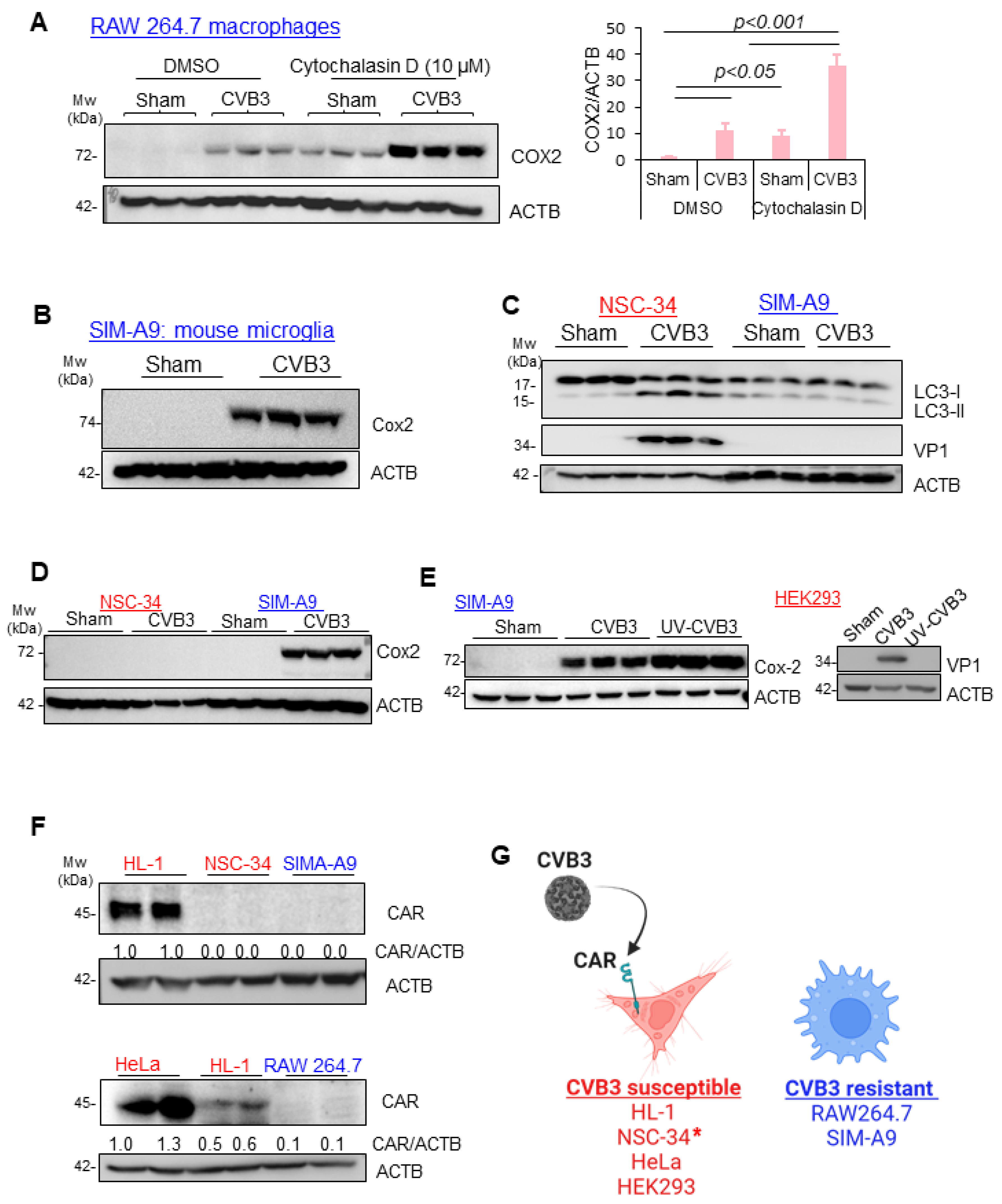
3.4. CVB3-Induced Macrophage Activation Is Independent of Viral Replication, CAR Expression, and Phagocytosis
3.5. CAR Expression Regulates Late, but Not Early, Response to CVB3 Infection
3.6. Viral-Induced LC3 Stimulates Macrophage Activation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newland, J.G.; Romero, J.R. Enteroviruses. In International Encyclopedia of Public Health; Heggenhougen, H.K., Ed.; Academic Press: Oxford, UK, 2008; pp. 347–352. [Google Scholar]
- Baggen, J.; Thibaut, H.J.; Strating, J.R.P.M.; van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Mohamud, Y.; Luo, H.L. The Intertwined Life Cycles of Enterovirus and Autophagy. Virulence 2019, 10, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015, 10, 629–653. [Google Scholar] [CrossRef] [PubMed]
- Jagdeo, J.M.; Dufour, A.; Klein, T.; Solis, N.; Kleifeld, O.; Kizhakkedathu, J.; Luo, H.; Overall, C.M.; Jan, E. N-Terminomics TAILS Identifies Host Cell Substrates of Poliovirus and Coxsackievirus B3 3C Proteinases That Modulate Virus Infection. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef]
- Saeed, M.; Kapell, S.; Hertz, N.T.; Wu, X.; Bell, K.; Ashbrook, A.W.; Mark, M.T.; Zebroski, H.A.; Neal, M.L.; Flodstrom-Tullberg, M.; et al. Defining the proteolytic landscape during enterovirus infection. PLoS Pathog. 2020, 16, e1008927. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004, 2, 109–122. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Mohamud, Y.; Shi, J.; Tang, H.; Xiang, P.; Xue, Y.C.; Liu, H.; Ng, C.S.; Luo, H. Coxsackievirus infection induces a non-canonical autophagy independent of the ULK and PI3K complexes. Sci. Rep. 2020, 10, 19068. [Google Scholar] [CrossRef]
- Brown, L.Y.; Dong, W.; Kantor, B. An Improved Protocol for the Production of Lentiviral Vectors. STAR Protoc. 2020, 1, 100152. [Google Scholar] [CrossRef]
- Fan, Y.M.; Zhang, Y.L.; Bahreyni, A.; Luo, H.; Mohamud, Y. Coxsackievirus Protease 2A Targets Host Protease ATG4A to Impair Autophagy. Viruses 2022, 14, 2026. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.M.; Zhang, Y.L.; Luo, H.; Mohamud, Y. Crosstalk between RNA viruses and DNA sensors: Role of the cGAS-STING signalling pathway. Rev. Med. Virol. 2022, 32, e2343. [Google Scholar] [CrossRef] [PubMed]
- Mohamud, Y.; Fu, C.; Fan, Y.M.; Zhang, Y.L.; Lin, J.F.C.; Hwang, S.W.; Wang, Z.C.; Luo, H. Activation of cGAS-STING suppresses coxsackievirus replication via interferon-dependent signaling. Antivir. Res. 2024, 222, 105811. [Google Scholar] [CrossRef] [PubMed]
- Casella, J.F.; Flanagan, M.D.; Lin, S. Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature 1981, 293, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Abernathy, E.; Mateo, R.; Majzoub, K.; van Buuren, N.; Bird, S.W.; Carette, J.E.; Kirkegaard, K. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 2019, 17, e2006926. [Google Scholar] [CrossRef]
- Mohamud, Y.; Shi, J.; Qu, J.; Poon, T.; Xue, Y.C.; Deng, H.; Zhang, J.; Luo, H. Enteroviral Infection Inhibits Autophagic Flux via Disruption of the SNARE Complex to Enhance Viral Replication. Cell Rep. 2018, 22, 3292–3303. [Google Scholar] [CrossRef]
- Jaquenod De Giusti, C.; Ure, A.E.; Rivadeneyra, L.; Schattner, M.; Gomez, R.M. Macrophages and galectin 3 play critical roles in CVB3-induced murine acute myocarditis and chronic fibrosis. J. Mol. Cell. Cardiol. 2015, 85, 58–70. [Google Scholar] [CrossRef]
- Shin, H.H.; Jeon, E.S.; Lim, B.K. Macrophage-Specific Coxsackievirus and Adenovirus Receptor Deletion Enhances Macrophage M1 Polarity in CVB3-Induced Myocarditis. Int. J. Mol. Sci. 2023, 24, 5309. [Google Scholar] [CrossRef]
- Dong, J.; Lu, J.; Cen, Z.; Tang, Q.; Li, Y.; Qin, L.; Yan, Y.; Lu, F.; Wu, W. Cardiac macrophages undergo dynamic changes after coxsackievirus B3 infection and promote the progression of myocarditis. J. Med. Virol. 2023, 95, e29004. [Google Scholar] [CrossRef]
- Bao, J.; Sun, T.; Yue, Y.; Xiong, S. Macrophage NLRP3 inflammasome activated by CVB3 capsid proteins contributes to the development of viral myocarditis. Mol. Immunol. 2019, 114, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xu, W.; Guo, Q.; Jiang, Z.; Wang, P.; Yue, Y.; Xiong, S. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ. Res. 2009, 105, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Shieh, J.T.C.; Pickles, R.J.; Okegawa, T.; Hsieh, J.T.; Bergelson, J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. USA 2001, 98, 15191–15196. [Google Scholar] [CrossRef] [PubMed]
- Geisler, A.; Hazini, A.; Heimann, L.; Kurreck, J.; Fechner, H. Coxsackievirus B3-Its Potential as an Oncolytic Virus. Viruses 2021, 13, 718. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T.; Luo, H.L. Development of Group B Coxsackievirus as an Oncolytic Virus: Opportunities and Challenges. Viruses 2021, 13, 1082. [Google Scholar] [CrossRef]
- Opavsky, M.A.; Martino, T.; Rabinovitch, M.; Penninger, J.; Richardson, C.; Petric, M.; Trinidad, C.; Butcher, L.; Chan, J.; Liu, P.P. Enhanced ERK-1/2 activation in mice susceptible to coxsackievirus-induced myocarditis. J. Clin. Investig. 2002, 109, 1561–1569. [Google Scholar] [CrossRef]
- Mena, I.; Perry, C.M.; Harkins, S.; Rodriguez, F.; Gebhard, J.; Whitton, J.L. The role of B lymphocytes in coxsackievirus B3 infection. Am. J. Pathol. 1999, 155, 1205–1215. [Google Scholar] [CrossRef]
- Lindner, D.; Li, J.; Savvatis, K.; Klingel, K.; Blankenberg, S.; Tschope, C.; Westermann, D. Cardiac fibroblasts aggravate viral myocarditis: Cell specific coxsackievirus B3 replication. Mediat. Inflamm. 2014, 2014, 519528. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Davola, M.E.; Cormier, O.; Vito, A.; El-Sayes, N.; Collins, S.; Salem, O.; Ask, K.; Revill, S.; Wan, Y.H.; Mossman, K. Oncolytic BHV-1 Is Sufficient to Induce Immunogenic Cell Death and Synergizes with Low-Dose Chemotherapy to Dampen Immunosuppressive T Regulatory Cells. Cancers 2023, 15, 1295. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.K.; Saulsbery, H.M.; Velazquez, A.F.C.; Jackson, W.T. Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit. Cell Rep. 2018, 22, 3304–3314. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Huang, L.; Yue, J. The interplay of autophagy and enterovirus. Semin. Cell Dev. Biol. 2020, 101, 12–19. [Google Scholar] [CrossRef]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef]
- Kirkegaard, K. Subversion of the cellular autophagy pathway by viruses. Curr. Top. Microbiol. Immunol. 2009, 335, 323–333. [Google Scholar]
- Sin, J.; McIntyre, L.; Stotland, A.; Feuer, R.; Gottlieb, R.A. Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J. Virol. 2017, 91, 10-1128. [Google Scholar] [CrossRef]
- McKnight, K.L.; Xie, L.; González-López, O.; Rivera-Serrano, E.E.; Chen, X.; Lemon, S.M. Protein composition of the hepatitis A virus quasi-envelope. Proc. Natl. Acad. Sci. USA 2017, 114, 6587–6592. [Google Scholar] [CrossRef]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Green, D.R. LC3-associated phagocytosis at a glance. J. Cell Sci. 2019, 132, jcs222984. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| 2A | GCTTTGCAGACATCCGTGATC | CAAGCTGTGTTCCACATAGTCCTTCA |
| Ifnb1 (mouse) | GCCTTTGCCATCCAAGAGATGC | ACACTGTCTGCTGGTGGAGTTC |
| IFNB (human) | CAACTTGCTTGGATTCCTACAAAG | TATTCAAGCCTCCCATTCAATTG |
| Il6 (mouse) | ACAACCACGGCCTTCCCTAC | TCTCATTTCCACGATTTCCCAG |
| IL6 (human) | ACTCACCTCTTCAGAACGAATTG | CCATCTTTGGAAGGTTCAGGTTG |
| Il1b (mouse) | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| IL-1B (human) | CCACAGACCTTCCAGGAGAATG | GTGCAGTTCAGTGATCGTACAGG |
| Tnfa (mouse) | GTCCCCAAAGGGATGAGAAGTT | GTTTGCTACGACGTGGGCTACA |
| TNFA (human) | CCTCTCTCTAATCAGCCCTCTG | GAGGACCTGGGAGTAGATGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamud, Y.; Lin, J.C.; Hwang, S.W.; Bahreyni, A.; Wang, Z.C.; Luo, H. Coxsackievirus B3 Activates Macrophages Independently of CAR-Mediated Viral Entry. Viruses 2024, 16, 1456. https://doi.org/10.3390/v16091456
Mohamud Y, Lin JC, Hwang SW, Bahreyni A, Wang ZC, Luo H. Coxsackievirus B3 Activates Macrophages Independently of CAR-Mediated Viral Entry. Viruses. 2024; 16(9):1456. https://doi.org/10.3390/v16091456
Chicago/Turabian StyleMohamud, Yasir, Jingfei Carly Lin, Sinwoo Wendy Hwang, Amirhossein Bahreyni, Zhihan Claire Wang, and Honglin Luo. 2024. "Coxsackievirus B3 Activates Macrophages Independently of CAR-Mediated Viral Entry" Viruses 16, no. 9: 1456. https://doi.org/10.3390/v16091456
APA StyleMohamud, Y., Lin, J. C., Hwang, S. W., Bahreyni, A., Wang, Z. C., & Luo, H. (2024). Coxsackievirus B3 Activates Macrophages Independently of CAR-Mediated Viral Entry. Viruses, 16(9), 1456. https://doi.org/10.3390/v16091456