
Multiplex PCR Approach for Rapid African Swine Fever Virus Genotyping
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral DNA
2.2. qPCR
2.3. Multiplex PCR
2.4. Size-Selection and Library Preparation
2.5. Bioinformatic Analysis
3. Results
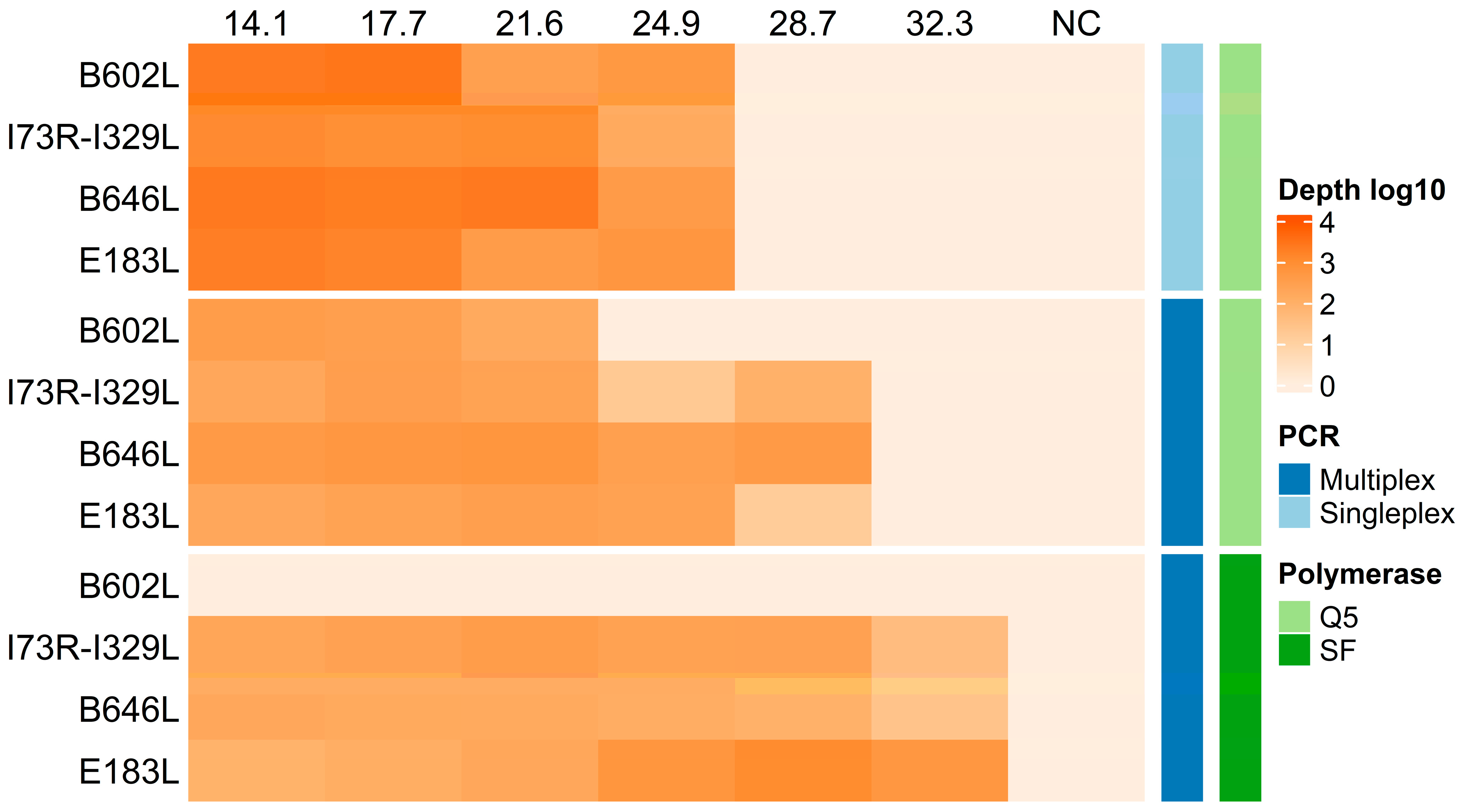
3.1. Multiplex PCR Calibration
3.2. Evaluation of Detection Sensitivity
3.3. Assessing Multiplex PCR Compatibility with Diverse ASFV Samples
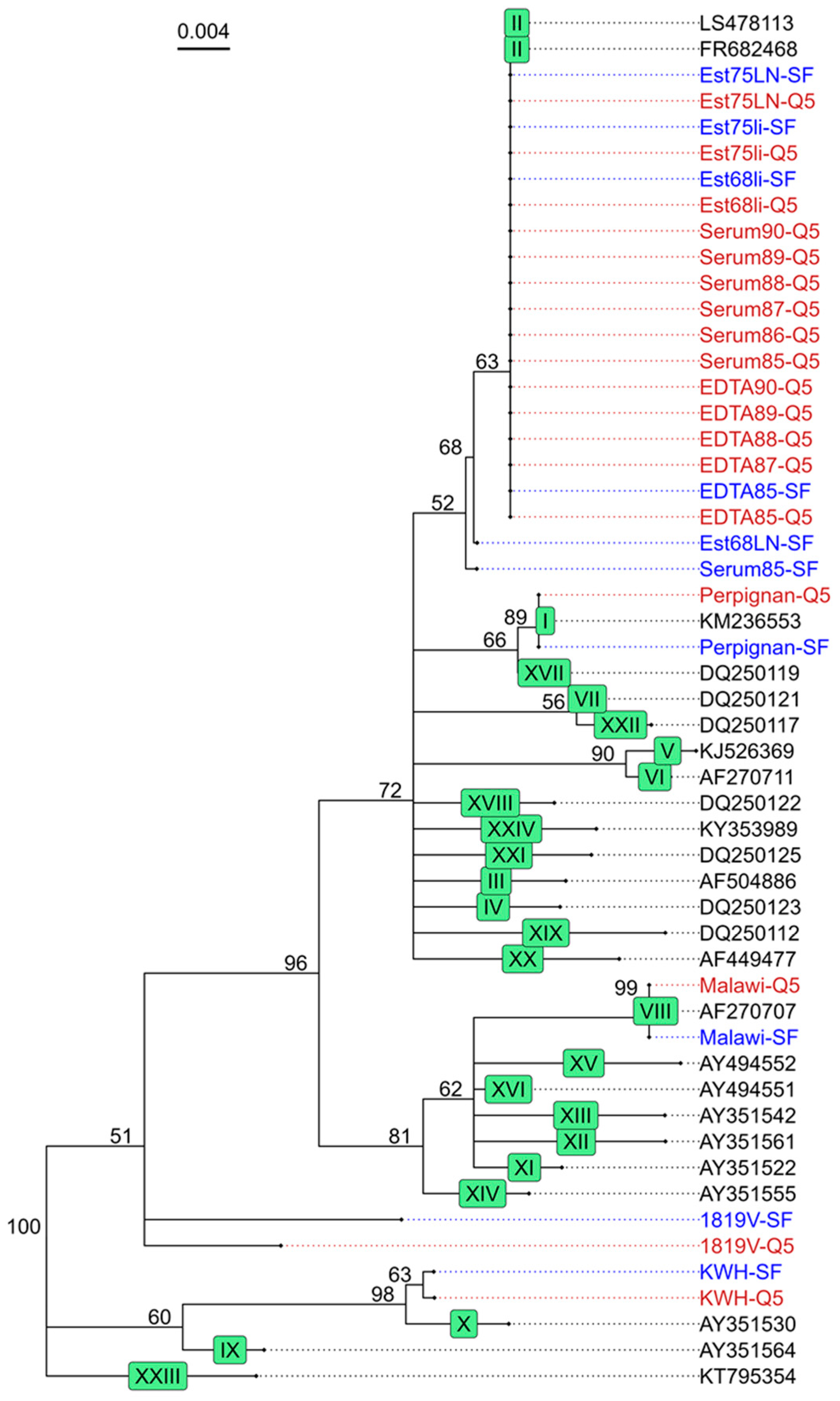
3.4. Phylogenetic Analysis of the Target Regions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blome, S.; Franzke, K.; Beer, M. African Swine Fever—A Review of Current Knowledge. Virus Res. 2020, 287, 198099. [Google Scholar] [CrossRef] [PubMed]
- Eustace Montgomery, R. On A Form of Swine Fever Occurring in British East Africa (Kenya Colony). J. Comp. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef]
- Penrith, M.L.; Kivaria, F.M. One Hundred Years of African Swine Fever in Africa: Where Have We Been, Where Are We Now, Where Are We Going? Transbound. Emerg. Dis. 2022, 69, e1179–e1200. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.; Huang, L.; Zhang, X.; Zhang, J.; Shen, D.; Zhang, Z.; Wang, Z.; Huo, H.; Wang, W.; Huangfu, H.; et al. Genotype I African Swine Fever Viruses Emerged in Domestic Pigs in China and Caused Chronic Infection. Emerg. Microbes Infect. 2021, 10, 2183. [Google Scholar] [CrossRef]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African Swine Fever Virus Isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Vizcaíno, J.M.; Mur, L.; Martínez-López, B. African Swine Fever (ASF): Five Years around Europe. Vet. Microbiol. 2013, 165, 45–50. [Google Scholar] [CrossRef]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic Variation among African Swine Fever Genotype II Viruses, Eastern and Central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef]
- Global Disease Monitoring Reports–Swine Health Information Center. Available online: https://www.swinehealth.org/global-disease-surveillance-reports/ (accessed on 19 June 2024).
- Zhou, X.; Li, N.; Luo, Y.; Liu, Y.; Miao, F.; Chen, T.; Zhang, S.; Cao, P.; Li, X.; Tian, K.; et al. Emergence of African Swine Fever in China, 2018. Transbound. Emerg. Dis. 2018, 65, 1482–1484. [Google Scholar] [CrossRef]
- Ito, S.; Kawaguchi, N.; Bosch, J.; Aguilar-Vega, C.; Sánchez-Vizcaíno, J.M. What Can We Learn from the Five-Year African Swine Fever Epidemic in Asia? Front. Vet. Sci. 2023, 10, 1273417. [Google Scholar] [CrossRef]
- Gonzales, W.; Moreno, C.; Duran, U.; Henao, N.; Bencosme, M.; Lora, P.; Reyes, R.; Núñez, R.; De Gracia, A.; Perez, A.M. African Swine Fever in the Dominican Republic. Transbound. Emerg. Dis. 2021, 68, 3018–3019. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, S.; Zhang, H.; Qin, Z.; Shan, H.; Cai, X. Vaccines for African Swine Fever: An Update. Front. Microbiol. 2023, 14, 1139494. [Google Scholar] [CrossRef]
- Spinard, E.; Dinhobl, M.; Tesler, N.; Birtley, H.; Signore, A.V.; Ambagala, A.; Masembe, C.; Borca, M.V.; Gladue, D.P. A Re-Evaluation of African Swine Fever Genotypes Based on P72 Sequences Reveals the Existence of Only Six Distinct P72 Groups. Viruses 2023, 15, 2246. [Google Scholar] [CrossRef] [PubMed]
- Quembo, C.J.; Jori, F.; Vosloo, W.; Heath, L. Genetic Characterization of African Swine Fever Virus Isolates from Soft Ticks at the Wildlife/Domestic Interface in Mozambique and Identification of a Novel Genotype. Transbound. Emerg. Dis. 2018, 65, 420–431. [Google Scholar] [CrossRef]
- Bastos, A.D.S.; Penrith, M.L.; Crucière, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping Field Strains of African Swine Fever Virus by Partial P72 Gene Characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrín, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a New Genotype of African Swine Fever Virus in Domestic Pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Nix, R.J.; Gallardo, C.; Hutchings, G.; Blanco, E.; Dixon, L.K. Molecular Epidemiology of African Swine Fever Virus Studied by Analysis of Four Variable Genome Regions. Arch. Virol. 2006, 151, 2475–2494. [Google Scholar] [CrossRef]
- Boshoff, C.I.; Bastos, A.D.S.; Gerber, L.J.; Vosloo, W. Genetic Characterisation of African Swine Fever Viruses from Outbreaks in Southern Africa (1973–1999). Vet. Microbiol. 2007, 121, 45–55. [Google Scholar] [CrossRef]
- Gallardo, C.; Mwaengo, D.M.; Macharia, J.M.; Arias, M.; Taracha, E.A.; Soler, A.; Okoth, E.; Martín, E.; Kasiti, J.; Bishop, R.P. Enhanced Discrimination of African Swine Fever Virus Isolates through Nucleotide Sequencing of the P54, P72, and pB602L (CVR) Genes. Virus Genes 2009, 38, 85–95. [Google Scholar] [CrossRef]
- Gallardo, C.; Casado, N.; Soler, A.; Djadjovski, I.; Krivko, L.; Madueño, E.; Nieto, R.; Perez, C.; Simon, A.; Ivanova, E.; et al. A Multi Gene-Approach Genotyping Method Identifies 24 Genetic Clusters within the Genotype II-European African Swine Fever Viruses Circulating from 2007 to 2022. Front. Vet. Sci. 2023, 10, 1112850. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Walczak, M.; Juszkiewicz, M.; Woźniakowski, G. The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences. Viruses 2020, 12, 1094. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Cho, K.H.; Mai, N.T.A.; Park, J.Y.; Trinh, T.B.N.; Jang, M.K.; Nguyen, T.T.H.; Vu, X.D.; Nguyen, T.L.; Nguyen, V.D.; et al. Multiple Variants of African Swine Fever Virus Circulating in Vietnam. Arch. Virol. 2022, 167, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Vilem, A.; Nurmoja, I.; Tummeleht, L.; Viltrop, A. Differentiation of African Swine Fever Virus Strains Isolated in Estonia by Multiple Genetic Markers. Pathogens 2023, 12, 720. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Liu, H.; Yin, Y.; Si, H.; Long, F.; Feng, S. Molecular Characterization of African Swine Fever Virus From 2019-2020 Outbreaks in Guangxi Province, Southern China. Front. Vet. Sci. 2022, 9, 912224. [Google Scholar] [CrossRef]
- Centro de Investigacion en Sanidad Animal (CISA-INIA) Procedure for the Genotyping of African Swine Fever Virus (ASFV) Isolates. Available online: https://asf-referencelab.info/asf-diagnosis/sops/ (accessed on 7 July 2022).
- Radulovic, E.; Mehinagic, K.; Wuthrich, T.; Hilty, M.; Posthaus, H.; Summerfield, A.; Ruggli, N.; Benarafa, C. The Baseline Immunological and Hygienic Status of Pigs Impact Disease Severity of African Swine Fever. PLoS Pathog. 2022, 18, e1010522. [Google Scholar] [CrossRef] [PubMed]
- Natale, V.A.I.; McCullough, K.C. Macrophage Culture: Influence of Species-Specific Incubation Temperature. J. Immunol. Methods 1998, 214, 165–174. [Google Scholar] [CrossRef]
- Haines, F.J.; Hofmann, M.A.; King, D.P.; Drew, T.W.; Crooke, H.R. Development and Validation of a Multiplex, Real-Time RT PCR Assay for the Simultaneous Detection of Classical and African Swine Fever Viruses. PLoS ONE 2013, 8, e71019. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Zhou, L.; Feng, T.; Xu, S.; Gao, F.; Lam, T.T.; Wang, Q.; Wu, T.; Huang, H.; Zhan, L.; Li, L.; et al. Ggmsa: A Visual Exploration Tool for Multiple Sequence Alignment and Associated Data. Brief. Bioinform. 2022, 23, bbac222. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z. Complex Heatmap Visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.M.; Gamaarachchi, H.; Nguyen, T.; Gollon, A.; Tong, S.; Aquilina-Reid, C.; Bowen-James, R.; Deveson, I.W. InterARTIC: An Interactive Web Application for Whole-Genome Nanopore Sequencing Analysis of SARS-CoV-2 and Other Viruses. Bioinformatics 2022, 38, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y. Next-Generation Sequencing Methods: Impact of Sequencing Accuracy on SNP Discovery. In Single Nucleotide Polymorphisms: Methods and Protocols; Komar, A.A., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 95–111. ISBN 978-1-60327-411-1. [Google Scholar]
- Nanopore Sequencing Accuracy. Available online: https://nanoporetech.com/platform/accuracy (accessed on 19 June 2024).
- Illumina Quality Scores for Next-Generation Sequencing. Available online: https://www.illumina.com/Documents/products/technotes/technote_Q-Scores.pdf (accessed on 19 June 2024).
- Forth, J.H.; Calvelage, S.; Fischer, M.; Hellert, J.; Sehl-Ewert, J.; Roszyk, H.; Deutschmann, P.; Reichold, A.; Lange, M.; Thulke, H.H.; et al. African Swine Fever Virus—Variants on the Rise. Emerg. Microbes Infect. 2023, 12, 2146537. [Google Scholar] [CrossRef]
- Sehl, J.; Pikalo, J.; Schäfer, A.; Franzke, K.; Pannhorst, K.; Elnagar, A.; Blohm, U.; Blome, S.; Breithaupt, A. Comparative Pathology of Domestic Pigs and Wild Boar Infected with the Moderately Virulent African Swine Fever Virus Strain “Estonia 2014”. Pathogens 2020, 9, 662. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qPCR | Q5 DNA Polymerase | Platinum SuperFi II DNA Polymerase | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ct Value | B646L | I73R-I329L | B602L | E183L | B646L | I73R-I329L | B602L | E183L | |
| Serum85 | 16 | ++ | ++ | ++ | ++ | + | ++ | - | + |
| Serum90 | 16.1 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| Serum86 | 17.4 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| Serum87 | 17.4 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| Serum88 | 17.5 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| Serum89 | 18.2 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| KWH | 19.1 | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| EDTA87 | 19.4 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| EDTA90 | 19.4 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| EDTA88 | 19.5 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| EDTA89 | 19.6 | ++ | ++ | ++ | ++ | ND | ND | ND | ND |
| EDTA85 | 19.7 | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| 1819V | 20.2 | ++ | ++ | ++ | ++ | ++ | ++ | + | ++ |
| Perpignan | 21.3 | ++ | ++ | ++ | ++ | ++ | ++ | - | ++ |
| Malawi | 21.6 | ++ | ++ | ++ | ++ | ++ | ++ | - | ++ |
| Est75li | 25.7 | ++ | ++ | ++ | - | ++ | ++ | + | + |
| Est68li | 26.3 | ++ | ++ | ++ | - | ++ | ++ | + | + |
| Est75LN | 29.9 | ++ | ++ | + | - | ++ | ++ | - | + |
| Est68LN | 31.1 | - | - | - | - | + | ++ | - | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Licheri, M.; Licheri, M.F.; Mehinagic, K.; Radulovic, E.; Ruggli, N.; Dijkman, R. Multiplex PCR Approach for Rapid African Swine Fever Virus Genotyping. Viruses 2024, 16, 1460. https://doi.org/10.3390/v16091460
Licheri M, Licheri MF, Mehinagic K, Radulovic E, Ruggli N, Dijkman R. Multiplex PCR Approach for Rapid African Swine Fever Virus Genotyping. Viruses. 2024; 16(9):1460. https://doi.org/10.3390/v16091460
Chicago/Turabian StyleLicheri, Matthias, Manon Flore Licheri, Kemal Mehinagic, Emilia Radulovic, Nicolas Ruggli, and Ronald Dijkman. 2024. "Multiplex PCR Approach for Rapid African Swine Fever Virus Genotyping" Viruses 16, no. 9: 1460. https://doi.org/10.3390/v16091460
APA StyleLicheri, M., Licheri, M. F., Mehinagic, K., Radulovic, E., Ruggli, N., & Dijkman, R. (2024). Multiplex PCR Approach for Rapid African Swine Fever Virus Genotyping. Viruses, 16(9), 1460. https://doi.org/10.3390/v16091460