
Extensive Diversity of Viruses in Millipedes Collected in the Dong Nai Biosphere Reserve (Vietnam)

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection, Homogenization, and Pooling of Millipedes
2.2. High-Throughput Sequencing Sample Preparation
2.3. Virus Discovery Pipeline
2.4. Estimation of Virus Assembly Quality and Virus Naming
2.5. Data Analysis and Visualization
3. Results
3.1. High-Throughput Sequencing
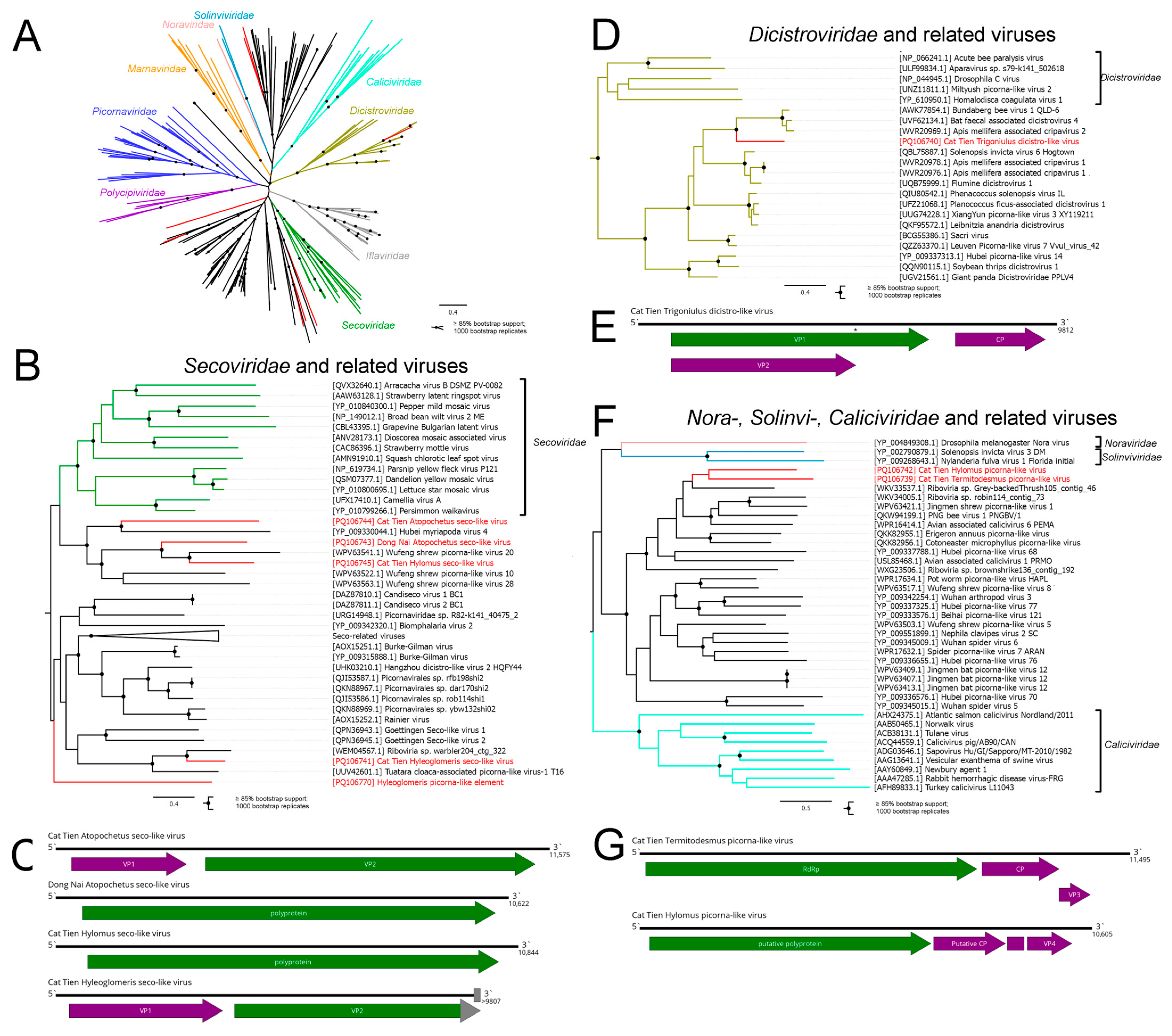
3.2. Picornavirales
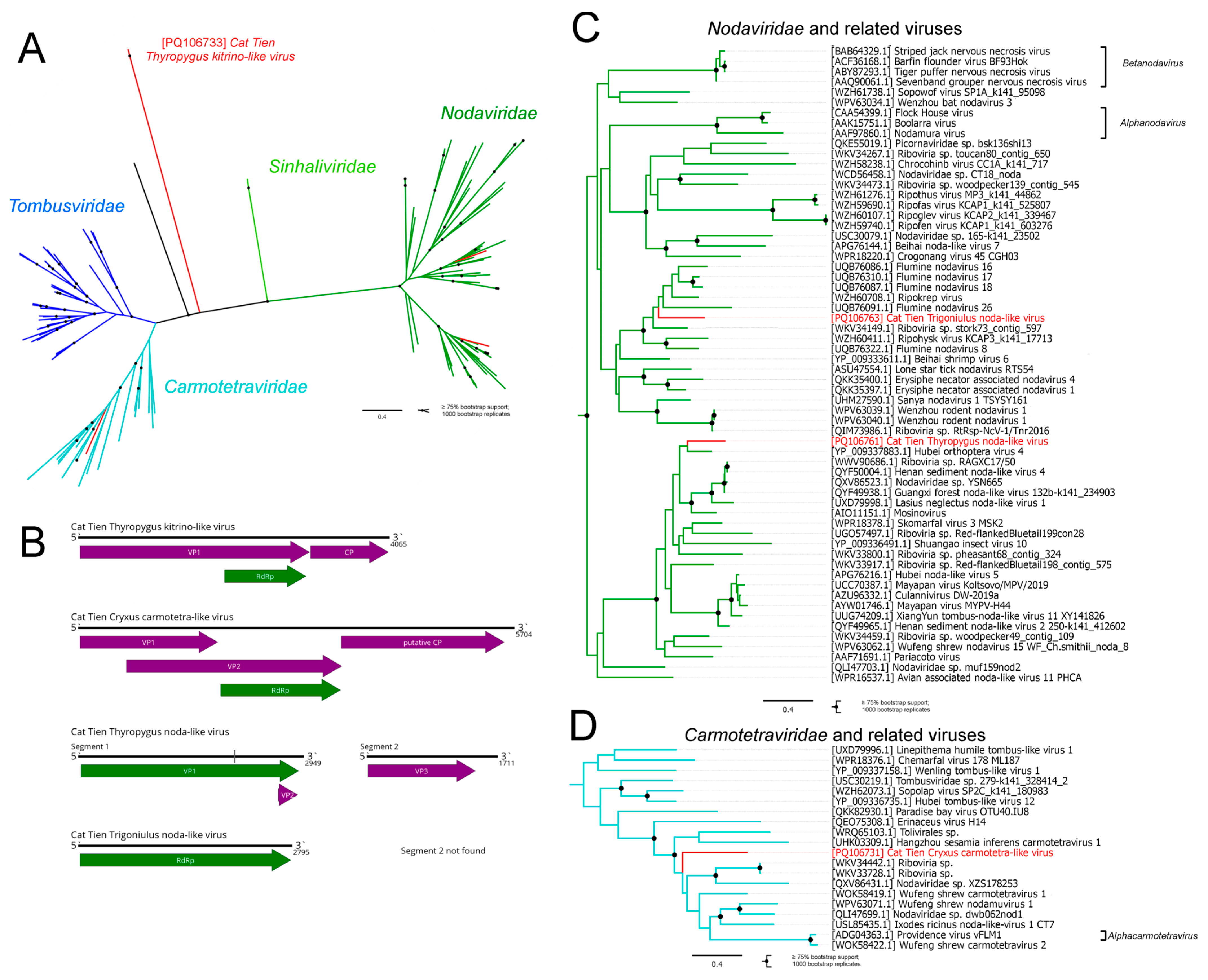
3.3. Tolivirales and Nodamuvirales
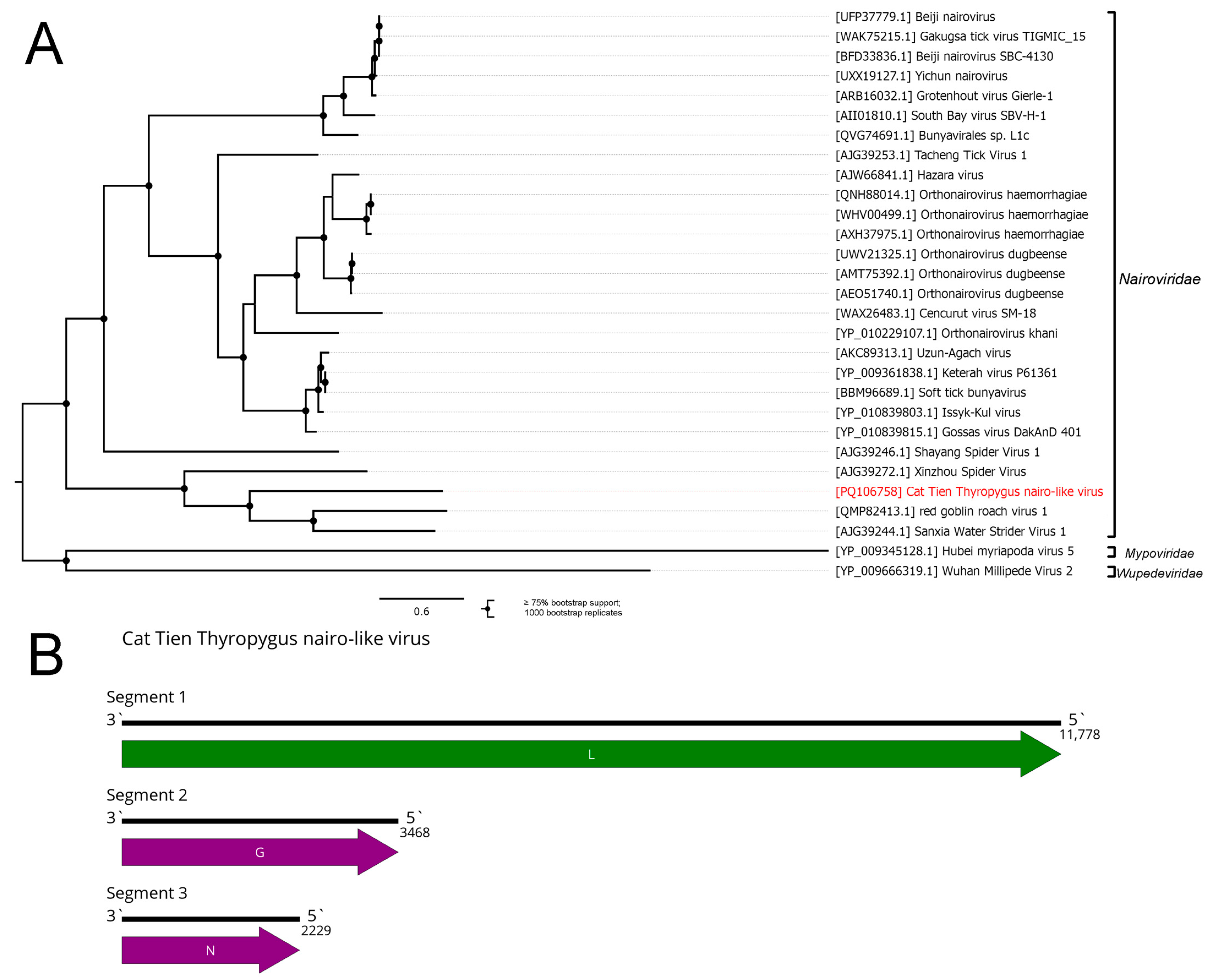
3.4. Bunyaviricetes
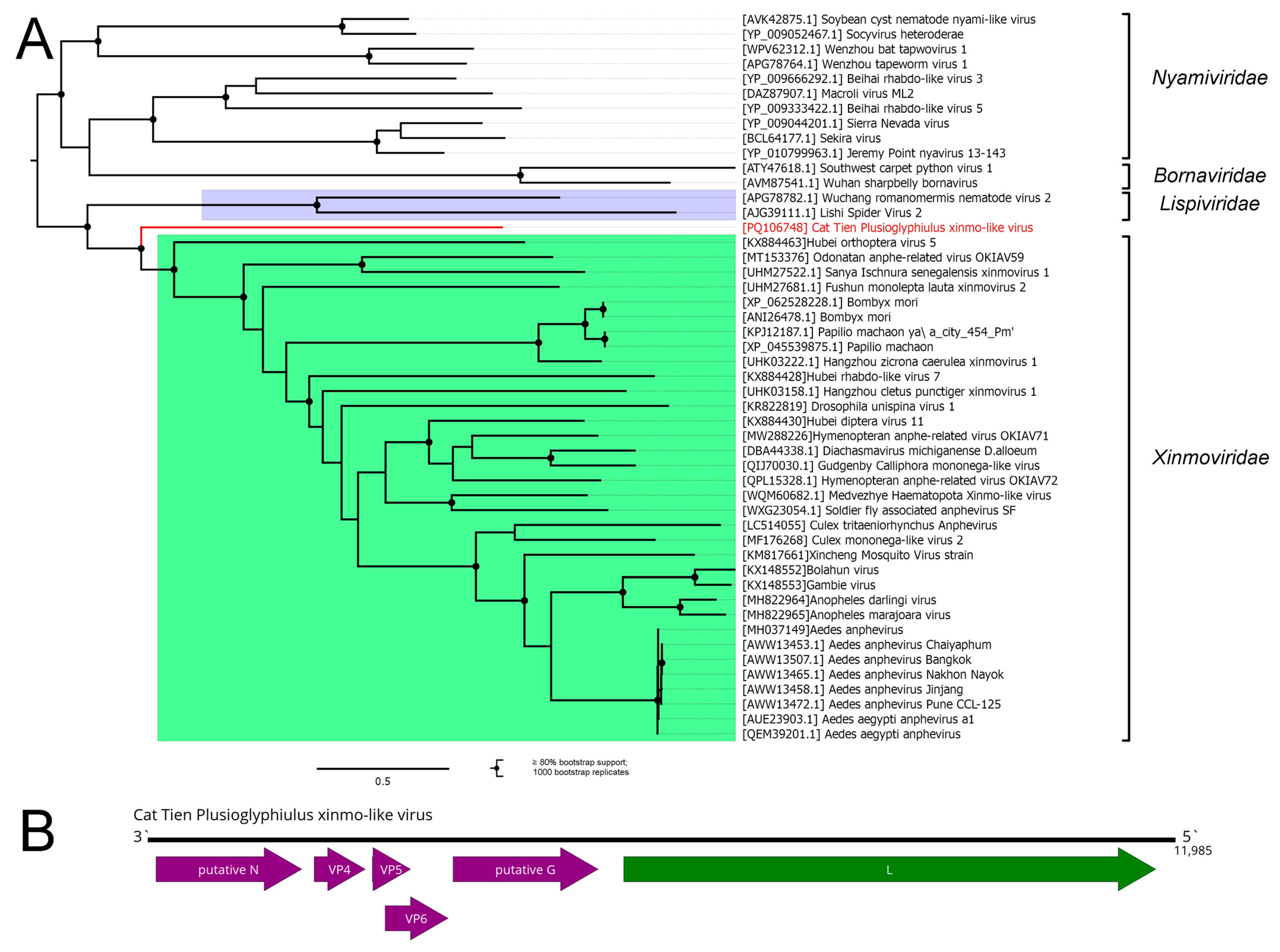
3.5. Mononegavirales
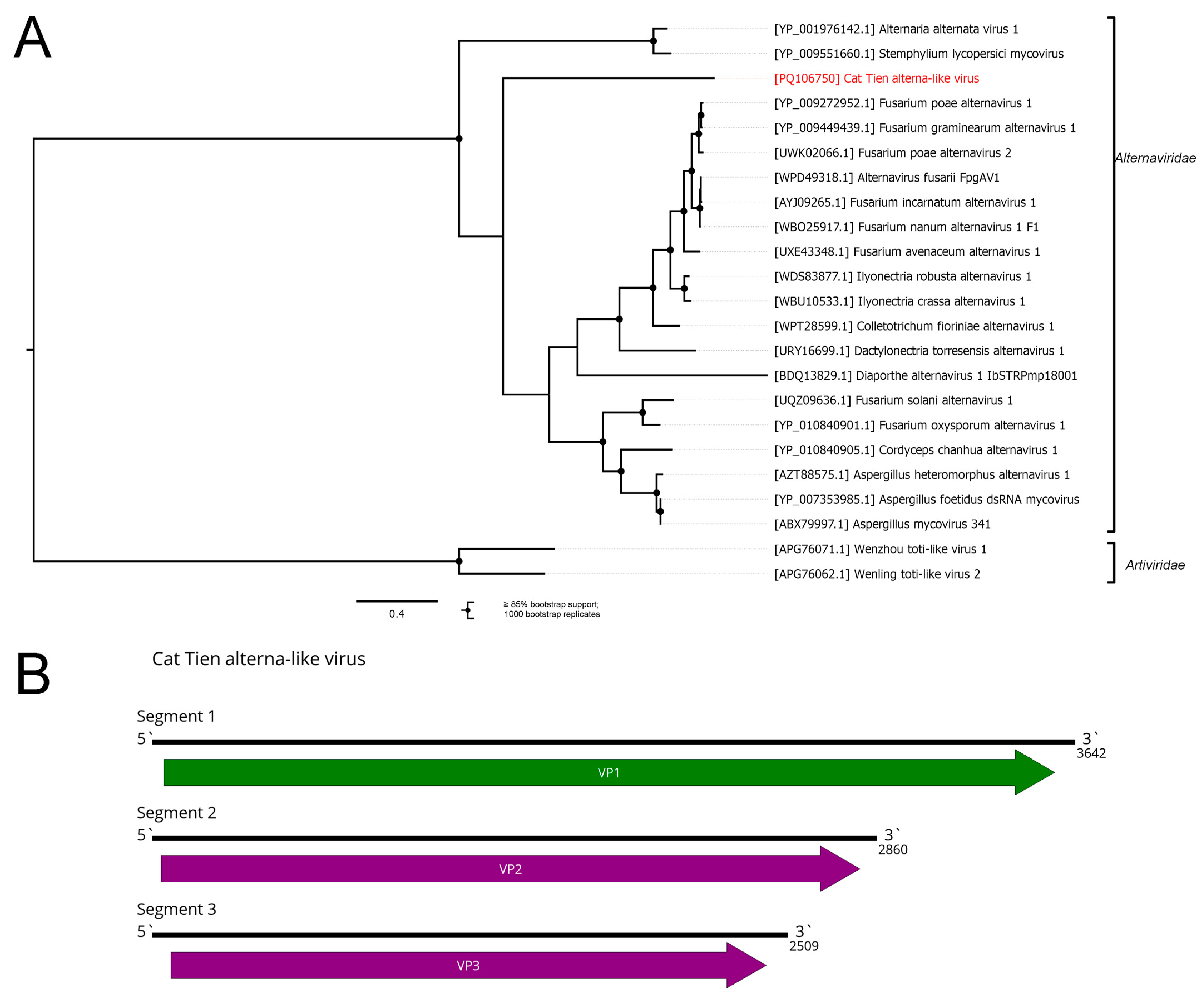
3.6. Ghabrivirales
3.7. Cryppavirales
3.8. Jingchuvirales
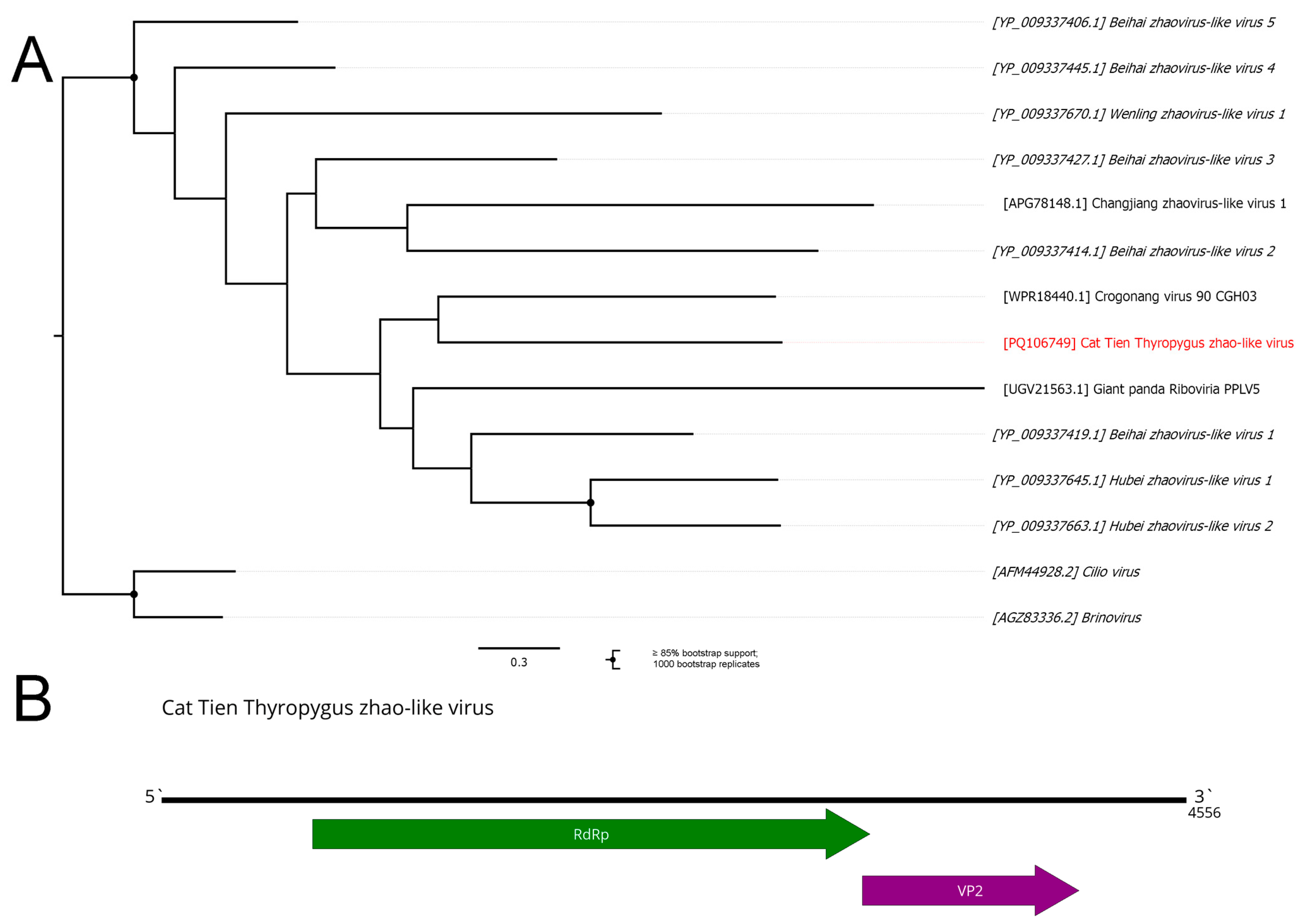
3.9. Zhaoviruses
3.10. Muvirales
3.11. Partial Virus Genomes
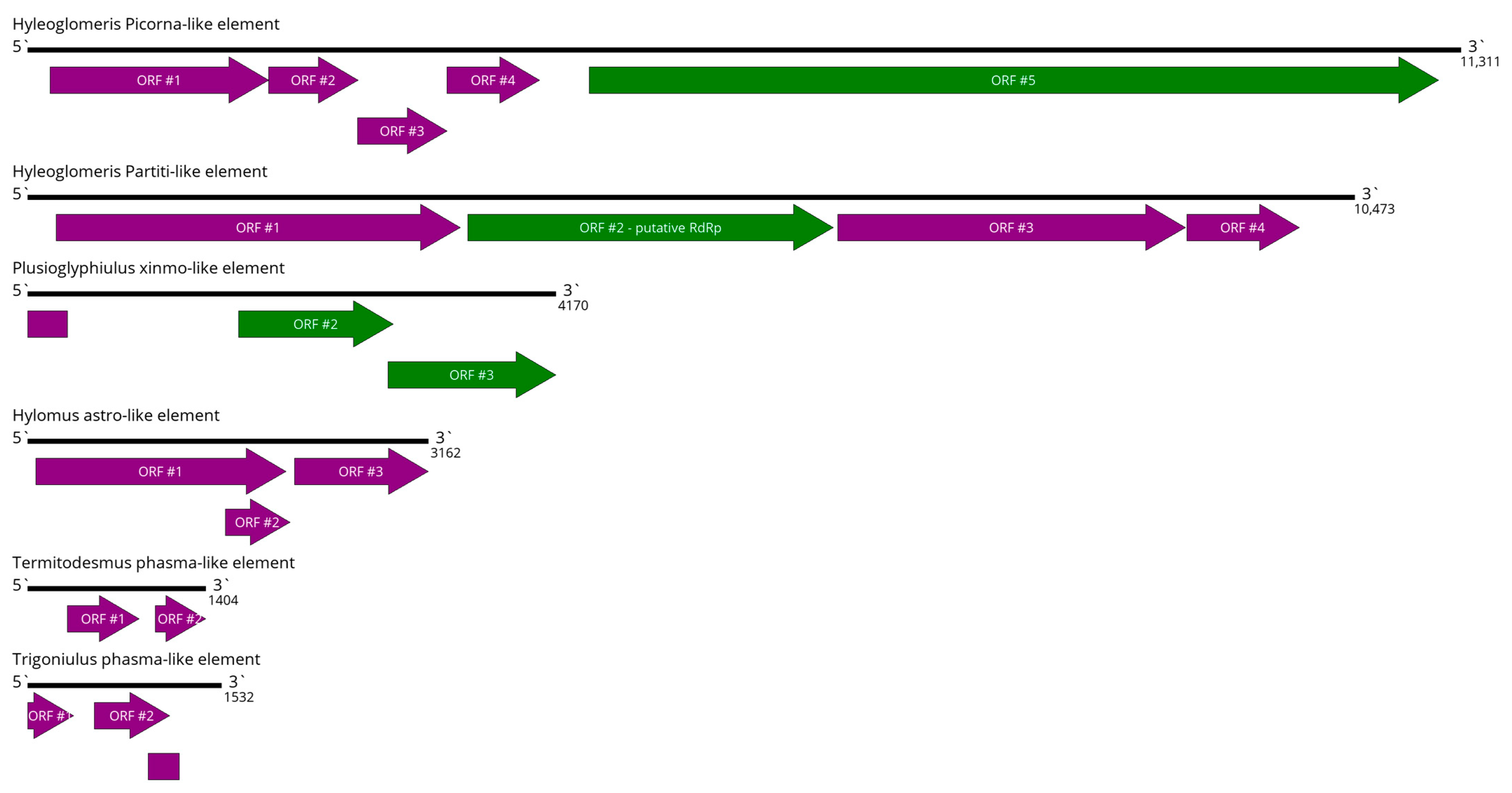
3.12. Virus-like Elements
3.13. Viral Genome Fragments
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shi, M.; Lin, X.; Tian, J.; Chen, L.; Chen, X.; Li, C.; Qin, X.; Li, J.; Cao, J.; Eden, J.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shi, M.; Tian, J.; Lin, X.; Kang, Y.; Chen, L.; Qin, X.; Xu, J.; Holmes, E.C.; Zhang, Y. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, S.; Ka, S.; Zirkel, F.; Donath, A.; Petersen, M.; Liu, S.; Zhou, X.; Drosten, C.; Misof, B.; Junglen, S. Viromics of extant insect orders unveil the evolution of the flavi-like superfamily. Virus Evol. 2021, 7, veab030. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, S.R.; Paraskevopoulou, S. ICTV Virus Taxonomy Profile: Xinmoviridae 2023. J. Gen. Virol. 2023, 104, 001906. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Koonin, E.V.; Krupovic, M.; Kuhn, J.H. ICTV Virus Taxonomy Profile: Qinviridae 2023. J. Gen. Virol. 2023, 104, 001905. [Google Scholar] [CrossRef]
- Hameed, M.; Wahaab, A.; Shan, T.; Wang, X.; Khan, S.; Di, D.; Xiqian, L.; Zhang, J.-J.; Anwar, M.N.; Nawaz, M.; et al. A Metagenomic Analysis of Mosquito Virome Collected From Different Animal Farms at Yunnan–Myanmar Border of China. Front. Microbiol. 2021, 11, 591478. [Google Scholar] [CrossRef]
- Shi, C.; Liu, Y.; Hu, X.; Xiong, J.; Zhang, B.; Yuan, Z. A Metagenomic Survey of Viral Abundance and Diversity in Mosquitoes from Hubei Province. PLoS ONE 2015, 10, e0129845. [Google Scholar] [CrossRef]
- Nguyen, P.T.T.; Culverwell, C.L.; Suvanto, M.T.; Korhonen, E.M.; Uusitalo, R.; Vapalahti, O.; Smura, T.; Huhtamo, E. Characterisation of the RNA Virome of Nine Ochlerotatus Species in Finland. Viruses 2022, 14, 1489. [Google Scholar] [CrossRef]
- Harvey, E.; Rose, K.; Eden, J.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive Diversity of RNA Viruses in Australian Ticks. J. Virol. 2019, 93, 3. [Google Scholar] [CrossRef]
- Ni, X.; Cui, X.; Liu, J.; Ye, R.; Chang, Q.; Zhao, L.; Han, X.; Ma, K.; Shen, S.; Zhang, M.-Z.; et al. Metavirome of 31 tick species provides a compendium of 1801 RNA virus genomes. Nat. Microbiol. 2023, 8, 162–173. [Google Scholar] [CrossRef]
- Roberts, J.M.K.; Anderson, D.L.; Durr, P.A. Metagenomic analysis of Varroa-free Australian honey bees (Apis mellifera) shows a diverse Picornavirales virome. J. Gen. Virol. 2018, 99, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Kadlečková, D.; Tachezy, R.; Erban, T.; Deboutte, W.; Nunvář, J.; Saláková, M.; Matthijnssens, J. The Virome of Healthy Honey Bee Colonies: Ubiquitous Occurrence of Known and New Viruses in Bee Populations. mSystems 2022, 7, e0007222. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, C.; Shi, M.; Bucek, A.; Bourguignon, T.; Lo, N.; Holmes, E.C. Unmapped RNA Virus Diversity in Termites and Their Symbionts. Viruses 2020, 12, 1145. [Google Scholar] [CrossRef] [PubMed]
- Van Eynde, B.; Christiaens, O.; Delbare, D.; Shi, C.; Vanhulle, E.; Yinda, C.K.; Matthijnssens, J.; Smaggh, G. Exploration of the virome of the European brown shrimp (Crangon crangon). J. Gen. Virol. 2020, 101, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Enghoff, H.; Golovatch, S.; Short, M.; Stoev, P.; Wesener, T. Diplopoda—Taxonomic overview. In Treatise on Zoology-Anatomy, Taxonomy, Biology-The Myriapoda; BRILL: Leiden, The Netherlands, 2015; Volume 2, pp. 363–453. [Google Scholar]
- Golovatch, S.I.; Kime, R.D. Millipede (Diplopoda) distributions: A review. Soil Org. 2009, 81, 565–597. [Google Scholar]
- Ebregt, E.; Struik, P.C.; Odongo, B.; Abidin, P. Pest damage in sweet potato, groundnut and maize in north-eastern Uganda with special reference to damage by millipedes (Diplopoda). NJAS Wagening. J. Life Sci. 2005, 53, 49–69. [Google Scholar] [CrossRef]
- Francisco, A.; Fontanetti, C.S. Diplopods and agrochemicals—A review. Water Air Soil Pollut. 2015, 226, 53. [Google Scholar] [CrossRef]
- Kania, G.; Klapec, T. Seasonal activity of millipedes (Diplopoda)–their economic and medical significance. Ann. Agric. Environ. Med. 2012, 19, 646–650. [Google Scholar]
- Shelley, R.M.; Lehtinen, P.T. Diagnoses, synonymies and occurrences of the pantropical millipeds, Leptogoniulus sorornus (Butler) and Trigoniulus corallinus (Gervais) (Spirobolida: Pachybolidae: Trigoniulinae). J. Nat. Hist. 1999, 33, 1379–1401. [Google Scholar] [CrossRef]
- Nieradko-Iwanicka, B.; Jung, M. Mass occurrences of millipedes in times of global climate change. Polish Hyperb. Res. 2020, 73, 81–88. [Google Scholar] [CrossRef]
- Voigtländer, K. Mass occurrences and swarming behaviour of millipedes (Diplopoda: Julidae) in Eastern Germany. Peckiana 2005, 4, 181–187. [Google Scholar]
- Kania, G. The Economic and Medical Significance of Millipedes (Diplopoda) with Emphasis on Ommatoiulus sabulosus. In Contributions to Soil Zoology in Central Europe III; Tajovsky, K., Schlagamersky, J., Pizl, V., Eds.; Institute of Soil Biology, Biology Centre, Academy of Sciences of the Czech Republic: Ceske Budejovice, Czech Republic, 2009; pp. 73–77. [Google Scholar]
- Semenyuk, I.I.; Tiunov, A.V. Foraging behaviour as a mechanism for trophic niche separation in a millipede community of southern Vietnam. Eur. J. Soil Biol. 2019, 90, 36–43. [Google Scholar] [CrossRef]
- de Sousa Antunes, L.F.; de Sousa Vaz, A.F.; Correia, M.E.F.; Cruvinel, F.F.; Martelleto, L.A.P. Millicompost: Sustainable substrate for the production of dragon fruit seedlings (Selenicereus undatus). Clean. Eng. Technol. 2021, 4, 100107. [Google Scholar] [CrossRef]
- Rosario, K.; Mettel, K.A.; Benner, B.E.; Johnson, R.; Scott, C.; Yusseff-vanegas, S.Z.; Baker, C.C.M.; Cassill, D.L.; Storer, C.; Varsani, A.; et al. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 2018, 6, e5761. [Google Scholar] [CrossRef]
- Dhandapani, G.; Woo, S.; Yeong, J.; Seong, N.; Jang, S.; Chan, M.; Hyun, K.; Gun, Y.; Dae, C.; Jeong, G.; et al. Detection of bat-associated circoviruses in Korean bats. Arch. Virol. 2021, 166, 3013–3021. [Google Scholar] [CrossRef]
- Jacquat, A.G.; Ulla, S.B.; Debat, H.J.; Muñoz-Adalia, E.J.; Theumer, M.G.; García-Pedrajas, M.D.; Dambolena, J.S. An in silico analysis revealed a novel evolutionary lineage of putative mitoviruses. Environ. Microbiol. 2022, 24, 6463–6475. [Google Scholar] [CrossRef]
- Tiunov, A.V. (Ed.) Structure and Functions of Soil Communities of a Monsoon Tropical Forest (Cat Tien National Park, Southern Vietnam); KMK Scientific Press: Moscow, Russia, 2011. (In Russian) [Google Scholar]
- Semenyuk, I.I.; Tiunov, A.V.; Golovatch, S.I. Structure of mandibles in relation to trophic niche differentiation in a tropical millipede community. Int. J. Myriap. 2011, 6, 37–49. [Google Scholar] [CrossRef]
- Litov, A.G.; Belova, O.A.; Kholodilov, I.S.; Gadzhikurbanov, M.N.; Gmyl, L.V.; Oorzhak, N.D.; Saryglar, A.A.; Ishmukhametov, A.A.; Karganova, G.G. Possible Arbovirus Found in Virome of Melophagus ovinus. Viruses 2021, 13, 2375. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wootton, J.C.; Federhen, S. Statistics of local complexity in amino acid sequences and sequence databases. Comput. Chem. 1993, 17, 149–163. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; Mcginnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2013, 9, 357–359. [Google Scholar] [CrossRef]
- Katoh, K.; Daron, M. Standley MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Le, O.; Peter, G.; Claude, C.; King, A.M.Q.; Knowles, N.J.; Nakashima, N.; Stanway, G.; Gorbalenya, A.E. Picornavirales, a proposed order of positive-sense single-stranded RNA viruses with a pseudo-T = 3 virion architecture. Arch. Virol. 2008, 153, 715–727. [Google Scholar]
- Fuchs, M.; Hily, J.-M.; Petrzik, K.; Sanfaçon, H.; Thompson, J.R.; van der Vlugt, R.; Wetzel, T.; Ictv Report Consortium. ICTV Virus Taxonomy Profile: Secoviridae 2022. J. Gen. Virol. 2022, 103, 001807. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Hu, S.-J.; Lin, X.-D.; Tian, J.-H.; Lv, J.-X.; Wang, M.-R.; Luo, X.-Q.; Pei, Y.-Y.; Hu, R.-X.; Song, Z.-G.; et al. Host traits shape virome composition and virus transmission in wild small mammals. Cell 2023, 186, 4662–4675.e12. [Google Scholar] [CrossRef] [PubMed]
- Waller, S.J.; Lamar, S.; Perry, B.J.; Grimwood, R.M.; Holmes, E.C.; Geoghegan, J.L. Cloacal virome of an ancient host lineage–The tuatara (Sphenodon punctatus)–Reveals abundant and diverse diet-related viruses. Virology 2022, 575, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Gu, D.M.A.; Hashimoto, Y.; Herrero, S.; De Miranda, J.R.; Ryabov, E.; Ictv Report Consortium. ICTV Virus Taxonomy Profile: Dicistroviridae. J. Gen. Virol. 2017, 98, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Brierley, I. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012, 93, 1385–1409. [Google Scholar] [CrossRef] [PubMed]
- Vinjé, J.; Estes, M.K.; Esteves, P.; Green, K.Y.; Katayama, K.; Knowles, N.J.; Homme, Y.L.; Martella, V.; Vennema, H.; White, P.A.; et al. ICTV Virus Taxonomy Profile: Caliciviridae. J. Gen. Virol. 2019, 100, 1469–1470. [Google Scholar] [CrossRef]
- Walter, C.T.; Pringle, F.M.; Nakayinga, R.; De, P.; Ryan, M.D.; Ball, L.A.; Dorrington, R.A. Genome organization and translation products of Providence virus: Insight into a unique tetravirus. J. Gen. Virol. 2010, 91, 2826–2835. [Google Scholar] [CrossRef]
- Hameed, A.S.S.; Ninawe, A.S.; Nakai, T.; Chi, S.C.; Johnson, K.L.; Ictv Report Consortium. ICTV Virus Taxonomy Profile: Nodaviridae. J. Gen. Virol. 2019, 100, 3–4. [Google Scholar] [CrossRef]
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the order Bunyavirales: Update 2019. Arch. Virol. 2020, 164, 1949–1965. [Google Scholar] [CrossRef]
- Kafer, S.; Paraskevopoulou, S.; Zirkel, F.; Wieseke, N.; Donath, A.; Petersen, M.; Jones, T.C.; Liu, S.; Zhou, X.; Middendorf, M.; et al. Re-assessing the diversity of negative strand RNA viruses in insects. PLoS Pathog. 2019, 15, e1008224. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Alkhovsky, S.V.; Avšič-županc, T.; Bergeron, É.; Burt, F.; Ergünay, K.; Garrison, A.R.; Marklewitz, M.; Mirazimi, A.; Papa, A.; et al. ICTV Virus Taxonomy Profile: Nairoviridae 2024. J. Gen. Virol. 2024, 105, 001974. [Google Scholar] [CrossRef]
- Amarasinghe, G.K.; Ayllón, M.A.; Bào, Y.; Basler, C.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Castón, J.; Hillman, B.; Kim, D.-H.; Kondo, H.; Nibert, M.; Lanza, D.; Sabanadzovic, S.; Stenger, D.; Wu, M.; et al. Reorganize the Order Ghabrivirales to Create Three New Suborders, 15 New Families, 12 New Genera, and 176 New Species. Available online: https://ictv.global/ictv/proposals/2023.015F.Ghabrivirales_reorg.zip (accessed on 23 May 2024).
- Kotta-Loizou, I.; Wu, C.; Moriyama, H.; Coutts, R. Create a New Family (Alternaviridae) Accommodating One New Genus (Alternavirus) and Five New Species. Available online: https://ictv.global/ictv/proposals/2022.001F.Alternaviridae_newfam.zip (accessed on 27 May 2024).
- Wolf, Y.; Kazlauskas, D.; Lucía-Sanz, A.; Kuhn, J.; Krupovic, M.; Dolja, V.; Koonin, E. Origins and Evolution of the Global RNA Virome. MBio 2018, 9, e02329-18. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L. Mitovirus UGA(Trp) codon usage parallels that of host mitochondria. Virology 2017, 507, 96–100. [Google Scholar] [CrossRef]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, X.; Yuan, H.; Shen, J.; Yang, M. Complete mitochondrial genome of Sellanucheza jaegeri Golovatch, 2013 by next generation sequencing (Polydesmida: Paradoxosomatidae) and phylogenetic analysis in Diplopoda. Mitochondrial DNA Part B Resour. 2018, 3, 603–604. [Google Scholar] [CrossRef] [PubMed]
- Donath, A.; Frank, J.; Al-arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Botella, L.; Manny, A.; Nibert, M.; Vainio, E. Create 100 New Species and Four New Genera (Cryppavirales: Mitoviridae). Available online: https://ictv.global/ictv/proposals/2021.003F.R.Mitoviridae_100nsp_4ngen.zip (accessed on 14 May 2024).
- Kuhn, J.H.; Dheilly, N.M.; Junglen, S.; Paraskevopoulou, S.; Shi, M.; di Paola, N. ICTV Virus Taxonomy Profile: Jingchuvirales 2023. J. Gen. Virol. 2023, 104, 001924. [Google Scholar] [CrossRef]
- French, R.K.; Anderson, S.H.; Cain, K.E.; Greene, T.C.; Minor, M.; Miskelly, C.M.; Montoya, J.M.; Wille, M.; Muller, C.G.; Taylor, M.W.; et al. Host phylogeny shapes viral transmission networks in an island ecosystem. Nat. Ecol. Evol. 2023, 7, 1834–1843. [Google Scholar] [CrossRef]
- Greninger, A.L.; DeRisi, J.L. Draft Genome Sequences of Ciliovirus and Brinovirus from San Francisco Wastewater. Genome Announc. 2015, 3, 3. [Google Scholar] [CrossRef]
- Richard, J.C.; Blevins, E.; Dunn, C.D.; Leis, E.M.; Goldberg, T.L. Viruses of Freshwater Mussels during Mass Mortality Events in Oregon and Washington, USA. Viruses 2023, 15, 1719. [Google Scholar] [CrossRef]
- Chiapello, M.; Bosco, L.; Ciuffo, M.; Ottati, S.; Salem, N.; Rosa, C.; Tavella, L.; Turina, M. Complexity and Local Specificity of the Virome Associated with Tospovirus-Transmitting Thrips Species. J. Virol. 2021, 95, e00597-21. [Google Scholar] [CrossRef] [PubMed]
- Viljakainen, L.; Holmberg, I.; Abri, S.; Jurvansuu, J. Viruses of invasive Argentine ants from the European Main supercolony: Characterization, interactions and evolution. J. Gen. Virol. 2018, 99, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Muscatt, G.; Kuhn, J.; Millard, A.; Bending, G.; Jameson, E. Create 418 New Genera and 1706 New Species in the Class Leviviricetes, Move and Rename 375 Species, and Abolish 114 Genera and 48 Species. Available online: https://ictv.global/filebrowser/download/15649?fid=15649#block-teamplus-page-title (accessed on 28 May 2024).
- Remnant, E.J.; Baty, J.W.; Bulgarella, M.; Dobelmann, J.; Quinn, O.; Gruber, M.A.M.; Lester, P.J. A Diverse Viral Community from Predatory Wasps in Their Native and Invaded Range, with a New Virus Infectious to Honey Bees. Viruses 2021, 13, 1431. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sadiq, S.; Tian, J.; Chen, X.; Lin, X.; Shen, J.; Chen, H.; Hao, Z.; Wille, M.; Zhou, Z.; et al. RNA viromes from terrestrial sites across China expand environmental viral diversity. Nat. Microbiol. 2022, 7, 1312–1323. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pool | Specimens in the Pool | Species |
|---|---|---|
| 1 | 4 | Antheromorpha festiva (Brölemann, 1896) |
| 2 | 4 | Atopochetus dollfusii (Pocock, 1893) |
| 3 | 6 | Cryxus ovalis (Linnaeus, 1758) |
| 4 | 1 | Helicorthomorpha cf. holstii (Pocock, 1895) |
| 5 | 5 | Hyleoglomeris cattienensis (Golovatch and Semenyuk, 2016) |
| 6 | 3 | Hylomus cattienensis (Nguyen, Golovatch, and Anichkin, 2005) |
| 7 | 4 | Hylomus pilosus (Attems, 1937) |
| 8 | 4 | Nedyopus dawydoffiae (Attems, 1953) |
| 9 | 13 | Orthomorpha rotundicollis (Attems, 1937) |
| 10 | 5 | Plusioglyphiulus ampullifer (Golovatch, Geoffroy, Mauriès, and VandenSpiegel, 2009) |
| 11 | 4 | Thyropygus carli (Attems, 1938) |
| 12 | 1 | Touranella moniliformis (Golovatch and Semenyuk, 2018) |
| 13 | 1 | Termitodesmus sp. |
| 14 | 1 | Trigoniulus corallinus (Gervais, 1842) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litov, A.G.; Semenyuk, I.I.; Belova, O.A.; Polienko, A.E.; Thinh, N.V.; Karganova, G.G.; Tiunov, A.V. Extensive Diversity of Viruses in Millipedes Collected in the Dong Nai Biosphere Reserve (Vietnam). Viruses 2024, 16, 1486. https://doi.org/10.3390/v16091486
Litov AG, Semenyuk II, Belova OA, Polienko AE, Thinh NV, Karganova GG, Tiunov AV. Extensive Diversity of Viruses in Millipedes Collected in the Dong Nai Biosphere Reserve (Vietnam). Viruses. 2024; 16(9):1486. https://doi.org/10.3390/v16091486
Chicago/Turabian StyleLitov, Alexander G., Irina I. Semenyuk, Oxana A. Belova, Alexandra E. Polienko, Nguyen Van Thinh, Galina G. Karganova, and Alexei V. Tiunov. 2024. "Extensive Diversity of Viruses in Millipedes Collected in the Dong Nai Biosphere Reserve (Vietnam)" Viruses 16, no. 9: 1486. https://doi.org/10.3390/v16091486