Abstract
The skin provides a life-sustaining interface between the body and the external environment. A dynamic communication among immune and non-immune cells in the skin is essential to ensure body homeostasis. Dysregulated cellular communication can lead to the manifestation of inflammatory skin conditions. In this review, we will focus on the following two key frontiers in the skin: innate immune sensors and cell death, as well as their cellular crosstalk in the context of skin homeostasis and inflammation. This review will highlight the recent advancements and mechanisms of how these pathways integrate signals and orchestrate skin immunity, focusing on inflammatory skin diseases and skin infections in mice and humans.
1. Skin Homeostasis and Immunity
1.1. Skin Structure
The skin consists of the epidermis, the dermis, and the hypodermis. The epidermis is the outermost layer of the skin. It mainly contains epithelial cells called keratinocytes, as well as other immune and non-immune cells such as melanocytes, Langerhans cells (LCs), and CD8+ T cells [1,2,3]. The mouse epidermis also harbours dendritic epidermal T cells (DETCs), which are absent in humans [4]. The dermis lies below the epidermis and harbours diverse groups of immune cells such as dendritic cells (dermal DCs, plasmacytoid DCs), mast cells, macrophages, T-lymphocytes (CD4+ T helper 1 (TH1), TH2, TH17 cells, γδ T cells), natural killer cells, and fibroblasts. They are integrated within a framework of extracellular matrices (collagen, elastic tissues, and reticular fibers) and lymphatic and blood vessels (Figure 1) [1,2,3].
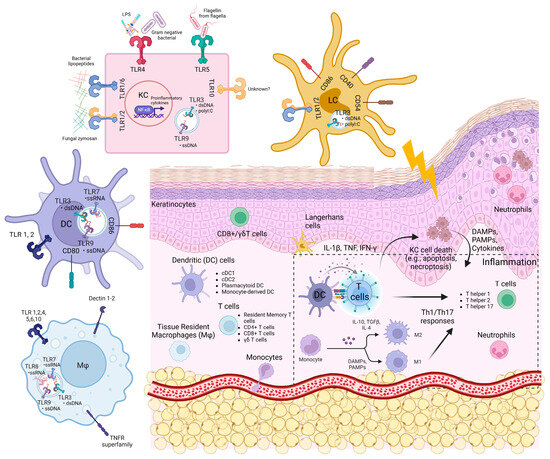
Figure 1.
The skin structure, skin cells, and expression of innate immune sensors.
Keratinocytes and innate immune cells are the key sentinels in the skin. They express pattern recognition receptors such as toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and nucleic acid sensors, which enable them to sense foreign pathogens and molecules such as PAMPs (pathogen-associated molecular patterns) and host DAMPs (damage-associated molecular patterns) (Figure 1 and Figure 2). They also possess molecular frameworks facilitating programmed cell death, such as apoptosis, necroptosis, and pyroptosis (Figure 2) [5,6,7].

Figure 2.
Programmed cell death, innate immune signalling, and their interactions. Interaction between the programmed cell death (PCD) pathways and NF-κB signalling central to the innate immune sensors in the skin cells. Different innate immune sensors such as TLRs, NLRs, nucleic acid sensors (AIM-2, cGAS-STING, RIG-I, MDA-5, and ZBP1), as well as cell death can directly or indirectly promote inflammatory responses and inflammation in the skin.
Epithelial–immune cell communication is essential for skin homeostasis [3,8,9]. The dysregulation of epithelial–immune cell communication can lead to the development of inflammatory skin conditions [3,10,11].
1.2. Skin Immunity
Skin homeostasis is maintained through a coordinated network of immune cells, non-immune cells, effector molecules (e.g., cytokines, chemokines), and immunomodulatory signalling pathways (e.g., NF-κB, AP-1). Innate immune cells, such as myeloid cells and adaptive immune cells, mainly lymphoid cells, are the major immune cell types in the skin. Both cellular pools are derived from the hematopoietic stem cell lineage [12].
Myeloid cells in the skin include DCs, LCs, macrophages, monocytes, and neutrophils. These cells actively engage in functions such as phagocytosis, antigen-presentation, antimicrobial peptide production, cytotoxic granules, cytokines and chemokines secretion, and provide an immediate response to stimuli. They also clear dead cells and tissue debris and promote tissue repair. These cells also coordinate the activation of the adaptive arm of skin immunity.
Lymphoid cells in the skin include CD4+ T helper cells, CD8+ cytotoxic T cells, regulatory T cells, resident memory T cells, gamma delta T cells (γδ T cells), and B lymphocytes. They are responsible for targeting specific immune responses and promoting long-term and rapid immunity upon the recurrence of diseases and infections [13].
Collectively, both myeloid and lymphoid immune cells facilitate a balanced and dynamic immune environment to respond to inflammatory stimuli and restore and maintain skin homeostasis.
Tightly regulated epithelial–immune signalling crosstalk is essential to maintain skin homeostasis. Keratinocytes and innate immune cells (e.g., macrophages, Langerhans cells, and dendritic cells) harbour several innate immune sensors (e.g., TLRs, NLRP inflammasomes, etc.) that help to mount immune responses. Dysregulated innate immune sensing in the skin is linked to cell death (e.g., pyroptosis and necroptosis) and autoimmune/inflammatory skin diseases (e.g., psoriasis and cutaneous lupus). DAMPs and PAMPs released from dead cells, particularly those from pyroptosis and necroptosis, can further amplify the responses of innate immune sensors in a positive feedback loop, exacerbating inflammation and death in the skin in unison with the production of pro-inflammatory cytokines such as TNF, IL-1β, IFN-γ, and others.
2. NF-κB, Programmed Cell Death, and Skin Immunity
Programmed cell death plays a key role in maintaining skin immunity [14,15,16,17]. Apoptosis, necroptosis, and pyroptosis are the most characterised types of programmed cell death. Figure 2 highlights these forms of cell death, their components, and interactions. Apoptosis is widely recognised as a non-inflammatory and immunologically silent form of cell death, where cellular components are enclosed in apoptotic bodies [18]. However, excessive apoptosis can drive inflammation in the skin [19,20,21,22,23]. In contrast to apoptosis, necroptosis, and pyroptosis are inflammatory forms of programmed cell death, in which the cells rupture and disrupt their plasma membrane integrity [14,15,16]. This occurs through the formation of mixed lineage kinase domain-like (MLKL) pores in necroptosis [24] and Gasdermin D pores in pyroptosis [25], resulting in the release of endogenous DAMPs such as high mobility group box 1 (HMGB-1) [26,27,28,29], heat shock proteins (HSPs) [30], and nucleic acids [31,32] thus exacerbating inflammation. A membrane protein, Ninjurin-1 (NINJ1), has also been found to be involved in plasma membrane rupture and the release of DAMPs in lytic cell death, including pyroptosis and necroptosis, hence promoting inflammation [33].
The relationship between programmed cell death and inflammation is a double-edged sword. Inflammation is essential to the mounting of immune responses during infections or tissue injuries, but it must also be controlled and resolved to restore skin homeostasis. If the interplay between cell death and inflammation is not properly regulated, this could drive excessive and persistent inflammation, which may contribute to the pathogenesis of chronic inflammatory skin diseases.
Programmed cell death has been associated with several inflammatory skin diseases such as psoriasis [34] and toxic epidermal necrolysis [35]. However, their relevance to skin diseases is not fully understood. Previous studies in mouse models have shown that dysregulated NF-κB signalling and programmed cell death in the keratinocytes drive skin inflammation [14,17,36,37]. Both the overactivation and inhibition of NF-κB in the epithelial cells of the skin led to the development of inflammatory skin phenotypes in mice [17,38]. Furthermore, mice with dysregulated programmed cell death such as the epidermal keratinocyte-specific deletion of apoptotic components—Fas-associated protein with death domain (FADDE-KO) [39] or caspase-8 (Caspase-8E-KO) [40,41]—or apoptotic/necroptotic components—receptor-interacting protein kinases (RIPKs) 1 (RIPK1E-KO) [42]—experienced the development of inflammatory skin lesions. The skin phenotype of FADDE-KO and RIPK1E-KO mice were characterised by epidermal hyperplasia, the upregulation of cytokines/chemokines, and the accumulation of immune cells and was driven by necroptosis. Crossing FADDE-KO or RIPK1E-KO mice with Ripk3 or Mlkl knockout mice prevented the development of skin lesions [39,42,43].
An overactivation of NF-κB in mice by deleting NF-κB inhibitor alpha (IκBα), a cytoplasmic inhibitor of NF-κB, resulted in a T cell-mediated severe skin inflammatory phenotype, leading to lethality at postnatal day (P) 8 [44]. Moreover, an overexpression of the inhibitor of NF-κB kinase (IKK2), a kinase activating NF-κB, in the epithelial cells also led to the development of inflammatory skin phenotype and the recruitment of macrophages and T cells in the skin by P14 [45].
The inhibition of NF-κB or the dysregulation of the NF-κB pathway components also led to the development of inflammatory skin disease. Keratinocyte-specific deletion of the gene encoding for the NF-κB essential modulator (NEMO), a regulatory subunit of the IKK complex in the NF-κB pathway, led to the development of inflammatory skin lesions at P2 and lethality at P6 [46]. The skin lesion development of the epidermis-specific NEMO knockout mice (NEMOE-KO) was partially dependent on TNFR1 signalling, as the genetic inhibition of TNFR1 prevented the development of skin lesions in these mice at an early stage [46]. However, the NEMOE-KO/Tnfr1−/− mice developed skin inflammation in adulthood [46]. NEMO deficiency in humans also causes a genetic disorder, incontinentia pigmenti, which is characterised by the development of skin lesions, as well as other complications, and is detrimental to males [47]. An overexpression of the degradation-resistant super--repressor of IκBα (K5-IκBαSR and K14-IκBαM) caused inflammation [48,49]. K5-IκBαSR mice developed squamous cell carcinoma in adulthood in a TNF-dependent manner [48,50], where the K14-IκBαM mice died by P5–P7. The inhibition of IKK2 in the keratinocytes resulted in microscopic inflammatory skin lesions at P3–P4, which reached severity and led to lethality at P7–P9 [51,52]. Crossing IKK2E-KO mice with full-body or epithelial cell-specific TNFR1-deficient mice rescued the skin lesion development, showing that TNFR1 signalling in keratinocytes drives the development of the inflammatory skin phenotype in these mice [51,52,53]. In subsequent studies, interleukin (IL)-24 signalling and cell death were identified as major drivers of the inflammatory skin phenotype of the IKK2E-KO mice [52,53]. The genetic inhibition of the components of necroptosis, RIPK3 or mixed lineage kinase domain-like pseudokinase (MLKL) (IKK2E-KO/Ripk3−/− or IKK2E-KO/Mlkl−/−), significantly ameliorated the severity of the skin disease [52], and the combined inhibition of both the necroptotic and apoptotic pathways by the global deletion of RIPK3 and FADD (IKK2E-KO/FADDE-KO/Ripk3−/−) completely prevented the inflammatory skin phenotype development in these mice [52]. Lastly, the deletion of both NF-κB subunits, RelA and c-Rel, in keratinocytes (RelAE-KO/cRelE-KO) led to the development of inflammatory skin lesions similar to the IKK2E-KO mice [52,54]. Crossing them with RIPK1 kinase mutated mice (Ripk1D138N/D138N) or Mlkl−/− mice prevented the inflammatory skin phenotypes observed in RelAE-KO/cRelE-KO mice at an early age, and they only developed mild lesions between the age 6–9 months or 3–4 months, respectively [52]. These data demonstrated that NF-κB acts as a checkpoint regulator to prevent epithelial cell death, and the inhibition of NF-κB signalling results in cell death and inflammation in the skin.
The cellular inhibitor of apoptosis protein (cIAP1) is a component of the NF-κB pathway [55,56,57,58]. Studies have shown that the inhibition of cIAP1 in the epidermis, as well as cIAP2, ubiquitously leads to skin inflammation and cell death [19]. Additionally, the deletion of tumour necrosis factor receptor-associated factor 2 (TRAF2) in keratinocytes (TRAF2E-KO) leads to a TNF-dependent epidermal thickening and skin inflammation [20]. The deletion of MLKL did not prevent the onset of skin inflammation in the TARF2E-KO mice [20]. However, the simultaneous deletion of MLKL and caspase-8 rescued the skin phenotype, suggesting that apoptosis, and not necroptosis, led to the inflammatory skin phenotype in these mice [20].
The global mutation of cpdm (encodes for SHANK-associated RH domain interactor (SHARPIN)), a component of the linear ubiquitin chain assembly complex (LUBAC), resulted in the spontaneous development of inflammatory skin lesions (Sharpincpdm) [21,22,23]. LUBAC consists of haem-oxidised iron-responsive element-binding protein 2 (IRP2), ubiquitin ligase-1 (HOIL-1), HOIL-1-interacting protein (HOIP), and SHARPIN, which adds linear ubiquitin to NF-κB signalling components such as NEMO to promote the nuclear translocation of NF-κB [59]. Further studies on this model showed the prevalence of massive cell death in the epidermis of Sharpincpdm mice. Moreover, the skin inflammation in these mice was driven by TNF, epithelial TNFR1 and RIPK1 kinases, FADD/ caspase-8-mediated cell death, and innate immune sensors, adaptors and components of inflammasomes [22,23,60,61,62,63,64,65].
Deficiency of the linear ubiquitin chain-specific deubiquitinase, OTULIN, causes OTULIN-related autoinflammatory syndrome (ORAS) in humans. Mice lacking Otulin (OTULINE-KO) also develop an inflammatory skin disease [66,67]. Similar to Sharpincpdm mice, the phenotype of OTULINE-KO mice is driven by TNFR1 and RIPK1 kinases. Moreover, RIPK3 or MLKL deficiency ameliorates skin lesion development, while the combined deletion of epidermal FADD and RIPK3 prevents skin lesion development [66,67].
In summary, these studies have shown that dysregulated NF-κB signalling and the programmed cell death pathway initiate the inflammatory skin phenotype in mice; their crucial roles in skin immunity have also been highlighted.
3. Innate Immune Sensors and Cell Death: Expression, Activation Signalling, and Their Contribution to Skin Inflammation and Immunity
The skin cells harbour several innate immune sensors that serve as the first line of defence to detect PAMPs and DAMPs. These sensors include TLRs, C-type lectin receptors, cytoplasmic sensors such as NOD-like receptors (NLRs), nucleic acid sensors such as RLRs, cyclic guanosine monophosphoate (GMP)–adenosine monophosphate (AMP) synthase-stimulator of interferon genes (cGAS-STING), interferon gamma-inducible protein 16 (IFI16), Z-DNA binding protein 1 (ZBP1), and absent in melanoma 2 (AIM2) [68,69,70,71]. In the skin, these pattern recognition receptors (PRRs) work in unison to mount immune responses and maintain skin homeostasis. Figure 1 shows the expression of these innate immune sensors on various cell types in the skin, and Figure 2 shows their downstream signalling and interactions with cell death pathways.
3.1. Toll-like Receptors (TLRs)
TLRs recognise the conserved molecular patterns of microbes, known as PAMPs, and promote the recruitment and activation of innate immune cells such as macrophages and DCs, which are essential to the coordination of innate and adaptive immunity. TLRs are type I integral glycoproteins with three domains—an N-terminus ligand recognition domain, a transmembrane domain, and a C-terminus cytoplasmic signalling domain. The recognition of microbial components and host stimuli through TLRs activates downstream signalling cascades, such as NF-κB and mitogen-activated protein kinase (MAP) kinases, resulting in the expression of cytokines and chemokines [72,73,74].
TLRs are grouped into two different categories based on their localisation. TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are localised at the cell surface membrane to sense microbial cell wall components, while TLR3, TLR7, TLR8, and TLR9 mainly detect nucleic acid components and are localised at the endosome of the cells [72,73,75]. Myeloid differentiation primary response 88 (MyD88) and toll/interleukin-1 receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF) are the two adaptor molecules in the TLR pathways. All TLRs can utilise MyD88-dependent signalling except TLR3. This initiates the formation of a complex consisting of IL1R-associated kinase (IRAK) family kinases such as IRAK1, IRAK3, IRAK4, and IRAK-M [72,76,77,78]. This leads to the recruitment of tumour necrosis factor receptor-associated factor 6 (TRAF6), which activates transforming growth factor beta-activated kinase 1 (TAK1) to phosphorylate the IKK complex. IKK2 then phosphorylates IκBα, resulting in the proteasome-mediated degradation of IκBα and the dissociation of NF-κB to translocate to the nucleus for the activation of the NF-κB-mediated genes. TAK1 can also activate the MAPK pathway to allow for the translocation of AP-1 to the nucleus to stimulate the pro-inflammatory factors [72,76,77,78].
TLR3 and TLR4 use TRIF to recruit TRAF6 and/or TRAF3 [76,77,78]. TRAF6 recruits RIPK1 to activate TAK1 for the activation of NF-κB and MAPK pathways. Meanwhile, TRAF3 recruits TANK-binding kinase 1 (TBK1) and inhibitor of κB kinase ϵ (IKKϵ), which phosphorylate and translocate interferon regulatory factor 3 (IRF3) and eventually lead to the production of interferon-β (IFNβ) [76,77,78].
Epidermal keratinocytes constitutively express TLR 1–6, 9, and 10 and play an important role in cutaneous inflammation and antiviral responses [9,79,80,81,82]. TLR1 can detect lipoteichoic acid, lipopeptides, and peptidoglycans from bacteria and zymosan from fungus and forms heterodimer with TLR2 or TLR6 [9,81,83]. TLR3 binds to dsRNA, which can be released from viruses and damaged host cells during cellular stress. A synthetic TLR3 ligand, polyinosinic–polycytidylic acid (polyI:C), has been shown to induce proinflammatory cytokines such as IL-6, IL-8, IL-1β, TNF, IFN-β, IL-23p19, IL-8, CCL5, and CCL20 in keratinocytes [84,85,86,87]. Additionally, TLR3 upregulates genes such as ATP-binding cassette, sub-family A, member 12 (ABCA12), glucocerebrosidase, acid sphinoglyelinase, and transglutaminase 1, which are involved in skin barrier recovery and wound healing in normal human epidermal keratinocytes (NHKs) [87].
TLR4 and TLR5 in keratinocytes are crucial players in the cutaneous defence against Gram-negative pathogens. TLR4 activation via lipopolysaccharide (LPS) stimulation regulates the proinflammatory cytokines and chemokines [79]. Cytokines like IFN-γ have been shown to increase the LPS-mediated expression of TLR2 and TLR4 genes in human keratinocytes in vitro [88]. TLR5 is expressed on the basal layer of the epidermis [89,90]. Stimulation of a primary air-lifted organotypic keratinocyte culture with bacterial flagellin and transforming growth factor (TGF)-α, a growth and differentiation factor important during wound healing and psoriasis, increased the level of IL-8 production in the keratinocytes [90]. Upon the engagement of TLR4 or TLR5 with the respective ligands, the NF-κB pathway was activated, which promoted the upregulation of IL-8, a chemoattractant for neutrophils and other immune cells to the site of infection and injury. TLR7 and 8 recognise single-stranded RNA but are not constitutively expressed in keratinocytes [79]. However, studies have shown that the imiquimod (TLR7/8 agonist) can induce NF-κB-mediated immune response in keratinocytes in the presence of calcium [80,91]. TLR9 binds to DNA with unmethylated CpG motifs released from damaged cells, bacteria, and viruses [92]. The stimulation of keratinocytes with CpG-oligodeoxynucleotides led to the production of CXCL8 and TNF in a concentration- and time-dependent manner through the phosphorylation of IκBα and the activation of NF-κB subunit p65 [79]. Moreover, the engagement of TLR9 can induce CXCL10 and IFNα in keratinocytes [79]. TLR10 is expressed in primary keratinocytes, but the PAMPs associated with the receptor in the skin have yet to be defined [80].
Immune cells in the skin also express a variety of TLRs (Figure 1). Human skin LCs express mRNA encoding TLR 1-3, TLR5, TLR6, and TLR10. LCs are highly responsive to TLR2 and TLR3 ligands, leading to IL-6, IL-8, and TNF-α production. However, poly(I:C)-stimulated LCs do not produce IFN-α/β [93,94]. In another study, poly(I:C) stimulation of LCs for two days led to the upregulation of co-stimulatory molecules (CD40, CD54, CD86), enhanced LC survival, and upregulated CXCL10, IL-6, and IL-12p40 [95]. LC-like DCs are derived from CD34+ progenitor cells and express comparable levels of TLR1-10 mRNA as monocyte-derived DCs [96]. They are responsive to TLR2 and TLR7/8 ligands, leading to the production of IL-8, IL-12, CCL3, CCL4, and TNF [96]. TLR3 stimulation of LC-like DCs also leads to IFN-β production, suggesting that LC might play a role in the antiviral activity within the skin [96]. Lastly, imiquimod and R-848 (TLR7/8 ligands)-treated human epidermal LCs promoted T cell proliferation in a primary mixed lymphocyte reaction, leading to T cell-mediated IFN-γ, IL-12, IL-1β, and TNF production [97].
Immature myeloid DCs express TLR 1–3, and their stimulation induces maturation as confirmed by the expression of co-stimulatory molecules, CD80 and CD86. The stimulation subsequently led to IL-12 production and the promotion of the Th1 immune response [98]. Plasmacytoid DCs express TLR7 and 9 [99,100], and the stimulation of TLR9 through CpG DNA induced type I IFN production [99,100]. Monocytes predominantly express TLR1, TLR2, TLR4, TLR5, and TLR8 [99,100]. Engaging these TLRs has been shown to enhance the phagocytic ability of macrophages and the release of TNF and IL-6 [101].
Mast cells in the skin mediate IgE allergic reactions and express TLR 1–7 and 9 mRNA transcripts. The activation of TLR3 and TLR9 by poly(I:C) and CpG DNA, respectively, inhibited mast cell adhesion to vitronectin and fibronectin in vitro [102]. Moreover, poly(I:C) stimulation of the mast cells resulted in the production of type I IFN. These studies suggest that mast cells may have a role in antiviral immunity [103]. T cells in the skin also express TLRs 1–10s [104]. Stimulation of T cells via the CD3 monoclonal antibody and TLRs 2, 5, 8, and 9 ligands leads to T cell proliferation and IFN-γ production [105,106,107,108,109].
Pyroptosis is an inflammatory form of cell death (Figure 2). TLRs and cytokines such as TNF act as a priming signal in pyroptosis, which activate NF-κB to induce the transcription of genes encoding for the inflammasome components and pro-IL-1β [110,111]. The presence of secondary signals such as DAMPs, potassium efflux, nigericin, or ATP then activates the formation of complexes, known as inflammasomes, which trigger pyroptosis and subsequently the release of proinflammatory factors like IL-1β [112,113,114]. Interestingly, DAMPs released during necrotic cell death such as high mobility group protein box 1 (HMGB1), a highly conserved nuclear protein, can also activate TLR2, 4, and 9 [115,116]. In the skin samples of patients with hidradenitis suppurativa (HS), a debilitating skin disease with limited therapeutic options [117], caspase-1 activation was observed in association with the heightened expression of NLRP3, IL1B, and IL18 [118]. In HS pathogenesis, the rupture of hair epidermal cysts releases endogenous DAMPs such as high-molecular-weight cornified keratin into the dermis [119], which has been shown to activate the NLRP3 inflammasome [120,121], suggesting that DAMPs and pyroptosis may play a role in exacerbating inflammation in HS.
Necroptosis is another form of programmed cell death (Figure 2). The stimulation of poly(I:C) or TLR4 to FADD-deprived Jurkat cells or mouse macrophages in the presence of caspase inhibitors has been shown to activate RIPK3-mediated necroptosis [122,123]. TLR3 engagement leads to the recruitment of TRIF and interaction with RIPK3 through RHIM domain, promoting the generation of reactive oxygen species (ROS) and necroptosis [123]. In human keratinocyte cell line, HaCaT, TLR3 agonist stimulation sensitised the cells towards RIPK1- and RIPK3-dependent necroptosis in the absence of cIAPs and caspases [124]. In mice injected with poly(I:C) or LPS with zVAD-fmk (pan-caspase inhibitor) led to necroptosis in macrophages and the increased production of IL-6, TNF, MCP-1, and IFN-γ [123]. When RIPK3-deficient mice or dominant negative mutant TrifLPS2/LPS2 were injected with poly(I:C) or LPS with zVAD-fmk, the death of macrophages was rescued [123]. This study suggests that RIPK3 and TRIF are involved in TLR-mediated necroptosis. It has also been suggested that DAMPs such as HMGB1 may be released during TLR4-mediated necroptosis as the TLR4 signalling pathway contributes to IRAK4-dependent HMGB1 secretion in macrophages [125]. Lastly, it has been shown that TLR3 activation through poly(I:C) is susceptible to the induction of cell death in HaCaT human keratinocytes, and this process requires the activation of caspase-3 by TLR3 and its adaptor, TRIF [126]. TLR4 and MyD88 have also been shown to facilitate UV irradiation-induced cell death in murine macrophages [127].
TLRs are critical components in innate immunity and play a central role in pathogen recognition during infections and maintaining homeostasis. A genetic screening study on 110 healthy human subjects showed that individuals with the TLR2 gene polymorphism (substitution of arginine to glutamine at gene position 753 [Arg753Gln]) had an increased risk of developing staphylococcal infection [128]. Moreover, atopic dermatitis patients with a TLR2 Arg753Gln gene polymorphism showed an increase in IL-6 and IL-12 levels as compared to patients with no mutation in the TLR2 gene upon TLR2 agonist stimulation of the monocytes [129]. In a mouse model of the Staphylococcus aureus (S. aureus) infection, the TLR2- and MyD88-deficient mice were highly susceptible, and the MyD88-deficient mice failed to recruit neutrophils to the infection site [130]. Therefore, TLR2 plays an important role in the pathogenesis of atopic dermatitis and may be involved in enhanced S. aureus skin infection [129].
Psoriasis is a well-characterised inflammatory skin disease with keratinocyte hyperproliferation and massive immune cell accumulation. T cells, innate immune cells, and keratinocytes are major contributors to the pathogenesis of psoriasis [131,132,133,134]. In psoriatic skin lesions, heat shock proteins were found to be overexpressed, which stimulated TLR4 on antigen-presenting cells such as the LCs [135]. TLR1 and TLR2 were elevated in the suprabasal keratinocyte layer in non-lesional and lesional psoriatic skin as compared to normal human skin, while TLR5 was downregulated in the basal keratinocyte layer of lesional psoriatic skin as compared to non-lesional psoriatic skin, suggesting that TLRs may play a role in the pathogenesis of psoriasis [89]. Similarly, other skin diseases such as HS have also been associated with the differential expression of TLRs [136,137,138]. In a mouse model of psoriasis, the inhibition of MyD88 in myeloid cells led to the amelioration of the skin phenotype, suggesting a critical role of TLR-mediating signalling during skin inflammation [139].
In several mouse models, which are driven by cell death, the importance of TLR and IL-1 signalling has been demonstrated, highlighting the crosstalk between cell death and TLRs in the disease context. In Sharpincpdm mice, in which inflammation is driven by cell death [22,23,60], the deficiency of MyD88 [64] or TLR3 [63] ameliorated skin lesion development. In RIPK1E-KO mice, in which the skin lesions are driven by necroptosis, the deficiency of TRIF mildly ameliorated inflammation [42]. In OTULINE-KO mice, where skin lesion development is driven by caspase-8-mediated cell death and necroptosis, the deficiency of Myd88 or injection with Anakinra, an IL-1 receptor inhibitor, strongly protected the development of skin lesions [66,67]. Single-cell RNA sequencing also showed an upregulation of Il1b and the genes involved in pyroptosis, such as Casp1, Pycard, and Nlrp3, in the lesional skin of OTULINE-KO mice [66], which suggests that pyroptosis may be implicated in the pathogenesis.
PTPN6 encoding tyrosine-protein phosphatase non-receptor type 6, which is also known as Src homology 2 (SH2) domain-containing cytosolic phosphatase 1 (SHP-1), is a key component controlling inflammation and cell death [140]. Several different mutations in the mouse Ptpn6 gene have been associated with neutrophilic dermatoses and autoimmune diseases [141,142]. For instance, functionally inactive PTPN6 (Ptpn6me/me mice) caused the mice to develop patchy hair loss and pigment in the skin before succumbing at 2–3 weeks of age [143,144,145,146,147]. Additionally, the insertion of the B2 element into exon 6 of mouse Ptpn6 (Ptpn6meB2/meB2) also led to the skin inflammation of the paws at 3–5 weeks of age [148]. Interestingly, mice that are homozygous recessive for the missense Y208N spontaneous inflammation (spin) in the carboxy terminus of Ptpn6 gene (Ptpn6spin mice) develop spontaneous skin lesions, chronic footpad swelling, and purulent inflammation at 8–16 weeks of age [149]. Neutrophils are the main driver of cutaneous inflammation in PTPN6-deficient mice [150].
The deletion of IL-1α, but not IL-1β, alleviates the Ptpn6spin-mediated skin phenotype; this is not mediated through NLRP3, as crossing these mice with the Nlrp3−/− or Casp1−/− did not rescue the skin phenotype [151]. However, the pharmacological inhibition of RIPK1 through necrostatin-1 and the genetic blockade of RIPK1, but not RIPK3 deletion, protected Ptpn6spin mice against wound-induced inflammation [151]. PTPN6 was found to inhibit the Syk-dependent MyD88 phosphorylation and engagement of RIPK1, TAK1, and apoptosis signal-regulating kinase (ASK) to prevent IL-1α signalling; hence, the absence of Ptpn6 led to inflammatory skin disease development [152,153]. PTPN6 deficiency in polymorphonuclear neutrophils (PMNs) Ptpn6ΔPMN also led to the inflammatory skin phenotype, which was prevented by the combined deletion of Casp8ΔPMN and either Ripk3−/− or Mlkl−/−, suggesting that PTPN6 maintains RIPK1’s function to prevent caspase-8 and RIPK3–MLKL-dependent cell death. PTPN6 is also a negative regulator of p38 MAPK, regulating TNF and IL-1α/β expression [154]. Additionally, the ablation of CARD9 in Ptpn6spin mice led to the downregulation of MAPK and NF-κB signalling, which dampened inflammation [155]. Therefore, the Ptpn6 murine model provides insights into the mechanisms controlling neutrophilic dermatosis. Clinical findings of these mice closely resemble those observed in human neutrophilic dermatoses, such as Sweet’s syndrome and pyoderma gangrenosum, opening therapeutic opportunities for human skin diseases [156,157,158].
These findings suggest the pro-inflammatory roles of TLRs either as an upstream regulator of inflammation or by acting as PAMP/DAMP receptors during stress. However, more investigation is needed to dissect the inflammation and cell death axes in the context of skin inflammation.
3.2. NOD-like Receptors (NLRs)
NOD-like receptors (NLRs) are present in the cytosols of immune and non-immune cells and sense intracellular DAMPs and PAMPs. NLRs are divided into five subfamilies based on different domains at the N-terminus. Here, we will focus on the NOD-like receptor CARD domain-containing (NLRC) family and the NOD-like receptor pyrin domain-containing (NLRP) family [159,160,161]. NLRC contains an N-terminal caspase activation and recruitment domain (CARD), a centrally located nucleotide-binding and oligomerisation domain (NACHT), and carboxy-terminal leucine-rich repeats (LRRs). The NLRPs contain a pyrin domain (PYD) at the N-terminus, the NACHT domain, and LRR domain at the C terminus [162]. Stimulation of NLRs drives the initiation of NF-κB and MAPK signalling pathways and subsequently promotes the expression of pro-inflammatory cytokines and chemokines that recruit immune cells, as well as the activation of inflammasomes that lead to programmed cell death known as pyroptosis (Figure 2) [159,160,161,163].
3.2.1. NLRC Subfamily
The NLRC subfamily is classified based on the presence of the CARD domain at the N-terminus. Amongst the NLRC family, NOD1, NOD2, and NLRC5 are expressed in keratinocytes and immune cells in the skin [163,164,165]. NOD1 and NLRC5 have one CARD domain, while NOD2 has two in tandem. Both NOD1 and NOD2 bind to peptidoglycans. NOD1 recognises γ-D-glutamyl-mesodiaminopimelic acid (iE-DAP), while NOD2 senses muramyl dipeptide (MDP) and single-stranded RNA from viruses (Figure 2) [166,167,168,169,170,171,172,173,174,175]. NLRC5 plays a regulatory role in the innate immune response [176].
Upon ligand recognition, NOD1 and NOD2 recruit RIPK2 via the CARD domain, which leads to the autophosphorylation and ubiquitination of RIPK2 [160,161,163,169,177,178,179,180]. This is further targeted by an X-linked inhibitor of apoptosis (XIAP) and other E3 ligases for non-degradative polyubiquitination, and this process further recruits TAK1 and IKK kinases to initiate the MAPK and NF-κB signalling pathway [169,177,178,179,180]. Through knockdown studies in human keratinocytes, it has been shown that NOD1 mediates Pseudomonas aeruginosa-induced CXCL8 secretion [181]. Moreover, the stimulation of primary keratinocytes with MDP results in the release of antimicrobial peptide human β-defensin-2 [182]. Furthermore, the knockdown of NOD2 in keratinocytes reduces S. aureus-induced IL-17C expression [183]. These suggest that NOD1 and NOD2 play an important role in the induction of cytokines and chemokines in keratinocytes and help in mounting immune responses against infections [184].
NLRC5 is expressed in keratinocytes and other immune cell types such as lymphocytes and macrophages [176,185]. IFN-γ can also induce NLRC5 and regulate major histocompatibility complex (MHC I) expression in keratinocytes [165,186]. In keloid fibroblasts, silencing NLRC5 inhibits extracellular matrix expression by inhibiting the transforming growth factor-beta1/Smad signalling pathway [187]. NLRC5 has also been shown to negatively regulate NF-κB activation, type I IFN, and inflammatory cytokine production [188]. Lastly, cytomegalovirus-infected human fibroblast showed the upregulation of NLRC5 and the production of IFN-γ-mediated by the Janus kinase-signal transducer and activator of transcription (JAK/STAT) signalling pathway, suggesting that NLRC5 contributes to the antiviral defence response in the skin [189]. Nonetheless, the role of NLRC5 in skin homeostasis and immunity is still unclear.
3.2.2. Nucleotide-Binding Oligomerisation Domain, Leucine-Rich Repeat, and Pyrin Domain-Containing Protein (NLRP)
The NLRP subfamily is classified based on the presence of the pyrin domain at the N-terminus, which allows for the recruitment of the inflammasome-activating scaffold protein, the apoptosis-associated speck-like protein containing a CARD (ASC) [190,191]. NLRP1, 3, and 10 are expressed in human skin [192,193]. Inflammasome activation facilitates the cleavage of caspase-1, pyroptotic cell death, and the processing and release of IL-1β and IL-18 [190,191,194]. Unstimulated human keratinocytes cultured in vitro have been shown to constitutively express pro-IL1B and IL18, suggesting that keratinocytes do not require priming for their production. [195,196,197,198].
NLRP1 is highly expressed in keratinocytes and is the predominant form of NLRPs in the skin [192,199]. Human NLRP1 is activated by ultraviolet (UV)B radiation [200]. However, UVB irradiation of mouse keratinocytes does not lead to the activation of inflammasomes, indicating that NLRP1 in human and mouse keratinocytes may not be highly conserved [200]. Other activators of human NLRP1 include long dsRNA, bacterial toxin (e.g., nigericin), and viral 3C proteases [201,202,203,204]. Poly(dA:dT), a synthetic dsDNA mimetic, has been shown to induce NLRP1 in human keratinocytes, leading to the activation of canonical inflammasomes (caspase-1, ASC) and IL-1β release [203]. Single-nucleotide polymorphisms in the NLRP1 gene has also been shown to be associated with several inflammatory skin diseases such as atopic dermatitis and psoriasis vulgaris [205,206,207].
NLRP3 is also expressed in skin and keratinocytes [192,208]. In human keratinocytes, NLRP3 can be activated by various signals such as dsRNA (poly I:C), contact sensitisers, mite allergens, UVB, and oxidative stress. These triggers activate the inflammasome, leading to the cleavage of caspase-1 and pyroptotic cell death [195,196,197,198]. NLRP3 also engages the non-canonical inflammasome, in which caspase-4 and caspase-5 cleave GSDMD to generate pore-forming N-terminal domain GSDMD-N [25,209,210,211,212]. This allows for the release of DAMPs and pro-inflammatory cytokines such as Il-1β and IL-18 [213,214]. Unlike conventionally released cytokines, which employ the endoplasmic reticulum (ER)/ Golgi pathways, IL-1β lacks a signal peptide and is secreted through the GSDMD-N pores [215]. Studies have shown that in UVB-irradiated keratinocytes, caspase-4 was indispensable for pro-IL-1β and pro-IL-18 maturation [216]. Nonetheless, this was dependent on caspase-1 expression as the knockdown of caspase-1 reduced the caspase-4-mediated unconventional secretion of matured IL-1β [216].
In Sharpincpdm mice, both canonical and non-canonical NLRP3 inflammasome were implicated [217]. Sharpincpdm bone marrow-derived macrophages (BMDMs) primed with LPS (TLR4 agonist) and Pam3CSK4 (TLR2 agonist) and activated with ATP showed decreased caspase-1 and IL-1β compared to the wildtype BMDMs [217]. Citrobacter rodentium activates the non-canonical NLRP3 inflammasome through caspase-11 [210]. Sharpincpdm BMDMs infected with C. rodentium showed reduced caspase-11, caspase-1, IL-1β, and IL-18 production compared to the wildtype BMDMs, showing that SHARPIN is essential for non-canonical NLRP3 inflammasome activation [217]. Sharpincpdm mice crossed with mice lacking interleukin-1β converting enzyme (Ice−/− mice), which are deficient in both caspase-1 and caspase-11, showed a delayed onset and alleviation of skin inflammation [218]. Additionally, reduced cleaved caspase-3/terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining and the decreased expression of both apoptosis and necroptosis effector proteins were observed in the Sharpincpdm; Ice−/− mice [218].
It has also been shown that the LUBAC complex, particularly HOIL-1L, is necessary for the activation of the NLRP3 inflammasome, as HOIL-1L−/− mice showed a reduction in IL-1β secretion upon NLRP3 stimulation and survived the lethal inflammation induced by LPS in vivo [219]. Interestingly, patients deficient in HOIL-1L were also highly susceptible to pyogenic bacterial infections [220], which may suggest that NLRP3 inflammasome assembly may be defective in these patients [219]. Overall, these studies suggest that linear ubiquitination is essential for NLRP3 inflammasome activation and governs the pathogenesis of Sharpincpdm mice [219]. In Sharpincpdm mice, systemic immune cells dysregulation such as the elevation of neutrophils and T cell activation precedes dermatitis [62]. When the Sharpincpdm mice were crossed with mice deficient in the IL-1 receptor (Il1r−/−), which mediates signals from both IL-1α and IL-1β, the absence of IL-1R delayed the progression of dermatitis in the Sharpincpdm mice [62]. Further investigation on Sharpincpdm Il1a−/− mice and Sharpincpdm Il1b−/− revealed that IL-1β, but not IL-1α, delayed the onset of dermatitis at the median age of 60 days as compared to Sharpincpdm mice at the median age of 42.5 days [62]. Clinically, NLRP3 has also been found to be associated with inflammatory skin diseases such as psoriasis, atopic dermatitis, urticaria, cryopyrin-associated periodic syndrome (CAPS), and vitiligo [194,205,221,222,223].
NLRP10 is another highly expressed NLR in the skin and keratinocytes, and it can be activated upon mitochondrial damage and cellular stress [193,224,225]. NLRP10 lacks the C-terminus leucine-rich repeat (LRR) domain, which is responsible for the recognition of DAMPs and PAMPs; therefore, it differs from other NLRPs [226]. NLRP10 has been shown to bind to the A20-binding inhibitor of NF-κB (ABIN1), a negative regulator of NF-κB via the NLRP10 NATCH domain [227]. Additionally, the pyrin domain of NLRP10 inhibits ASC-mediated NF-κB activation, cell death, and caspase-1-mediated IL-1β maturation [224,228]. In genome-wide association studies, NLRP10 has been linked to several inflammatory skin diseases, including atopic dermatitis and contact hypersensitivity [229,230,231]. The role of NLRP10 has also been shown in the contact hypersensitivity response in mice, where the keratinocyte-specific knockout of NLRP10 resulted in less inflammation [231]. NLRP10 regulates the innate immune response by modulating the p38 and NF-κB signalling pathways in the dermal fibroblast and epithelial cells upon infection with Shigella flexneri [193]. This effect was dependent on interactions with NOD1 and its binding partners, RIPK2, TAK1, and NEMO [193]. In a cutaneous infection with the West Nile virus, LCs migrate to the lymph node, and the neutralisation with anti-IL1β, but not anti-TNF, inhibited this migration [232]. The mechanism of IL-1β-dependent LC migration in a cutaneous West Nile virus infection is unclear; however, the involvement of inflammasomes can be speculated. These studies highlight the crucial roles of NLRs in skin homeostasis, inflammation, and infection, and further research is needed to explore their exploitation as therapeutic targets.
3.3. Nucleic Acid Sensors
3.3.1. Absence in Melanoma 2 (AIM2)
The absence in melanoma 2 (AIM2) is a cytoplasmic DNA sensor which recognises dsDNA and is best known for its defence role against bacteria and viruses [233,234,235,236,237,238]. The binding of cytosolic DNA to AIM2 leads to the self-assembly of inflammasome adaptor protein ASC [233,236,237]. This allows for the recruitment of pro-caspase-1 to the inflammasome, leading to the processing of caspase-1 and the maturation of pro-IL-1β and pro-IL-18 [233,237,239]. However, AIM2 lacks sequence specificity in recognising DNA; therefore, it can also detect endogenous self-nucleic acid that could be released during cellular stress or cell death [238,240]. Consequently, this can have significant implications for autoimmune diseases as the recognition of self-DNA would activate autoreactive immune cells, break self-tolerance, and amplify autoimmune response and tissue damage.
Interestingly, AIM2 has been shown to crosstalk with the apoptosis pathway during infection. The AIM2/ASC complex activated caspase-8-mediated cell death in caspase-1 knockout macrophages upon infection with intracellular bacteria, Francisella novicida [241]. Moreover, the inhibition of caspase-8 or caspase-9 or the overexpression of Bcl-2 or Bcl-XL rescued caspase-8-mediated cell death in caspase-1 knockout macrophages, indicating that the mitochondrial intrinsic pathway is involved in F. novicida infection [241]. In a separate study, the electroporation of CT DNA (ds homopolymer of Poly(dA:dT) in bone marrow-derived macrophages from caspase-1 knockout mice led to caspase-1-dependent pyroptosis and caspase-1-independent cell death mediated by ASC [242]. Additionally, both AIM2 and NLRP3 inflammasomes were shown to activate caspase-8 and initiate caspase-8-dependent cell death and pyroptosis [242]. Moreover, in primary human monocyte-derived macrophages, Toxoplasma infection induced AIM2-dependent apoptosis [243]. Infection of AIM2-knockdown dermal fibroblast with Chikungunya or the West Nile virus led to a decrease in IL-1β production [244]. AIM2 expression was also elevated in primary human skin fibroblasts infected with the Zika virus, which resulted in IL-1β production [245]. These studies emphasise the adaptability of the AIM2 inflammasome in eliciting diverse immune responses depending on the circumstances.
AIM2 has been implicated in inflammatory skin diseases such as psoriasis, atopic and contact dermatitis [205,240,246,247,248,249], and viral skin infections [250]. For example, herpes simplex virus type 1 (HSV-1) activated both AIM2 and NLRP3 inflammasomes in IFN-γ-stimulated human primary keratinocytes [250]. Human keratinocytes primed with IFN-γ and TNF followed by stimulation with Poly(dA:dT) enhanced the release of IL-1β as compared to unprimed keratinocytes [246,250]. The knockdown of AIM2 in human keratinocytes diminished the IFN-γ-primed enhanced responses and IL-1β release upon HSV-1 infection [250]. Interestingly, the anti-microbial cathelicidin peptide, LL-37, is increased in psoriatic skin and has been proposed to be a physiologic inhibitor of AIM2 inflammasome activation [246]. In this study, LL-37 reduced the AIM2-dependent release of IL-1β in human keratinocytes [246]. In another study, the AIM2 inflammasome was expressed in the entire epidermis of both the healthy and lesional skin of the AD and psoriasis patients [251]. Furthermore, in response to Poly(dA:dT) transfection, healthy human keratinocytes released IL-1β without IFN-γ stimulation [251]. These studies and the existing discrepancies highlight the need for more studies on the role of AIM2 in skin diseases and infection [246,251].
3.3.2. Cyclic GMP-AMP Synthase (cGAS) and Interferon-γ Inducible Protein 16 (IFI16)
cGAS recognises cytosolic dsDNA, and its enzymatic domain catalyses the synthesis of 2′ 3′ cyclic GMP-AMP (cGAMP), a second messenger, which binds and activates the ER membrane protein, stimulator of interferon genes (STING) [252,253,254,255]. cGAMP binding to STING results in confirmational changes that allows for the binding of TBK1 and interferon regulatory factor 3 (IRF3) [252,253,254,255]. TBK1 phosphorylates IRF3, which allows for its dimerisation and nuclear translocation to induce type I interferon and interferon-stimulated genes (ISGs) [252,253,254,255].
STING also engages the NF-κB pathway and induces the expression of inflammatory cytokines such as TNF, IL-1β, and IL-6 [252,253,254,255]. TBK1 has been shown to be dispensable for NF-κB activation, and the TBK1 homolog, IKKε, plays a redundant role with TBK1 to initiate STING-induced NF-κB activation [256]. NF-κB activation has been demonstrated to promote STING signalling. The deletion of p65 in THP-1, a human monocytic cell line, completely blocks the activation of NF-κB by TLRs, interleukin-1 receptors, TNFRs, protein kinase C, and growth factor receptors, thus preventing the enhancement of STING signalling [257]. Moreover, NF-κB activation prevents STING degradation, enabling prolonged and robust STING signalling [257]. The cGAS-STING pathway is associated with ER stress and the NLRP3 pathway. HEK293T cells transfected with cGAS and STING upregulate the markers of ER stress and unfolded protein response [258,259]. Moreover, cGAS-STING has been shown to interact with Ca2+ sensor stromal interaction molecule 1 (STIM1) [258,260]. After HSV-1 infection or cytosolic DNA stimulation, STING binds to NLRP3 and promotes the activation of the NLRP3 inflammasome through NLRP3 localisation in the ER [261].
Interferon-γ-inducible protein 16 (IFI16) and cGAS-STING signalling pathway play a critical role in keratinocyte inflammatory responses [262]. IFI16 is expressed in human cells, and mice possess its orthologue, p204. IFI16 shuttles between the nucleus and the cytosol [236]. Under a steady state, it is predominantly localised in the nucleus but is also a well-known cytosolic DNA sensor [262,263]. During a vaccinia virus infection in the human keratinocyte cell line, HaCaT, the expression of IFI16 was detected in both the nucleus and cytoplasm, which led to the elevated expression of IFNB, ISG56, and IL6 mRNA [263]. IFI16 interaction with cGAS was required for the DNA-induced activation of STING and IRF3 in HaCaT cells [263]. The mouse orthologue of IFI16, p204, has been shown to interact with STING and promote IRF3 and NF-κB activation in mouse macrophages [264]. Additionally, UVB-induced DNA damage in HaCaT cells activated the cGAS-STING and NF-κB and IRF3-IFN I pathways, leading to apoptosis of the cells [265].
In recent years, CRISPR/Cas9 technology has been suggested for the treatment of congenital skin disorders. However, it has been proposed that the human keratinocyte cell line (N/TERT) exhibits low transfection efficiency with CRISPR/Cas9 transfection due to poor plasmid uptake and rapid degradation [266]. The study showed that in recognition of the CRISPR plasmid, STING was activated, triggering a response specific to IFN-κ, a predominant type I IFN expressed by basal keratinocytes [266]. The production of IFN-κ directly affected cytidine deaminase APOBEC3G expression, which led to the degradation of the intracellular CRISPR plasmid thus reducing transfection efficiency in the keratinocytes [266]. Nevertheless, regardless of the target gene, suppressed IFNK and ISG mRNA expression was observed in the successfully CRISPR/Cas9-generated KO keratinocytes [266]. Moreover, IFNK deficiency in the keratinocytes interrupted the IFN autocrine signalling, which led to the improvement in CRISPR/Cas9 transfection efficiency [266]. This study highlighted the mechanism by which STING plays a role in mediating CRISPR/Cas9 transfection resistance in keratinocytes [266].
STING and IFI16 have been associated with skin diseases such as psoriasis [267,268,269]. The lesional skin of psoriasis patients has shown increased mRNA expression of gene encoding for STING (TMEM173) and its downstream targets, such as TBK1, NFKB1, NFKB2, and IL6 [267]. In a mouse model of imiquimod-induced psoriasis-like inflammation, the application of H-151, a STING antagonist, alleviated the inflammatory skin phenotype [267], while in Tmem173gt mice, the imiquimod-induced inflammation was attenuated [269]. Additionally, mice with selective loss of STING in the dendritic cells (CD11c-Cre-Sting−/−) showed an attenuated skin phenotype [268]. Mechanistically, in vitro studies have shown that H-151 inhibits STING/NF-κB signalling in THP-1 monocytic cells [267]. In another study, cytosolic DNA synergised with TNF and induced the expression of Il1b mRNA in keratinocytes through the STING-dependent NF-κB pathway [269]. Moreover, in vitro stimulation with cytosolic DNA and TNF showed the reduced expression of Ccl20 and Cxcl10, chemokines associated with psoriasis, in STING-deficient keratinocytes [269].
3.3.3. Retinoic Acid-Inducible Gene (RIG)-I-like Receptors (RLR), RIG-I, and Melanoma Differentiation-Associated Protein 5 (MDA5)
RIG-I and MDA5 are protein sensors of viral dsRNA [270,271]. Upon binding to dsRNA, RIG-I and MDA5 interact with the mitochondrial antiviral signalling protein (MAVS) [272,273,274]. This allows for the formation of molecular complexes, which eventually leads to an interaction with TBK1 and IKKϵ. TBK1 and IKKϵ then phosphorylate and translocate the transcription factors IRF3 and IRF7 into the nucleus, where they activate the type I interferon genes, IFNα and IFNβ [270,275,276,277]. In addition, both RIG-I and MDA5 activate the NF-κB pathway in a respiratory syncytial virus (RSV) infection, whereby RIG-I acts upstream of the canonical NF-κB pathway and mediates the translocation of the p50 and p65 NF-κB subunits into the nucleus [278]. The mechanisms of how MDA5 influences the NF-κB pathway are unclear, though it is believed that it acts independently of IκBα [279]. Both RIG-I and MDA5 induce the NF-κB-mediated production of IL-6 and pro-IL-1β through the interaction of CARD9 and BCL10 [280,281].
RIG-I is constitutively expressed in the cytosol of oral keratinocytes (RT7) and HaCaT cells [282,283]. Moreover, the transfection of Poly(I:C) induced the mRNA expression of IFNB and CXCL10 via the phosphorylation of IRF3 and STAT1 in the RT7 cells [282]. In a separate study, the incubation of RT7 cells with LL-37 increased necrotic cell supernatant-induced CXCL10 mRNA expression, in which its expression was significantly reduced upon dsRNA cleavage with RNase III digestion [284]. The LL37- and Poly(I:C)-induced level of CXCL10 mRNA was attenuated by the inhibition of the NF-κB pathway using the inhibitor, Bay11-7082, suggesting that NF-κB signalling regulates the viral immune response in oral keratinocytes [284]. These studies emphasise the role of RIG-I in the detection of dsRNA and its coordination with NF-κB in the modulation of the immune response to a viral infection in the oral keratinocytes. In HaCaT cells, IFN-γ or TNF stimulation increased RIG-I protein expression as compared to untreated cells [283]. Furthermore, the expression of RIGI, IFIH1 (encodes for MDA5), IRF3, IRF7, and IRF-dependent antiviral genes in Chikungunya virus-challenged primary human keratinocytes were found to be elevated as compared to those with the mock treatment, indicating that the innate immune response was activated in keratinocytes during an infection [285].
In mice with streptozotocin-induced diabetes and wound samples of diabetic feet, where wound healing was impaired, decreased RIG-I expression was observed [286]. NF-κB signalling was further suggested to facilitate RIG-I function, promoting keratinocyte proliferation and skin wound repair through tissue inhibitor of metalloproteinase-1 (TIMP-1). This suggests that RIG-I may be a biomarker and therapeutic target for skin injuries [286]. RIG-I protein expression was also highly upregulated in the spinous and basal keratinocyte skin layer of psoriasis vulgaris samples as compared to healthy skin, suggesting that RIG-I may be involved in psoriasis pathogenesis [283,287]. Indeed, RIG-I has been found to induce IL-23 production by myeloid dendritic cells [287].
In human patients with progressive vitiligo, the MDA5 level was found to be elevated as compared to the healthy control [288]. Moreover, the study found an association between cytomegalovirus infection and progressive vitiligo, in which MDA5 plays a key role in promoting the secretion of chemokines, CXCL10 and CXCL16, in the epidermis and the infiltration of CD8+ T cells in the skin tissues [289]. The mechanistic study showed that poly(I:C) stimulation of normal human epidermal keratinocyte (NHK) cells also led to an increase in IFN-β secretion, mediated through the MDA5-MAVS-NF-κB/IRF3 signalling pathway [289]. This consequently led to the production of CXCL16 mediated by the MDA5-IRF3 pathway and CXCL10 secretion via the JAK1/STAT1 pathway [289]. Lastly, single-cell RNA sequencing data showed that the RIG-I pathway genes were elevated in the lesional skin of systemic lupus erythematosus (SLE) and discoid lupus erythematosus (DLE) patients [290,291,292,293,294]. In addition, UVB irradiation of HaCaT cells led to elevated levels of human endogenous retrovirus dsRNA transcription and activated the RIG-I/MDA5/ IRF7 pathway, leading to ISG expression, which further promoted UVB-induced cell death and the inhibition of HaCaT cells proliferation [295].
3.3.4. Z-DNA Binding Protein (ZBP1)
Z-DNA binding protein (ZBP1) is an innate immune sensor that recognises the Z-form of double-stranded nucleic acids and adopts a left-handed double-helical structure [296]. ZBP1 contains two N-terminal Zα domains that bind to Z-form nucleic acid and has three C-terminal receptor-interacting protein (RIP) homotypic interaction motifs (RHIMs) that allow for interactions with other RHIM-containing proteins such as RIPK1, RIPK3, and TRIF [297]. ZBP1 is involved in inflammation and different forms of programmed cell death, apoptosis, necroptosis, and PANoptosis [298,299,300,301]. PANoptosis is an inflammatory form of programmed cell death regulated by PANoptosomes, which serve as a central signalling hub integrating different programmed cell death such as apoptosis, necroptosis, and pyroptosis [5,299,302]. This process is orchestrated by caspases and RIPKs through the assembly of PANoptosomes, which are multiprotein complexes formed in response to the recognition of PAMPs and DAMPs by cytosolic innate immune sensors [5,299,302].
The sensing of Z-nucleic acid activates ZBP1 and enables the formation of a complex with RIPK3 via an RHIM-mediated interaction [303]. This interaction promotes the RIPK3-mediated phosphorylation of MLKL to execute necroptosis. Moreover, the ZBP1, RIPK3, and caspase-8 assembly triggers NLRP3 inflammasome activation to facilitate pyroptosis [304]. ZBP1 has also been shown to act as an adaptor for the multiprotein assembly facilitating PANoptosis [305]. Within TLR signalling, the assembly of ZBP1, RIPK1, RIPK3, and TRIF also facilitates cell death [306]. In mice deficient in caspase-8 and TNFR1, ZBP1 and TRIF are capable of initiating necroptosis [307]. In the absence of RIPK3, ZBP1 can also induce apoptosis by interacting with RIPK1 and activating caspase-8-dependent apoptosis, leading to a non-lytic form of cell death [43]. Lastly, when RIPK1 is absent in murine embryonic fibroblasts, ZBP1 can drive RIPK3-mediated cell death induced by IFNs [308]. Deletion of Zbp1 or the core IFN signalling components prolonged the survival of Ripk1−/− mice [308].
The role of ZBP1 in viral infections, such as influenza and cytomegalovirus, has been well investigated [304,309,310,311]. For instance, in murine cytomegalovirus infection, ZBP1 interacts with RIPK3 to mediate virus-induced necrosis [309]. However, viruses possess proteins that mimic the host’s cell death proteins [311]. For instance, MCMV M45 contains a viral inhibitor of RIP activation (vIRA), which is a viral RHIM that mimics host RHIM. The interruption of ZBP1-RIPK3 interaction via vIRA eventually inhibits necroptosis [309,311]. HSV-1 and HSV-2 encode for ICP6 and ICP10, respectively. These viral encoded proteins contain viral RHIMs that inhibit RIPK3-mediated necroptosis in human cells [312,313]. Additionally, the varicella zoster virus that causes chickenpox and shingles also produces open reading frame 20, a capsid triplex protein that contains RHIM [314]. The interaction of RHIM with ZBP1 sequesters ZBP1 into decoy amyloid assemblies and prevents ZBP1-driven apoptosis during a varicella zoster virus infection of the human colorectal adenocarcinoma cell line, HT-29 [314]. Other viruses, such as the vaccinia virus also produce vaccinia virus protein E3 to compete for Z-form RNA through an N-terminal Zα domain thus blocking ZBP1/RIPK3/MLKL-dependent necroptosis during infection [315].
ZBP1 is a central player in the influenza A host defence. Zbp1−/− and wildtype mice exhibit different levels of susceptibility towards influenza A infection. Moreover, the different domains of ZBP1 have also been shown to control in vitro cell death [303,304,316,317,318,319]. The ZBP1 Zα domain has also been shown to be a key PANoptosome sensor and interacts with RIPK3, caspase-8, caspase-6, and RIPK1 to form the ZBP-1 PANoptosome complex and activate PANoptosis [305,320,321]. In recent years, studies have shown that ZBP1 is a key mediator of PANoptosis and drives NLRP3-inflammasome activation during an influenza infection, which subsequently mediates cytokine release [299,302]. A deeper understanding of the molecular mechanisms regulating ZBP1 is still required to explore ZBP1’s function in managing viral skin infections.
In addition to its role in viral sensing, ZBP1 also plays an important role in autoinflammation [301]. During skin homeostasis, the RHIM domain of RIPK1 prevented ZBP1-mediated necroptosis in mice [322]. In the full-body RIPK1 knockout mice (Ripk1−/−) or the mice expressing mutant RIPK1, the mutation of the RHIM residues IQIG to AAAA (Ripk1mRHIM/mRHIM) led to perinatal lethality due to RIPK3-MLKL-mediated necroptosis [319,322,323]. Mice lacking epidermal RIPK1 (RIPK1E-KO) or the RIPK1 RHIM domain (RIPK1mRHIM/E-KO) developed progressive inflammatory skin lesions, which were triggered by RIPK3-ZBP1-MLKL-dependent necroptosis [42,322]. The skin phenotype of RIPK1E-KO mice was partially dependent on TNFR1 [42]. In the subsequent studies, crossing RIPK1E-KO mice with the knock-in mice expressing ZBP1 with a deletion (Zbp1∆Zα/∆Zα) or mutation of both Zα domains (Zbp1mZα1–2/mZα1–2) or Zα2 alone (Zbp1mZα2/mZα2), substituting key amino acids (Zα1:N46D, Y50A; Zα2: N122D, Y126A) that are essential for Z-NA binding, prevented severe skin lesion development until up to 18–42 weeks of age [319]. In another study, the Zα domains of ZBP1 were also shown to trigger the inflammatory skin phenotype in the RIPK1E-KO mice [31]. Additionally, the RIPK1E-KO mice crossed with the Zbp1mR1/mR1 mice expressing ZBP1 with four amino acid substitutions disrupting its core RHIM sequence (192IQIG to 192AAAA) developed only a mild lesion by the age of 40 weeks [319]. Furthermore, the treatment of RIPK1E-KO mice with retroviral drugs ameliorated the skin phenotype, suggesting the role of endogenous retroviral elements in skin inflammation [319]. Moreover, the embryonic lethality of Ripk1mRHIM/mRHIM was also dependent on ZBP1 [319]. ZBP-1 also has an autoinhibitory function governed by RHIM-B, RHIM-C, and the C-terminal end of ZBP1 [43]. The truncated C-terminus of mouse ZBP1 (ZBP1ca) which led to the loss of 203 amino acid residues, consisting of the Zα domains and RHIM-A, was shown to be constitutively active [43]. The expression of ZBP1ca triggered keratinocyte cell death and led to the development of inflammation skin lesions, suggesting that ZBP1ca could drive skin inflammation [43]. Moreover, the abrogation of RIPK3-MLKL-dependent necroptosis could only partially rescue the inflammation skin phenotype [43]. Nonetheless, the combination of the inhibition of RIPK3-MLKL-dependent necroptosis and RIPK1-caspase-8-driven cell death could fully prevent skin inflammation mediated by ZBP1ca [43]. Lastly, the inhibition of RIPK1 kinase activity and mutation of the MLKL phosphorylation sites did not prevent caspase-8-mediated inflammation in ZBP1caE-het mice, suggesting that ZBP1 induces RIPK1 kinase activity independent cell death [43].
In another study, ZBP1-mediated necroptosis in human keratinocytes was dependent on IFNγ secretion, as intervening with IFNγ signalling through JAK inhibition led to the inhibition of cell death [324]. Mice lacking epidermal FADD developed necroptosis-mediated skin inflammation [39]. Neither TNFR1 deficiency nor ZBP1 ablation alone was sufficient to inhibit skin inflammation; however, the double deficiency of ZBP1 and TNFR1 prevented skin inflammation [39,43,322]. These studies show that ZBP1-mediated necroptosis drives TNFR-1-independent skin inflammation in mice lacking epidermal FADD. This suggests that dysregulated ZBP1 signalling may disrupt skin homeostasis and contribute to inflammatory skin diseases. Further studies are required to illustrate the impact of ZBP1 on the inflammatory landscape of human skin diseases.
4. Concluding Remarks
In this review, we have outlined the innate immune sensors and cell death pathways, their relationships, and their potential relevance to skin inflammation and infection in mice and humans. Although there is functional redundancy in the recognition of nucleic acids amongst the different innate immune sensors within the keratinocytes and immune cells of the skin, this genetic redundancy enables the host to cope with foreign and host molecules, including pathogen evasions. Moreover, the convergence of multiple downstream signalling pathways could facilitate PAMPs or DAMPs recognition by different innate immune sensors in keratinocytes and induce a robust immune response to maintain skin homeostasis. Nevertheless, innate immune sensors could also be a double-edged sword in host immunity as their dysregulation could lead to imbalanced epithelial–immune cell communication and ultimately result in inflammatory skin diseases. Further studies in mice and humans on the regulation of innate immune sensors and cell death pathways, as well as their cellular interaction in the skin, are required to explore their therapeutic potentials during inflammatory skin conditions.
Author Contributions
Y.M.S. and S.K.: writing—original draft preparation, Y.M.S. and S.L.S.: figure generation, Y.M.S., S.L.S. and S.K.: conceptualisation, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
S. K acknowledges support from the Frazer Institute, the University of Queensland, and Y.M.S acknowledges support from the University of Queensland for the Research Training Scholarship and the Dr. Jian Zhou Memorial Scholarship.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Nguyen, A.V.; Soulika, A.M. The Dynamics of the Skin’s Immune System. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef]
- Abdallah, F.; Mijouin, L.; Pichon, C. Skin Immune Landscape: Inside and Outside the Organism. Mediat. Inflamm. 2017, 2017, 5095293. [Google Scholar] [CrossRef]
- Nestle, F.O.; Di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef]
- Castillo-Gonzalez, R.; Cibrian, D.; Sanchez-Madrid, F. Dissecting the complexity of gammadelta T-cell subsets in skin homeostasis, inflammation, and malignancy. J. Allergy Clin. Immunol. 2021, 147, 2030–2042. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.D. Innate immune sensing of cell death in disease and therapeutics. Nat. Cell Biol. 2024, 26, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gullett, J.M.; Tweedell, R.E.; Kanneganti, T.D. Innate immune inflammatory cell death: PANoptosis and PANoptosomes in host defense and disease. Eur. J. Immunol. 2023, 53, e2250235. [Google Scholar] [CrossRef]
- Fang, Y.; Peng, K. Regulation of innate immune responses by cell death-associated caspases during virus infection. FEBS J. 2022, 289, 4098–4111. [Google Scholar] [CrossRef]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin barrier immunity and ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Kahlenberg, J.M.; Gudjonsson, J.E. Cytokinocytes: The diverse contribution of keratinocytes to immune responses in skin. JCI Insight 2020, 5, e142067. [Google Scholar] [CrossRef]
- Campione, E.; Lanna, C.; Diluvio, L.; Cannizzaro, M.V.; Grelli, S.; Galluzzo, M.; Talamonti, M.; Annicchiarico-Petruzzelli, M.; Mancini, M.; Melino, G.; et al. Skin immunity and its dysregulation in atopic dermatitis, hidradenitis suppurativa and vitiligo. Cell Cycle 2020, 19, 257–267. [Google Scholar] [CrossRef]
- Richmond, J.M.; Harris, J.E. Immunology and skin in health and disease. Cold Spring Harb. Perspect. Med. 2014, 4, a015339. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Hong, S.H. Hematopoietic Stem Cells and Their Roles in Tissue Regeneration. Int. J. Stem Cells 2020, 13, 1–12. [Google Scholar] [CrossRef]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Anderton, H.; Alqudah, S. Cell death in skin function, inflammation, and disease. Biochem. J. 2022, 479, 1621–1651. [Google Scholar] [CrossRef]
- Yuan, J.; Ofengeim, D. A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 2024, 25, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Shao, Y.; Li, C. Different types of cell death and their shift in shaping disease. Cell Death Discov. 2023, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Pasparakis, M. Epithelial Cell Death and Inflammation in Skin. Curr. Top. Microbiol. Immunol. 2017, 403, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Anderton, H.; Rickard, J.A.; Varigos, G.A.; Lalaoui, N.; Silke, J. Inhibitor of Apoptosis Proteins (IAPs) Limit RIPK1-Mediated Skin Inflammation. J. Investig. Dermatol. 2017, 137, 2371–2379. [Google Scholar] [CrossRef]
- Etemadi, N.; Chopin, M.; Anderton, H.; Tanzer, M.C.; Rickard, J.A.; Abeysekera, W.; Hall, C.; Spall, S.K.; Wang, B.; Xiong, Y.; et al. TRAF2 regulates TNF and NF-kappaB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1. eLife 2015, 4, e10592. [Google Scholar] [CrossRef] [PubMed]
- HogenEsch, H.; Gijbels, M.J.; Offerman, E.; van Hooft, J.; van Bekkum, D.W.; Zurcher, C. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol. 1993, 143, 972–982. [Google Scholar] [PubMed]
- Rickard, J.A.; Anderton, H.; Etemadi, N.; Nachbur, U.; Darding, M.; Peltzer, N.; Lalaoui, N.; Lawlor, K.E.; Vanyai, H.; Hall, C.; et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 2014, 3, e03464. [Google Scholar] [CrossRef]
- Kumari, S.; Redouane, Y.; Lopez-Mosqueda, J.; Shiraishi, R.; Romanowska, M.; Lutzmayer, S.; Kuiper, J.; Martinez, C.; Dikic, I.; Pasparakis, M.; et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 2014, 3, e03422. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.W.; Jiang, W.; Reich, C.F., 3rd; Pisetsky, D.S. The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef]
- Negroni, A.; Colantoni, E.; Pierdomenico, M.; Palone, F.; Costanzo, M.; Oliva, S.; Tiberti, A.; Cucchiara, S.; Stronati, L. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Dig. Liver Dis. 2017, 49, 1201–1210. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Binder, R.J.; Suto, R.; Anderson, K.M.; Srivastava, P.K. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 2000, 12, 1539–1546. [Google Scholar] [CrossRef]
- Devos, M.; Tanghe, G.; Gilbert, B.; Dierick, E.; Verheirstraeten, M.; Nemegeer, J.; de Reuver, R.; Lefebvre, S.; De Munck, J.; Rehwinkel, J.; et al. Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J. Exp. Med. 2020, 217, e20191913. [Google Scholar] [CrossRef]
- Toussaint, M.; Jackson, D.J.; Swieboda, D.; Guedan, A.; Tsourouktsoglou, T.D.; Ching, Y.M.; Radermecker, C.; Makrinioti, H.; Aniscenko, J.; Bartlett, N.W.; et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat. Med. 2017, 23, 681–691. [Google Scholar] [CrossRef]
- Shen, J.; Chen, R.; Duan, S. NINJ1: Bridging lytic cell death and inflammation therapy. Cell Death Dis. 2024, 15, 831. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Liu, X.; Liu, N.; Huang, Y.; Jin, Z.; Zhang, S.; Ming, Z.; Chen, H. Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis. 2020, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Kim, W.J.; Yoon, J.H.; Ji, J.H.; Morgan, M.J.; Cho, H.; Kim, Y.C.; Kim, Y.S. Upregulated RIP3 Expression Potentiates MLKL Phosphorylation-Mediated Programmed Necrosis in Toxic Epidermal Necrolysis. J. Investig. Dermatol. 2015, 135, 2021–2030. [Google Scholar] [CrossRef]
- Pasparakis, M.; Haase, I.; Nestle, F.O. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 2014, 14, 289–301. [Google Scholar] [CrossRef]
- Kondylis, V.; Kumari, S.; Vlantis, K.; Pasparakis, M. The interplay of IKK, NF-kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 2017, 277, 113–127. [Google Scholar] [CrossRef]
- Wullaert, A.; Bonnet, M.C.; Pasparakis, M. NF-kappaB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011, 21, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.C.; Preukschat, D.; Welz, P.S.; van Loo, G.; Ermolaeva, M.A.; Bloch, W.; Haase, I.; Pasparakis, M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 2011, 35, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Kim, J.C.; Kang, T.B.; Rajput, A.; Bogdanov, K.; Dittrich-Breiholz, O.; Kracht, M.; Brenner, O.; Wallach, D. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J. Exp. Med. 2009, 206, 2161–2177. [Google Scholar] [CrossRef]
- Lee, P.; Lee, D.J.; Chan, C.; Chen, S.W.; Ch’en, I.; Jamora, C. Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature 2009, 458, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Dannappel, M.; Vlantis, K.; Kumari, S.; Polykratis, A.; Kim, C.; Wachsmuth, L.; Eftychi, C.; Lin, J.; Corona, T.; Hermance, N.; et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 2014, 513, 90–94. [Google Scholar] [CrossRef]
- Koerner, L.; Wachsmuth, L.; Kumari, S.; Schwarzer, R.; Wagner, T.; Jiao, H.; Pasparakis, M. ZBP1 causes inflammation by inducing RIPK3-mediated necroptosis and RIPK1 kinase activity-independent apoptosis. Cell Death Differ. 2024, 31, 938–953. [Google Scholar] [CrossRef]
- Rebholz, B.; Haase, I.; Eckelt, B.; Paxian, S.; Flaig, M.J.; Ghoreschi, K.; Nedospasov, S.A.; Mailhammer, R.; Debey-Pascher, S.; Schultze, J.L.; et al. Crosstalk between keratinocytes and adaptive immune cells in an IkappaBalpha protein-mediated inflammatory disease of the skin. Immunity 2007, 27, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Navarro, M.; Garin, M.; Perez, P.; Casanova, M.L.; Moreno, R.; Jorcano, J.L.; Cascallana, J.L.; Bravo, A.; Ramirez, A. IKKbeta leads to an inflammatory skin disease resembling interface dermatitis. J. Investig. Dermatol. 2010, 130, 1598–1610. [Google Scholar] [CrossRef]
- Nenci, A.; Huth, M.; Funteh, A.; Schmidt-Supprian, M.; Bloch, W.; Metzger, D.; Chambon, P.; Rajewsky, K.; Krieg, T.; Haase, I.; et al. Skin lesion development in a mouse model of incontinentia pigmenti is triggered by NEMO deficiency in epidermal keratinocytes and requires TNF signaling. Hum. Mol. Genet. 2006, 15, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Smahi, A.; Courtois, G.; Vabres, P.; Yamaoka, S.; Heuertz, S.; Munnich, A.; Israel, A.; Heiss, N.S.; Klauck, S.M.; Kioschis, P.; et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 2000, 405, 466–472. [Google Scholar] [CrossRef] [PubMed]
- van Hogerlinden, M.; Rozell, B.L.; Ahrlund-Richter, L.; Toftgard, R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999, 59, 3299–3303. [Google Scholar] [PubMed]
- Seitz, C.S.; Lin, Q.; Deng, H.; Khavari, P.A. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc. Natl. Acad. Sci. USA 1998, 95, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.H.; Rozell, B.; Wallin, R.P.; van Hogerlinden, M.; Ljunggren, H.G.; Toftgard, R.; Sur, I. Tumor necrosis factor receptor 1-mediated signaling is required for skin cancer development induced by NF-kappaB inhibition. Proc. Natl. Acad. Sci. USA 2004, 101, 4972–4977. [Google Scholar] [CrossRef]
- Pasparakis, M.; Courtois, G.; Hafner, M.; Schmidt-Supprian, M.; Nenci, A.; Toksoy, A.; Krampert, M.; Goebeler, M.; Gillitzer, R.; Israel, A.; et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 2002, 417, 861–866. [Google Scholar] [CrossRef]
- Kumari, S.; Van, T.M.; Preukschat, D.; Schuenke, H.; Basic, M.; Bleich, A.; Klein, U.; Pasparakis, M. NF-kappaB inhibition in keratinocytes causes RIPK1-mediated necroptosis and skin inflammation. Life Sci. Alliance 2021, 4, e202000956. [Google Scholar] [CrossRef]
- Kumari, S.; Bonnet, M.C.; Ulvmar, M.H.; Wolk, K.; Karagianni, N.; Witte, E.; Uthoff-Hachenberg, C.; Renauld, J.C.; Kollias, G.; Toftgard, R.; et al. Tumor necrosis factor receptor signaling in keratinocytes triggers interleukin-24-dependent psoriasis-like skin inflammation in mice. Immunity 2013, 39, 899–911. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Dainichi, T.; Oh, H.; Heise, N.; Klein, U.; Schmid, R.M.; Hayden, M.S.; Ghosh, S. Cutting edge: NF-kappaB p65 and c-Rel control epidermal development and immune homeostasis in the skin. J. Immunol. 2015, 194, 2472–2476. [Google Scholar] [CrossRef]
- Graber, T.E.; Holcik, M. Distinct roles for the cellular inhibitors of apoptosis proteins 1 and 2. Cell Death Dis. 2011, 2, e135. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Vucic, D. (Un)expected roles of c-IAPs in apoptotic and NFkappaB signaling pathways. Cell Cycle 2008, 7, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Wong, W.W.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Taraborrelli, L.; Walczak, H. Linear ubiquitination in immunity. Immunol. Rev. 2015, 266, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef]
- Berger, S.B.; Kasparcova, V.; Hoffman, S.; Swift, B.; Dare, L.; Schaeffer, M.; Capriotti, C.; Cook, M.; Finger, J.; Hughes-Earle, A.; et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 2014, 192, 5476–5480. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Sharma, B.R.; Kanneganti, T.D. Distinct role of IL-1beta in instigating disease in Sharpin(cpdm) mice. Sci. Rep. 2016, 6, 36634. [Google Scholar] [CrossRef] [PubMed]
- Zinngrebe, J.; Rieser, E.; Taraborrelli, L.; Peltzer, N.; Hartwig, T.; Ren, H.; Kovacs, I.; Endres, C.; Draber, P.; Darding, M.; et al. LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J. Exp. Med. 2016, 213, 2671–2689. [Google Scholar] [CrossRef]
- Sharma, B.R.; Karki, R.; Lee, E.; Zhu, Q.; Gurung, P.; Kanneganti, T.D. Innate immune adaptor MyD88 deficiency prevents skin inflammation in SHARPIN-deficient mice. Cell Death Differ. 2019, 26, 741–750. [Google Scholar] [CrossRef]
- Laurien, L.; Nagata, M.; Schunke, H.; Delanghe, T.; Wiederstein, J.L.; Kumari, S.; Schwarzer, R.; Corona, T.; Kruger, M.; Bertrand, M.J.M.; et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 2020, 11, 1747. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.; Lecomte, K.; Annusver, K.; Vandamme, N.; Roels, J.; Maschalidi, S.; Verboom, L.; Vikkula, H.K.; Sze, M.; Van Hove, L.; et al. OTULIN maintains skin homeostasis by controlling keratinocyte death and stem cell identity. Nat. Commun. 2021, 12, 5913. [Google Scholar] [CrossRef] [PubMed]
- Schunke, H.; Gobel, U.; Dikic, I.; Pasparakis, M. OTULIN inhibits RIPK1-mediated keratinocyte necroptosis to prevent skin inflammation in mice. Nat. Commun. 2021, 12, 5912. [Google Scholar] [CrossRef]
- Baccala, R.; Gonzalez-Quintial, R.; Lawson, B.R.; Stern, M.E.; Kono, D.H.; Beutler, B.; Theofilopoulos, A.N. Sensors of the innate immune system: Their mode of action. Nat. Rev. Rheumatol. 2009, 5, 448–456. [Google Scholar] [CrossRef]
- Briard, B.; Place, D.E.; Kanneganti, T.D. DNA Sensing in the Innate Immune Response. Physiology 2020, 35, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.Z.; Kim, S.M.; Wang, C.; Lee, S.Y.; Oh, S.C.; Lee, S.; Jo, S.; Kim, T.D. Understanding nucleic acid sensing and its therapeutic applications. Exp. Mol. Med. 2023, 55, 2320–2331. [Google Scholar] [CrossRef]
- Wicherska-Pawlowska, K.; Wrobel, T.; Rybka, J. Toll-Like Receptors (TLRs), NOD-Like Receptors (NLRs), and RIG-I-Like Receptors (RLRs) in Innate Immunity. TLRs, NLRs, and RLRs Ligands as Immunotherapeutic Agents for Hematopoietic Diseases. Int. J. Mol. Sci. 2021, 22, 13397. [Google Scholar] [CrossRef] [PubMed]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed. Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef]
- Botos, I.; Segal, D.M.; Davies, D.R. The structural biology of Toll-like receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef]
- Behzadi, P.; Garcia-Perdomo, H.A.; Karpinski, T.M. Toll-Like Receptors: General Molecular and Structural Biology. J. Immunol. Res. 2021, 2021, 9914854. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Lebre, M.C.; van der Aar, A.M.; van Baarsen, L.; van Capel, T.M.; Schuitemaker, J.H.; Kapsenberg, M.L.; de Jong, E.C. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J. Investig. Dermatol. 2007, 127, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kollisch, G.; Kalali, B.N.; Voelcker, V.; Wallich, R.; Behrendt, H.; Ring, J.; Bauer, S.; Jakob, T.; Mempel, M.; Ollert, M. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 2005, 114, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Piipponen, M.; Li, D.; Landen, N.X. The Immune Functions of Keratinocytes in Skin Wound Healing. Int. J. Mol. Sci. 2020, 21, 8790. [Google Scholar] [CrossRef]
- Hari, A.; Flach, T.L.; Shi, Y.; Mydlarski, P.R. Toll-like receptors: Role in dermatological disease. Mediators Inflamm. 2010, 2010, 437246. [Google Scholar] [CrossRef]
- Lee, C.C.; Avalos, A.M.; Ploegh, H.L. Accessory molecules for Toll-like receptors and their function. Nat. Rev. Immunol. 2012, 12, 168–179. [Google Scholar] [CrossRef]
- Grimstad, O.; Husebye, H.; Espevik, T. TLR3 mediates release of IL-1beta and cell death in keratinocytes in a caspase-4 dependent manner. J. Dermatol. Sci. 2013, 72, 45–53. [Google Scholar] [CrossRef]
- Grimstad, O.; Pukstad, B.; Stenvik, J.; Espevik, T. Oligodeoxynucleotides inhibit Toll-like receptor 3 mediated cytotoxicity and CXCL8 release in keratinocytes. Exp. Dermatol. 2012, 21, 7–12. [Google Scholar] [CrossRef]
- Grimstad, O.; Sandanger, O.; Ryan, L.; Otterdal, K.; Damaas, J.K.; Pukstad, B.; Espevik, T. Cellular sources and inducers of cytokines present in acute wound fluid. Wound Repair. Regen. 2011, 19, 337–347. [Google Scholar] [CrossRef]
- Borkowski, A.W.; Park, K.; Uchida, Y.; Gallo, R.L. Activation of TLR3 in keratinocytes increases expression of genes involved in formation of the epidermis, lipid accumulation, and epidermal organelles. J. Investig. Dermatol. 2013, 133, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Pivarcsi, A.; Bodai, L.; Rethi, B.; Kenderessy-Szabo, A.; Koreck, A.; Szell, M.; Beer, Z.; Bata-Csorgoo, Z.; Magocsi, M.; Rajnavolgyi, E.; et al. Expression and function of Toll-like receptors 2 and 4 in human keratinocytes. Int. Immunol. 2003, 15, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.S.; Ovigne, J.M.; Powles, A.V.; Corcoran, S.; Fry, L. Normal keratinocytes express Toll-like receptors (TLRs) 1, 2 and 5: Modulation of TLR expression in chronic plaque psoriasis. Br. J. Dermatol. 2003, 148, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; Sorensen, O.E.; Liu, P.T.; Jalian, H.R.; Eshtiaghpour, D.; Behmanesh, B.E.; Chung, W.; Starner, T.D.; Kim, J.; Sieling, P.A.; et al. TGF-alpha regulates TLR expression and function on epidermal keratinocytes. J. Immunol. 2005, 174, 6137–6143. [Google Scholar] [CrossRef]
- Li, Z.J.; Sohn, K.C.; Choi, D.K.; Shi, G.; Hong, D.; Lee, H.E.; Whang, K.U.; Lee, Y.H.; Im, M.; Lee, Y.; et al. Roles of TLR7 in activation of NF-kappaB signaling of keratinocytes by imiquimod. PLoS ONE 2013, 8, e77159. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Furio, L.; Billard, H.; Valladeau, J.; Peguet-Navarro, J.; Berthier-Vergnes, O. Poly(I:C)-Treated human langerhans cells promote the differentiation of CD4+ T cells producing IFN-gamma and IL-10. J. Investig. Dermatol. 2009, 129, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Flacher, V.; Bouschbacher, M.; Verronese, E.; Massacrier, C.; Sisirak, V.; Berthier-Vergnes, O.; de Saint-Vis, B.; Caux, C.; Dezutter-Dambuyant, C.; Lebecque, S.; et al. Human Langerhans cells express a specific TLR profile and differentially respond to viruses and Gram-positive bacteria. J. Immunol. 2006, 177, 7959–7967. [Google Scholar] [CrossRef]
- Berthier-Vergnes, O.; Bermond, F.; Flacher, V.; Massacrier, C.; Schmitt, D.; Peguet-Navarro, J. TNF-alpha enhances phenotypic and functional maturation of human epidermal Langerhans cells and induces IL-12 p40 and IP-10/CXCL-10 production. FEBS Lett. 2005, 579, 3660–3668. [Google Scholar] [CrossRef]
- Renn, C.N.; Sanchez, D.J.; Ochoa, M.T.; Legaspi, A.J.; Oh, C.K.; Liu, P.T.; Krutzik, S.R.; Sieling, P.A.; Cheng, G.; Modlin, R.L. TLR activation of Langerhans cell-like dendritic cells triggers an antiviral immune response. J. Immunol. 2006, 177, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.P., Jr.; Ferbel, B.; Tomai, M.; Miller, R.; Gaspari, A.A. The imidazoquinolines, imiquimod and R-848, induce functional, but not phenotypic, maturation of human epidermal Langerhans’ cells. Clin. Immunol. 2000, 94, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Schnare, M.; Barton, G.M.; Holt, A.C.; Takeda, K.; Akira, S.; Medzhitov, R. Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2001, 2, 947–950. [Google Scholar] [CrossRef]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Liu, Y.J.; Kadowaki, N. Functional diversity and plasticity of human dendritic cell subsets. Int. J. Hematol. 2005, 81, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Medzhitov, R. Regulation of phagosome maturation by signals from toll-like receptors. Science 2004, 304, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Kulka, M.; Metcalfe, D.D. TLR3 activation inhibits human mast cell attachment to fibronectin and vitronectin. Mol. Immunol. 2006, 43, 1579–1586. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Supajatura, V.; Ushio, H.; Nakao, A.; Akira, S.; Okumura, K.; Ra, C.; Ogawa, H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J. Clin. Investig. 2002, 109, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef]
- Bendigs, S.; Salzer, U.; Lipford, G.B.; Wagner, H.; Heeg, K. CpG-oligodeoxynucleotides co-stimulate primary T cells in the absence of antigen-presenting cells. Eur. J. Immunol. 1999, 29, 1209–1218. [Google Scholar] [CrossRef]
- Caron, G.; Duluc, D.; Fremaux, I.; Jeannin, P.; David, C.; Gascan, H.; Delneste, Y. Direct stimulation of human T cells via TLR5 and TLR7/8: Flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J. Immunol. 2005, 175, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Guo, Z.; Kiniwa, Y.; Voo, K.S.; Peng, W.; Fu, T.; Wang, D.Y.; Li, Y.; Wang, H.Y.; Wang, R.F. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 2005, 309, 1380–1384. [Google Scholar] [CrossRef] [PubMed]
- Crellin, N.K.; Garcia, R.V.; Hadisfar, O.; Allan, S.E.; Steiner, T.S.; Levings, M.K. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J. Immunol. 2005, 175, 8051–8059. [Google Scholar] [CrossRef] [PubMed]
- Gelman, A.E.; LaRosa, D.F.; Zhang, J.; Walsh, P.T.; Choi, Y.; Sunyer, J.O.; Turka, L.A. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity 2006, 25, 783–793. [Google Scholar] [CrossRef]
- Nystrom, S.; Antoine, D.J.; Lundback, P.; Lock, J.G.; Nita, A.F.; Hogstrand, K.; Grandien, A.; Erlandsson-Harris, H.; Andersson, U.; Applequist, S.E. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 2013, 32, 86–99. [Google Scholar] [CrossRef]
- Xing, Y.; Yao, X.; Li, H.; Xue, G.; Guo, Q.; Yang, G.; An, L.; Zhang, Y.; Meng, G. Cutting Edge: TRAF6 Mediates TLR/IL-1R Signaling-Induced Nontranscriptional Priming of the NLRP3 Inflammasome. J. Immunol. 2017, 199, 1561–1566. [Google Scholar] [CrossRef]
- Hsu, C.G.; Chavez, C.L.; Zhang, C.; Sowden, M.; Yan, C.; Berk, B.C. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ. 2022, 29, 1790–1803. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef]
- Krueger, J.G.; Frew, J.; Jemec, G.B.E.; Kimball, A.B.; Kirby, B.; Bechara, F.G.; Navrazhina, K.; Prens, E.; Reich, K.; Cullen, E.; et al. Hidradenitis suppurativa: New insights into disease mechanisms and an evolving treatment landscape. Br. J. Dermatol. 2024, 190, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.; Hughes, R.; McGarry, T.; van den Born, M.; Adamzik, K.; Fitzgerald, R.; Lawlor, C.; Tobin, A.M.; Sweeney, C.M.; Kirby, B. Dysregulated cytokine expression in lesional and nonlesional skin in hidradenitis suppurativa. Br. J. Dermatol. 2015, 173, 1431–1439. [Google Scholar] [CrossRef]
- Melnik, B.C.; Plewig, G. Impaired Notch signalling: The unifying mechanism explaining the pathogenesis of hidradenitis suppurativa (acne inversa). Br. J. Dermatol. 2013, 168, 876–878. [Google Scholar] [CrossRef] [PubMed]
- van der Zee, H.H.; de Ruiter, L.; van den Broecke, D.G.; Dik, W.A.; Laman, J.D.; Prens, E.P. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: A rationale for targeting TNF-alpha and IL-1beta. Br. J. Dermatol. 2011, 164, 1292–1298. [Google Scholar] [CrossRef]
- van der Zee, H.H.; Laman, J.D.; Boer, J.; Prens, E.P. Hidradenitis suppurativa: Viewpoint on clinical phenotyping, pathogenesis and novel treatments. Exp. Dermatol. 2012, 21, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Kalai, M.; Van Loo, G.; Vanden Berghe, T.; Meeus, A.; Burm, W.; Saelens, X.; Vandenabeele, P. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ. 2002, 9, 981–994. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lu, Z.; Hawkes, M.; Yang, H.; Kain, K.C.; Liles, W.C. Fas (CD95) induces rapid, TLR4/IRAK4-dependent release of pro-inflammatory HMGB1 from macrophages. J. Inflamm. 2010, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Kirejczyk, Z.; Besch, R.; Potthoff, S.; Leverkus, M.; Hacker, G. Proapoptotic signalling through Toll-like receptor-3 involves TRIF-dependent activation of caspase-8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ. 2010, 17, 942–951. [Google Scholar] [CrossRef]
- Harberts, E.; Fishelevich, R.; Liu, J.; Atamas, S.P.; Gaspari, A.A. MyD88 mediates the decision to die by apoptosis or necroptosis after UV irradiation. Innate Immun. 2014, 20, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, E.; Mira, J.P.; Cornish, K.L.; Arbour, N.C.; Schwartz, D.A. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect. Immun. 2000, 68, 6398–6401. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Langnickel, J.; Draing, C.; Renz, H.; Kapp, A.; Werfel, T. Dysregulation of toll-like receptor-2 (TLR-2)-induced effects in monocytes from patients with atopic dermatitis: Impact of the TLR-2 R753Q polymorphism. Allergy 2008, 63, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; O’Connell, R.M.; Gutierrez, M.A.; Pietras, E.M.; Shahangian, A.; Gross, C.E.; Thirumala, A.; Cheung, A.L.; Cheng, G.; Modlin, R.L. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 2006, 24, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Dai, C.; Zeng, F. Cellular Mechanisms of Psoriasis Pathogenesis: A Systemic Review. Clin. Cosmet. Investig. Dermatol. 2023, 16, 2503–2515. [Google Scholar] [CrossRef] [PubMed]
- Kamata, M.; Tada, Y. Dendritic Cells and Macrophages in the Pathogenesis of Psoriasis. Front. Immunol. 2022, 13, 941071. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: From keratinocyte perspective. Cell Death Dis. 2022, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Rendon, A.; Schakel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef]
- Curry, J.L.; Qin, J.Z.; Bonish, B.; Carrick, R.; Bacon, P.; Panella, J.; Robinson, J.; Nickoloff, B.J. Innate immune-related receptors in normal and psoriatic skin. Arch. Pathol. Lab. Med. 2003, 127, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Lai, X.; Zhang, C.; Wang, Z.; Yang, Y.; Wang, B.; Yan, Y. Upregulation of TLR2 in keratinocytes activates the MAPK pathway and plays a role in the pathogenesis of FHS. J. Dermatol. Sci. 2024, 117, 8–17. [Google Scholar] [CrossRef]
- Hunger, R.E.; Surovy, A.M.; Hassan, A.S.; Braathen, L.R.; Yawalkar, N. Toll-like receptor 2 is highly expressed in lesions of acne inversa and colocalizes with C-type lectin receptor. Br. J. Dermatol. 2008, 158, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Dreno, B.; Khammari, A.; Brocard, A.; Moyse, D.; Blouin, E.; Guillet, G.; Leonard, F.; Knol, A.C. Hidradenitis suppurativa: The role of deficient cutaneous innate immunity. Arch. Dermatol. 2012, 148, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Marini, O.; Bevilacqua, D.; DeFranco, A.L.; Hou, B.; Lonardi, S.; Vermi, W.; Rodegher, P.; Panato, A.; Tagliaro, F.; et al. Role of MyD88 signaling in the imiquimod-induced mouse model of psoriasis: Focus on innate myeloid cells. J. Leukoc. Biol. 2017, 102, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Kruger, J.; Butler, J.R.; Cherapanov, V.; Dong, Q.; Ginzberg, H.; Govindarajan, A.; Grinstein, S.; Siminovitch, K.A.; Downey, G.P. Deficiency of Src homology 2-containing phosphatase 1 results in abnormalities in murine neutrophil function: Studies in motheaten mice. J. Immunol. 2000, 165, 5847–5859. [Google Scholar] [CrossRef] [PubMed]
- Kiratikanon, S.; Chattipakorn, S.C.; Chattipakorn, N.; Kumfu, S. The regulatory effects of PTPN6 on inflammatory process: Reports from mice to men. Arch. Biochem. Biophys. 2022, 721, 109189. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, W.J.; Pulido, R. Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochim. Biophys. Acta 2013, 1832, 1673–1696. [Google Scholar] [CrossRef] [PubMed]
- Green, M.C.; Shultz, L.D. Motheaten, an immunodeficient mutant of the mouse. I. Genetics and pathology. J. Hered. 1975, 66, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Schweitzer, P.A.; Rajan, T.V.; Yi, T.; Ihle, J.N.; Matthews, R.J.; Thomas, M.L.; Beier, D.R. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell 1993, 73, 1445–1454. [Google Scholar] [CrossRef]
- Tsui, H.W.; Siminovitch, K.A.; de Souza, L.; Tsui, F.W. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat. Genet. 1993, 4, 124–129. [Google Scholar] [CrossRef]
- Sidman, C.L.; Shultz, L.D.; Unanue, E.R. The mouse mutant “motheaten.” II. Functional studies of the immune system. J. Immunol. 1978, 121, 2399–2404. [Google Scholar] [CrossRef] [PubMed]
- Sidman, C.L.; Shultz, L.D.; Unanue, E.R. The mouse mutant “motheaten”. I. Development of lymphocyte populations. J. Immunol. 1978, 121, 2392–2398. [Google Scholar] [CrossRef]
- Nesterovitch, A.B.; Szanto, S.; Gonda, A.; Bardos, T.; Kis-Toth, K.; Adarichev, V.A.; Olasz, K.; Ghassemi-Najad, S.; Hoffman, M.D.; Tharp, M.D.; et al. Spontaneous insertion of a b2 element in the ptpn6 gene drives a systemic autoinflammatory disease in mice resembling neutrophilic dermatosis in humans. Am. J. Pathol. 2011, 178, 1701–1714. [Google Scholar] [CrossRef]
- Croker, B.A.; Lawson, B.R.; Rutschmann, S.; Berger, M.; Eidenschenk, C.; Blasius, A.L.; Moresco, E.M.; Sovath, S.; Cengia, L.; Shultz, L.D.; et al. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc. Natl. Acad. Sci. USA 2008, 105, 15028–15033. [Google Scholar] [CrossRef]
- Abram, C.L.; Roberge, G.L.; Pao, L.I.; Neel, B.G.; Lowell, C.A. Distinct roles for neutrophils and dendritic cells in inflammation and autoimmunity in motheaten mice. Immunity 2013, 38, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Vogel, P.; Johnson, G.R.; Kelliher, M.A.; Iwakura, Y.; Lamkanfi, M.; Kanneganti, T.D. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature 2013, 498, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Fan, G.; Lukens, J.R.; Vogel, P.; Tonks, N.K.; Kanneganti, T.D. Tyrosine Kinase SYK Licenses MyD88 Adaptor Protein to Instigate IL-1alpha-Mediated Inflammatory Disease. Immunity 2017, 46, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Tartey, S.; Gurung, P.; Dasari, T.K.; Burton, A.; Kanneganti, T.D. ASK1/2 signaling promotes inflammation in a mouse model of neutrophilic dermatosis. J. Clin. Investig. 2018, 128, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Speir, M.; Nowell, C.J.; Chen, A.A.; O’Donnell, J.A.; Shamie, I.S.; Lakin, P.R.; D’Cruz, A.A.; Braun, R.O.; Babon, J.J.; Lewis, R.S.; et al. Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation. Nat. Immunol. 2020, 21, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Tartey, S.; Gurung, P.; Samir, P.; Burton, A.; Kanneganti, T.D. Cutting Edge: Dysregulated CARD9 Signaling in Neutrophils Drives Inflammation in a Mouse Model of Neutrophilic Dermatoses. J. Immunol. 2018, 201, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Brydges, S.; Kastner, D.L. The systemic autoinflammatory diseases: Inborn errors of the innate immune system. Curr. Top. Microbiol. Immunol. 2006, 305, 127–160. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, N.; Furukawa, F. Autoinflammatory syndromes with a dermatological perspective. J. Dermatol. 2007, 34, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Farasat, S.; Aksentijevich, I.; Toro, J.R. Autoinflammatory diseases: Clinical and genetic advances. Arch. Dermatol. 2008, 144, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Warner, N.; Viani, K.; Nunez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [PubMed]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Yeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef]
- Chou, W.C.; Jha, S.; Linhoff, M.W.; Ting, J.P. The NLR gene family: From discovery to present day. Nat. Rev. Immunol. 2023, 23, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Almeida-da-Silva, C.L.C.; Savio, L.E.B.; Coutinho-Silva, R.; Ojcius, D.M. The role of NOD-like receptors in innate immunity. Front. Immunol. 2023, 14, 1122586. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Wolf, N.; Lavelle, E.C. Innate immune receptors. In NLR Proteins. Methods in Molecular Biology NLR Proteins: Methods and Protocols; Di Virgilio, F., Pelegrín, P., Eds.; Humana Press: Totowa, NJ, USA, 2016; Volume 1417, pp. 1–43. [Google Scholar] [CrossRef]
- Danis, J.; Mellett, M. Nod-Like Receptors in Host Defence and Disease at the Epidermal Barrier. Int. J. Mol. Sci. 2021, 22, 4677. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef]
- Chamaillard, M.; Hashimoto, M.; Horie, Y.; Masumoto, J.; Qiu, S.; Saab, L.; Ogura, Y.; Kawasaki, A.; Fukase, K.; Kusumoto, S.; et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 2003, 4, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.; Antignac, A.; Jehanno, M.; Viala, J.; Tedin, K.; Taha, M.K.; Labigne, A.; Zahringer, U.; et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujimoto, Y.; Lucas, P.C.; Nakano, H.; Fukase, K.; Nunez, G.; Inohara, N. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008, 27, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Koseki, T.; del Peso, L.; Hu, Y.; Yee, C.; Chen, S.; Carrio, R.; Merino, J.; Liu, D.; Ni, J.; et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J. Biol. Chem. 1999, 274, 14560–14567. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Ogura, Y.; Chen, F.F.; Muto, A.; Nunez, G. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J. Biol. Chem. 2001, 276, 2551–2554. [Google Scholar] [CrossRef]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef]
- Masumoto, J.; Yang, K.; Varambally, S.; Hasegawa, M.; Tomlins, S.A.; Qiu, S.; Fujimoto, Y.; Kawasaki, A.; Foster, S.J.; Horie, Y.; et al. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J. Exp. Med. 2006, 203, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Inohara, N.; Benito, A.; Chen, F.F.; Yamaoka, S.; Nunez, G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 2001, 276, 4812–4818. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Benko, S.; Magalhaes, J.G.; Philpott, D.J.; Girardin, S.E. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 2010, 185, 1681–1691. [Google Scholar] [CrossRef]
- Goncharov, T.; Hedayati, S.; Mulvihill, M.M.; Izrael-Tomasevic, A.; Zobel, K.; Jeet, S.; Fedorova, A.V.; Eidenschenk, C.; deVoss, J.; Yu, K.; et al. Disruption of XIAP-RIP2 Association Blocks NOD2-Mediated Inflammatory Signaling. Mol. Cell 2018, 69, 551–565.e7. [Google Scholar] [CrossRef]
- Krieg, A.; Correa, R.G.; Garrison, J.B.; Le Negrate, G.; Welsh, K.; Huang, Z.; Knoefel, W.T.; Reed, J.C. XIAP mediates NOD signaling via interaction with RIP2. Proc. Natl. Acad. Sci. USA 2009, 106, 14524–14529. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, Y.G.; McDonald, C.; Kanneganti, T.D.; Hasegawa, M.; Body-Malapel, M.; Inohara, N.; Nunez, G. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J. Immunol. 2007, 178, 2380–2386. [Google Scholar] [CrossRef]
- Pellegrini, E.; Desfosses, A.; Wallmann, A.; Schulze, W.M.; Rehbein, K.; Mas, P.; Signor, L.; Gaudon, S.; Zenkeviciute, G.; Hons, M.; et al. RIP2 filament formation is required for NOD2 dependent NF-kappaB signalling. Nat. Commun. 2018, 9, 4043. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, R.; de Souza, P.M.; Sriskandan, S.; Duffin, C.; Paul-Clark, M.J.; Mitchell, J.A. Pattern recognition receptors and interleukin-8 mediate effects of Gram-positive and Gram-negative bacteria on lung epithelial cell function. Br. J. Pharmacol. 2008, 154, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Voss, E.; Wehkamp, J.; Wehkamp, K.; Stange, E.F.; Schroder, J.M.; Harder, J. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J. Biol. Chem. 2006, 281, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.A.; Simanski, M.; Rademacher, F.; Schroder, L.; Harder, J. The pattern recognition receptor NOD2 mediates Staphylococcus aureus-induced IL-17C expression in keratinocytes. J. Investig. Dermatol. 2014, 134, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Ju, P.; Kou, W.; Zhai, M.; Zeng, Y.; Maimaitiaili, N.; Shi, Y.; Xu, X.; Zhao, Y.; Jian, W.; et al. Macrophage-Specific NLRC5 Protects From Cardiac Remodeling Through Interaction With HSPA8. JACC Basic. Transl. Sci. 2023, 8, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.S.; van den Elsen, P.J. NLRC5: A key regulator of MHC class I-dependent immune responses. Nat. Rev. Immunol. 2012, 12, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.L.; Zhao, X.F.; Chen, G.Z.; Fang, R.H.; Zhang, F.R. Silencing NLRC5 inhibits extracellular matrix expression in keloid fibroblasts via inhibition of transforming growth factor-beta1/Smad signaling pathway. Biomed. Pharmacother. 2016, 83, 1016–1021. [Google Scholar] [CrossRef]
- Cui, J.; Zhu, L.; Xia, X.; Wang, H.Y.; Legras, X.; Hong, J.; Ji, J.; Shen, P.; Zheng, S.; Chen, Z.J.; et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010, 141, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Kuenzel, S.; Till, A.; Winkler, M.; Hasler, R.; Lipinski, S.; Jung, S.; Grotzinger, J.; Fickenscher, H.; Schreiber, S.; Rosenstiel, P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 2010, 184, 1990–2000. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Burian, M.; Yazdi, A.S. NLRP1 Is the Key Inflammasome in Primary Human Keratinocytes. J. Investig. Dermatol. 2018, 138, 2507–2510. [Google Scholar] [CrossRef]
- Lautz, K.; Damm, A.; Menning, M.; Wenger, J.; Adam, A.C.; Zigrino, P.; Kremmer, E.; Kufer, T.A. NLRP10 enhances Shigella-induced pro-inflammatory responses. Cell. Microbiol. 2012, 14, 1568–1583. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Schroder, K. Inflammasome components as new therapeutic targets in inflammatory disease. Nat. Rev. Immunol. 2024, 25, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.L.; Mamai, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverenyi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, L.; Keller, M.; Niklaus, G.; Hohl, D.; Werner, S.; Beer, H.D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr. Biol. 2007, 17, 1140–1145. [Google Scholar] [CrossRef]
- Naik, S.M.; Cannon, G.; Burbach, G.J.; Singh, S.R.; Swerlick, R.A.; Wilcox, J.N.; Ansel, J.C.; Caughman, S.W. Human keratinocytes constitutively express interleukin-18 and secrete biologically active interleukin-18 after treatment with pro-inflammatory mediators and dinitrochlorobenzene. J. Investig. Dermatol. 1999, 113, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Fenini, G.; Karakaya, T.; Hennig, P.; Di Filippo, M.; Beer, H.D. The NLRP1 Inflammasome in Human Skin and Beyond. Int. J. Mol. Sci. 2020, 21, 4788. [Google Scholar] [CrossRef] [PubMed]
- Sand, J.; Haertel, E.; Biedermann, T.; Contassot, E.; Reichmann, E.; French, L.E.; Werner, S.; Beer, H.D. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis. 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Tsu, B.V.; Beierschmitt, C.; Ryan, A.P.; Agarwal, R.; Mitchell, P.S.; Daugherty, M.D. Diverse viral proteases activate the NLRP1 inflammasome. eLife 2021, 10, e60609. [Google Scholar] [CrossRef]
- Robinson, K.S.; Teo, D.E.T.; Tan, K.S.; Toh, G.A.; Ong, H.H.; Lim, C.K.; Lay, K.; Au, B.V.; Lew, T.S.; Chu, J.J.H.; et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 2020, 370, eaay2002. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Sarkar, M.K.; Okamura, K.; Harris, J.E.; Gudjonsson, J.E.; Fitzgerald, K.A. Activation of the NLRP1 inflammasome in human keratinocytes by the dsDNA mimetic poly(dA:dT). Proc. Natl. Acad. Sci. USA 2023, 120, e2213777120. [Google Scholar] [CrossRef]
- Rozario, P.; Pinilla, M.; Gorse, L.; Vind, A.C.; Robinson, K.S.; Toh, G.A.; Firdaus, M.J.; Martinez, J.F.; Kerk, S.K.; Lin, Z.; et al. Mechanistic basis for potassium efflux-driven activation of the human NLRP1 inflammasome. Proc. Natl. Acad. Sci. USA 2024, 121, e2309579121. [Google Scholar] [CrossRef]
- Ciazynska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibinska, M.; Lesiak, A. The Role of NLRP1, NLRP3, and AIM2 Inflammasomes in Psoriasis: Review. Int. J. Mol. Sci. 2021, 22, 5898. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Hao, S.; Zheng, H.; Zhao, X.; Li, Y. Association of NLRP1 and NLRP3 Polymorphisms with Psoriasis Vulgaris Risk in the Chinese Han Population. Biomed. Res. Int. 2018, 2018, 4714836. [Google Scholar] [CrossRef] [PubMed]
- Bivik, C.; Verma, D.; Winge, M.C.; Lieden, A.; Bradley, M.; Rosdahl, I.; Soderkvist, P. Genetic variation in the inflammasome and atopic dermatitis susceptibility. J. Investig. Dermatol. 2013, 133, 2486–2489. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Tohyama, M.; Murakami, M.; Sayama, K. Epidermal keratinocytes sense dsRNA via the NLRP3 inflammasome, mediating interleukin (IL)-1beta and IL-18 release. Exp. Dermatol. 2017, 26, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Cozzolino, F.; Talio, M.; Sitia, R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990, 9, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Strittmatter, G.E.; Kistowska, M.; French, L.E.; Beer, H.D. Caspase-4 is required for activation of inflammasomes. J. Immunol. 2012, 188, 1992–2000. [Google Scholar] [CrossRef]
- Gurung, P.; Lamkanfi, M.; Kanneganti, T.D. Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J. Immunol. 2015, 194, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.; Champagne, C.; Morizot, A.; Lapointe, J.M.; Saleh, M. The Inflammatory Caspases-1 and -11 Mediate the Pathogenesis of Dermatitis in Sharpin-Deficient Mice. J. Immunol. 2015, 195, 2365–2373. [Google Scholar] [CrossRef]
- Rodgers, M.A.; Bowman, J.W.; Fujita, H.; Orazio, N.; Shi, M.; Liang, Q.; Amatya, R.; Kelly, T.J.; Iwai, K.; Ting, J.; et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 2014, 211, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israel, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef]
- Li, S.; Kang, P.; Zhang, W.; Jian, Z.; Zhang, Q.; Yi, X.; Guo, S.; Guo, W.; Shi, Q.; Li, B.; et al. Activated NLR family pyrin domain containing 3 (NLRP3) inflammasome in keratinocytes promotes cutaneous T-cell response in patients with vitiligo. J. Allergy Clin. Immunol. 2020, 145, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yao, L.; Zhou, Y.; Gu, X.; Wang, C.; Bao, K.; Sun, Y.; Hong, M. A novel function of NLRP3 independent of inflammasome as a key transcription factor of IL-33 in epithelial cells of atopic dermatitis. Cell Death Dis. 2021, 12, 871. [Google Scholar] [CrossRef] [PubMed]
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Imamura, R.; Wang, Y.; Kinoshita, T.; Suzuki, M.; Noda, T.; Sagara, J.; Taniguchi, S.; Okamoto, H.; Suda, T. Anti-inflammatory activity of PYNOD and its mechanism in humans and mice. J. Immunol. 2010, 184, 5874–5884. [Google Scholar] [CrossRef]
- Prochnicki, T.; Vasconcelos, M.B.; Robinson, K.S.; Mangan, M.S.J.; De Graaf, D.; Shkarina, K.; Lovotti, M.; Standke, L.; Kaiser, R.; Stahl, R.; et al. Mitochondrial damage activates the NLRP10 inflammasome. Nat. Immunol. 2023, 24, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, B.; Tweedell, R.E.; Prasanth Kumar, S.; Kanneganti, T.D. The NLR family of innate immune and cell death sensors. Immunity 2024, 57, 674–699. [Google Scholar] [CrossRef]
- Mirza, N.; Sowa, A.S.; Lautz, K.; Kufer, T.A. NLRP10 Affects the Stability of Abin-1 To Control Inflammatory Responses. J. Immunol. 2019, 202, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hasegawa, M.; Imamura, R.; Kinoshita, T.; Kondo, C.; Konaka, K.; Suda, T. PYNOD, a novel Apaf-1/CED4-like protein is an inhibitor of ASC and caspase-1. Int. Immunol. 2004, 16, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Takahashi, A.; Kubo, M.; Tsunoda, T.; Tomita, K.; Sakashita, M.; Yamada, T.; Fujieda, S.; Tanaka, S.; Doi, S.; et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 2012, 44, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, F.; Nothnagel, M.; Parwez, Q.; Petrasch-Parwez, E.; Bechara, F.G.; Epplen, J.T.; Hoffjan, S. Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp. Dermatol. 2007, 16, 692–698. [Google Scholar] [CrossRef]
- Damm, A.; Giebeler, N.; Zamek, J.; Zigrino, P.; Kufer, T.A. Epidermal NLRP10 contributes to contact hypersensitivity responses in mice. Eur. J. Immunol. 2016, 46, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.N.; Halliday, G.M.; Johnston, L.J.; King, N.J. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Investig. Dermatol. 2001, 117, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Burckstummer, T.; Baumann, C.; Bluml, S.; Dixit, E.; Durnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Kumari, P.; Russo, A.J.; Shivcharan, S.; Rathinam, V.A. AIM2 in health and disease: Inflammasome and beyond. Immunol. Rev. 2020, 297, 83–95. [Google Scholar] [CrossRef]
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tian, Y.; Yin, Q. AIM2 Inflammasome Assembly and Signaling. Adv. Exp. Med. Biol. 2019, 1172, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, X.; Cheng, H.; Zhou, F. AIM2 and Psoriasis. Front. Immunol. 2023, 14, 1085448. [Google Scholar] [CrossRef]
- Pierini, R.; Juruj, C.; Perret, M.; Jones, C.L.; Mangeot, P.; Weiss, D.S.; Henry, T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012, 19, 1709–1721. [Google Scholar] [CrossRef]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Fisch, D.; Bando, H.; Clough, B.; Hornung, V.; Yamamoto, M.; Shenoy, A.R.; Frickel, E.M. Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 2019, 38, e100926. [Google Scholar] [CrossRef]
- Ekchariyawat, P.; Hamel, R.; Bernard, E.; Wichit, S.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; Choumet, V.; Yssel, H.; Despres, P.; et al. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect. Genet. Evol. 2015, 32, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Goss, C.; Anz, D.; Simanski, M.; Glaser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef]
- de Koning, H.D.; Bergboer, J.G.; van den Bogaard, E.H.; van Vlijmen-Willems, I.M.; Rodijk-Olthuis, D.; Simon, A.; Zeeuwen, P.L.; Schalkwijk, J. Strong induction of AIM2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp. Dermatol. 2012, 21, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gao, J.; Huang, C.; Jeong, S.; Ko, R.; Shen, X.; Chen, C.; Zhong, W.; Zou, Y.; Yu, B.; et al. Roles of AIM2 Gene and AIM2 Inflammasome in the Pathogenesis and Treatment of Psoriasis. Front. Genet. 2022, 13, 929162. [Google Scholar] [CrossRef]
- Uresti-Rivera, E.E.; Garcia-Hernandez, M.H. AIM2-inflammasome role in systemic lupus erythematous and rheumatoid arthritis. Autoimmunity 2022, 55, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, G.E.; Sand, J.; Sauter, M.; Seyffert, M.; Steigerwald, R.; Fraefel, C.; Smola, S.; French, L.E.; Beer, H.D. IFN-gamma Primes Keratinocytes for HSV-1-Induced Inflammasome Activation. J. Investig. Dermatol. 2016, 136, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Kopfnagel, V.; Wittmann, M.; Werfel, T. Human keratinocytes express AIM2 and respond to dsDNA with IL-1beta secretion. Exp. Dermatol. 2011, 20, 1027–1029. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhuang, Z.; Li, J.; Feng, Z. Significance of the cGAS-STING Pathway in Health and Disease. Int. J. Mol. Sci. 2023, 24, 13316. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Fei, C.J.; Hu, Y.; Wu, X.Y.; Nie, L.; Chen, J. Current understanding of the cGAS-STING signaling pathway: Structure, regulatory mechanisms, and related diseases. Zool. Res. 2023, 44, 183–218. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKepsilon Act Redundantly to Mediate STING-Induced NF-kappaB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, X.; Wang, Z.; Liu, P.; Hou, Y.; Xu, Y.; Su, H.; Koci, M.D.; Yin, H.; Zhang, C. NF-kappaB activation enhances STING signaling by altering microtubule-mediated STING trafficking. Cell Rep. 2023, 42, 112185. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Liu, A.; Xia, N.; Chen, N.; Meurens, F.; Zhu, J. How the Innate Immune DNA Sensing cGAS-STING Pathway Is Involved in Apoptosis. Int. J. Mol. Sci. 2023, 24, 3029. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Y.J.; Dobbs, N.; Sakai, T.; Liou, J.; Miner, J.J.; Yan, N. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med. 2019, 216, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Woo, J.S.; Wu, B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C.; et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 2019, 20, 152–162. [Google Scholar] [CrossRef]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef]
- Cao, T.; Shao, S.; Li, B.; Jin, L.; Lei, J.; Qiao, H.; Wang, G. Up-regulation of Interferon-inducible protein 16 contributes to psoriasis by modulating chemokine production in keratinocytes. Sci. Rep. 2016, 6, 25381. [Google Scholar] [CrossRef]
- Almine, J.F.; O’Hare, C.A.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, W.; Wang, F.; Hayashi, T.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Ikejima, T. DNA damage-triggered activation of cGAS-STING pathway induces apoptosis in human keratinocyte HaCaT cells. Mol. Immunol. 2021, 131, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.K.; Uppala, R.; Zeng, C.; Billi, A.C.; Tsoi, L.C.; Kidder, A.; Xing, X.; Perez White, B.E.; Shao, S.; Plazyo, O.; et al. Keratinocytes sense and eliminate CRISPR DNA through STING/IFN-kappa activation and APOBEC3G induction. J. Clin. Investig. 2023, 133, e159393. [Google Scholar] [CrossRef]
- Pan, Y.; You, Y.; Sun, L.; Sui, Q.; Liu, L.; Yuan, H.; Chen, C.; Liu, J.; Wen, X.; Dai, L.; et al. The STING antagonist H-151 ameliorates psoriasis via suppression of STING/NF-kappaB-mediated inflammation. Br. J. Pharmacol. 2021, 178, 4907–4922. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, L.; Wang, J.; Luo, X.; Wang, M.; Wang, C.; Chen, J.; Zhou, Y.; Yin, H.; Song, Y.; et al. Targeting STING in dendritic cells alleviates psoriatic inflammation by suppressing IL-17A production. Cell Mol. Immunol. 2024, 21, 738–751. [Google Scholar] [CrossRef]
- Yu, Y.; Xue, X.; Tang, W.; Su, L.; Zhang, L.; Zhang, Y. Cytosolic DNA–Mediated STING-Dependent Inflammation Contributes to the Progression of Psoriasis. J. Investig. Dermatol. 2022, 142, 898–906.e4. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef]
- Vazquez, C.; Horner, S.M. MAVS Coordination of Antiviral Innate Immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef]
- Kouwaki, T.; Okamoto, M.; Tsukamoto, H.; Fukushima, Y.; Matsumoto, M.; Seya, T.; Oshiumi, H. Zyxin stabilizes RIG-I and MAVS interactions and promotes type I interferon response. Sci. Rep. 2017, 7, 11905. [Google Scholar] [CrossRef]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Tenoever, B.R.; Ng, S.L.; Chua, M.A.; McWhirter, S.M.; Garcia-Sastre, A.; Maniatis, T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef] [PubMed]
- Kasumba, D.M.; Grandvaux, N. Therapeutic Targeting of RIG-I and MDA5 Might Not Lead to the Same Rome. Trends Pharmacol. Sci. 2019, 40, 116–127. [Google Scholar] [CrossRef]
- Bertin, J.; Guo, Y.; Wang, L.; Srinivasula, S.M.; Jacobson, M.D.; Poyet, J.L.; Merriam, S.; Du, M.Q.; Dyer, M.J.; Robison, K.E.; et al. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-kappa B. J. Biol. Chem. 2000, 275, 41082–41086. [Google Scholar] [CrossRef] [PubMed]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschlager, N.; Schlee, M.; Rothenfusser, S.; Barchet, W.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010, 11, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Fukui, A.; Shigeishi, H.; Ishida, Y.; Nishi, H.; Tobiume, K.; Takechi, M.; Kamata, N. Expression and function of RIG-I in oral keratinocytes and fibroblasts. Cell Physiol. Biochem. 2014, 34, 1556–1565. [Google Scholar] [CrossRef]
- Kitamura, H.; Matsuzaki, Y.; Kimura, K.; Nakano, H.; Imaizumi, T.; Satoh, K.; Hanada, K. Cytokine modulation of retinoic acid-inducible gene-I (RIG-I) expression in human epidermal keratinocytes. J. Dermatol. Sci. 2007, 45, 127–134. [Google Scholar] [CrossRef]
- Kato, H.; Ohta, K.; Akagi, M.; Fukada, S.; Sakuma, M.; Naruse, T.; Nishi, H.; Shigeishi, H.; Takechi, M.; Aikawa, T. LL-37-dsRNA Complexes Modulate Immune Response via RIG-I in Oral Keratinocytes. Inflammation 2023, 46, 808–823. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Hamel, R.; Neyret, A.; Ekchariyawat, P.; Moles, J.P.; Simmons, G.; Chazal, N.; Despres, P.; Misse, D.; Briant, L. Human keratinocytes restrict chikungunya virus replication at a post-fusion step. Virology 2015, 476, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, Q.; Huang, Q.; Yang, H.; Zheng, J.; Xie, R.; Han, D.; Wei, Q. RIG-I contributes to keratinocyte proliferation and wound repair by inducing TIMP-1 expression through NF-kappaB signaling pathway. J. Cell Physiol. 2023, 238, 1876–1890. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lou, F.; Yin, Q.; Gao, Y.; Sun, Y.; Bai, J.; Xu, Z.; Liu, Z.; Cai, W.; Ke, F.; et al. RIG-I antiviral signaling drives interleukin-23 production and psoriasis-like skin disease. EMBO Mol. Med. 2017, 9, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Spritz, R.A.; Andersen, G.H. Genetics of Vitiligo. Dermatol. Clin. 2017, 35, 245–255. [Google Scholar] [CrossRef]
- Zhuang, T.; Yi, X.; Chen, J.; Kang, P.; Chen, X.; Chen, J.; Cui, T.; Chang, Y.; Ye, Z.; Ni, Q.; et al. Intracellular virus sensor MDA5 exacerbates vitiligo by inducing the secretion of chemokines in keratinocytes under virus invasion. Cell Death Dis. 2020, 11, 453. [Google Scholar] [CrossRef]
- Scholtissek, B.; Zahn, S.; Maier, J.; Klaeschen, S.; Braegelmann, C.; Hoelzel, M.; Bieber, T.; Barchet, W.; Wenzel, J. Immunostimulatory Endogenous Nucleic Acids Drive the Lesional Inflammation in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2017, 137, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Chen, Q.; Feng, Y.; Wang, Y.; Wang, J.; Liang, J.; Xu, J. Bioinformatic analysis of key biomarkers and immune filtration of skin biopsy in discoid lupus erythematosus. Lupus 2021, 30, 807–817. [Google Scholar] [CrossRef]
- Dong, Q.; Chen, K.; Xie, J.; Han, H.; Feng, Y.; Lu, J.; Wang, W. Identification of key genes and pathways in discoid lupus skin via bioinformatics analysis. Medicine 2021, 100, e25433. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Hu, Z.; Mei, X.; Ouyang, L.; Song, Y.; Zhou, W.; Kong, Y.; Wu, R.; Rao, S.; Long, H.; et al. Single-cell sequencing shows cellular heterogeneity of cutaneous lesions in lupus erythematosus. Nat. Commun. 2022, 13, 7489. [Google Scholar] [CrossRef]
- Vorwerk, G.; Zahn, S.; Bieber, T.; Wenzel, J. NKG2D and its ligands as cytotoxic factors in cutaneous lupus erythematosus. Exp. Dermatol. 2021, 30, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Zheng, M.; Yu, Y.; Wu, J.; Kuang, Q.; Hu, Z.; Ouyang, L.; Lu, S.; Zhao, M. Ultraviolet light induces HERV expression to activate RIG-I signalling pathway in keratinocytes. Exp. Dermatol. 2022, 31, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kanneganti, T.D. ZBP1: Innate Sensor Regulating Cell Death and Inflammation. Trends Immunol. 2018, 39, 123–134. [Google Scholar] [CrossRef]
- Shi, C.; Cao, P.; Wang, Y.; Zhang, Q.; Zhang, D.; Wang, Y.; Wang, L.; Gong, Z. PANoptosis: A Cell Death Characterized by Pyroptosis, Apoptosis, and Necroptosis. J. Inflamm. Res. 2023, 16, 1523–1532. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. PANoptosome signaling and therapeutic implications in infection: Central role for ZBP1 to activate the inflammasome and PANoptosis. Curr. Opin. Immunol. 2023, 83, 102348. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Lee, S. Recent advances in ZBP1-derived PANoptosis against viral infections. Front. Immunol. 2023, 14, 1148727. [Google Scholar] [CrossRef]
- Mishra, S.; Dey, A.A.; Kesavardhana, S. Z-Nucleic Acid Sensing and Activation of ZBP1 in Cellular Physiology and Disease Pathogenesis. Immunol. Rev. 2025, 329, e13437. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Qi, Z.; Zhang, H.; Wang, N. Nucleic Acid Sensor-Mediated PANoptosis in Viral Infection. Viruses 2024, 16, 966. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef]
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, R.K.S.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S.; et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 237. [Google Scholar] [CrossRef]
- Muendlein, H.I.; Connolly, W.M.; Magri, Z.; Jetton, D.; Smirnova, I.; Degterev, A.; Balachandran, S.; Poltorak, A. ZBP1 promotes inflammatory responses downstream of TLR3/TLR4 via timely delivery of RIPK1 to TRIF. Proc. Natl. Acad. Sci. USA 2022, 119, e2113872119. [Google Scholar] [CrossRef] [PubMed]
- Solon, M.; Ge, N.; Hambro, S.; Haller, S.; Jiang, J.; Baca, M.; Preston, J.; Maltzman, A.; Wickliffe, K.E.; Liang, Y.; et al. ZBP1 and TRIF trigger lethal necroptosis in mice lacking caspase-8 and TNFR1. Cell Death Differ. 2024, 31, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.P.; Thapa, R.J.; Fisher, A.; Tummers, B.; Zhang, T.; Yin, C.; Rodriguez, D.A.; Guo, H.; Lane, R.; Williams, R.; et al. ZBP1/DAI Drives RIPK3-Mediated Cell Death Induced by IFNs in the Absence of RIPK1. J. Immunol. 2019, 203, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Brune, W.; Andoniou, C.E. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses 2017, 9, 249. [Google Scholar] [CrossRef]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef]
- Guo, H.; Gilley, R.P.; Fisher, A.; Lane, R.; Landsteiner, V.J.; Ragan, K.B.; Dovey, C.M.; Carette, J.E.; Upton, J.W.; Mocarski, E.S.; et al. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis. 2018, 9, 816. [Google Scholar] [CrossRef]
- Steain, M.; Baker, M.; Pham, C.L.L.; Shanmugam, N.; Gambin, Y.; Sierecki, E.; McSharry, B.P.; Avdic, S.; Slobedman, B.; Sunde, M.; et al. Varicella zoster virus encodes a viral decoy RHIM to inhibit cell death. PLoS Pathog. 2020, 16, e1008473. [Google Scholar] [CrossRef] [PubMed]
- Koehler, H.; Cotsmire, S.; Zhang, T.; Balachandran, S.; Upton, J.W.; Langland, J.; Kalman, D.; Jacobs, B.L.; Mocarski, E.S. Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe 2021, 29, 1266–1276.e5. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Lee, S.; Mall, R.; Pandian, N.; Wang, Y.; Sharma, B.R.; Malireddi, R.S.; Yang, D.; Trifkovic, S.; Steele, J.A.; et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci. Immunol. 2022, 7, eabo6294. [Google Scholar] [CrossRef]
- Momota, M.; Lelliott, P.; Kubo, A.; Kusakabe, T.; Kobiyama, K.; Kuroda, E.; Imai, Y.; Akira, S.; Coban, C.; Ishii, K.J. ZBP1 governs the inflammasome-independent IL-1alpha and neutrophil inflammation that play a dual role in anti-influenza virus immunity. Int. Immunol. 2020, 32, 203–212. [Google Scholar] [CrossRef]
- Jiao, H.; Wachsmuth, L.; Kumari, S.; Schwarzer, R.; Lin, J.; Eren, R.O.; Fisher, A.; Lane, R.; Young, G.R.; Kassiotis, G.; et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 2020, 580, 391–395. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Malireddi, R.K.S.; Burton, A.R.; Porter, S.N.; Vogel, P.; Pruett-Miller, S.M.; Kanneganti, T.D. The Zalpha2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J. Biol. Chem. 2020, 295, 8325–8330. [Google Scholar] [CrossRef]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Strasser, A.; Pham, V.C.; Lill, J.R.; Roose-Girma, M.; Warming, S.; Solon, M.; et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 2016, 540, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Freund, L.; Oehrl, S.; Schwingen, J.; Haeberle, S.; Dobel, T.; Lee, P.D.H.; Meisel, S.; Mihalceanu, S.; Russwurm, M.; Luft, T.; et al. IFNgamma Causes Keratinocyte Necroptosis in Acute Graft-Versus-Host Disease. J. Investig. Dermatol. 2023, 143, 1746–1756.e9. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).