
Genetic Diversity and Molecular Evolution of Hepatitis E Virus Within the Genus Chirohepevirus in Bats


Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Collection
2.2. Sequence Analyses
2.3. Phylogenetic and Evolutionary Analyses
3. Results
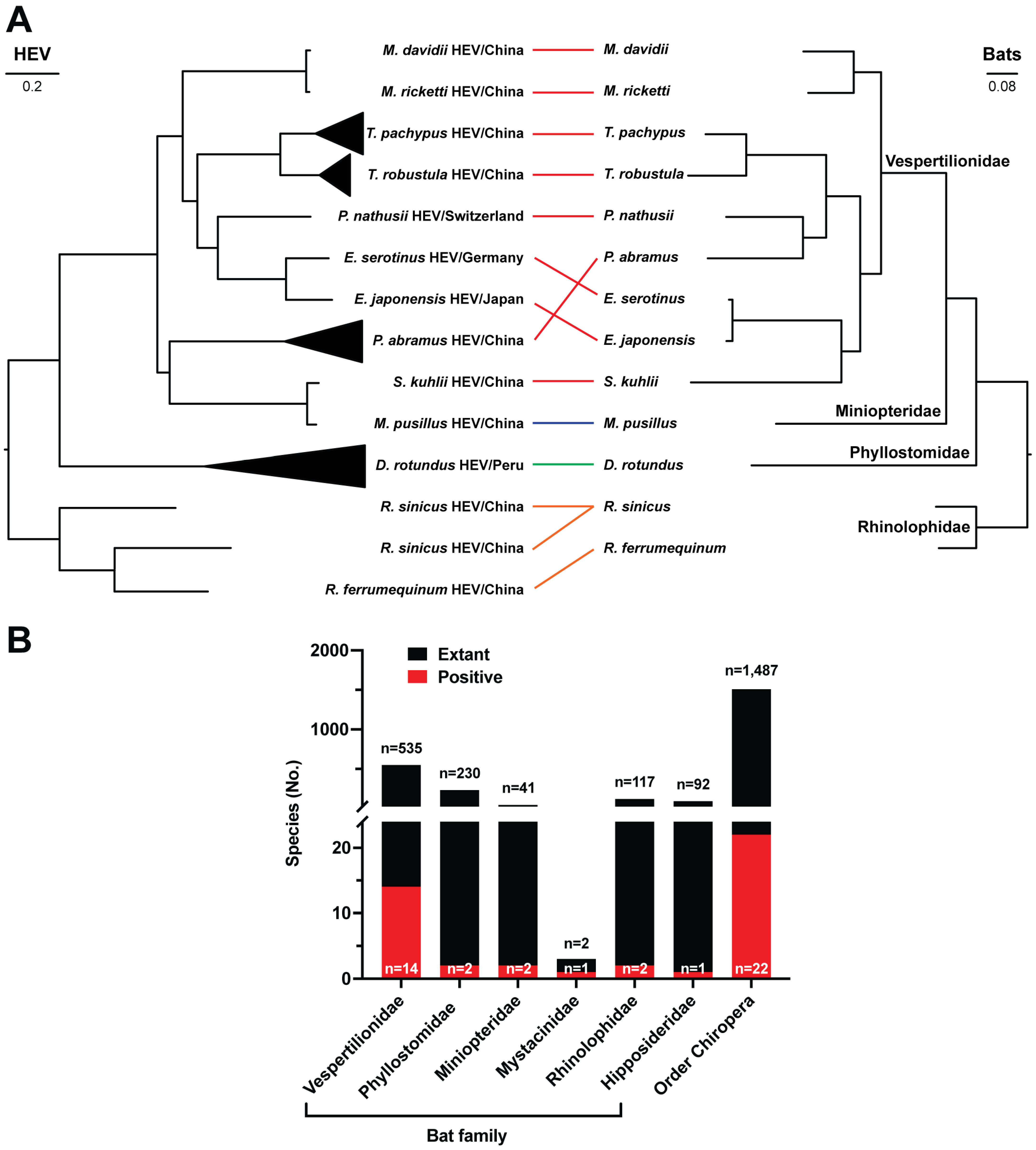
3.1. Detection and Distribution of Chirohepeviruses
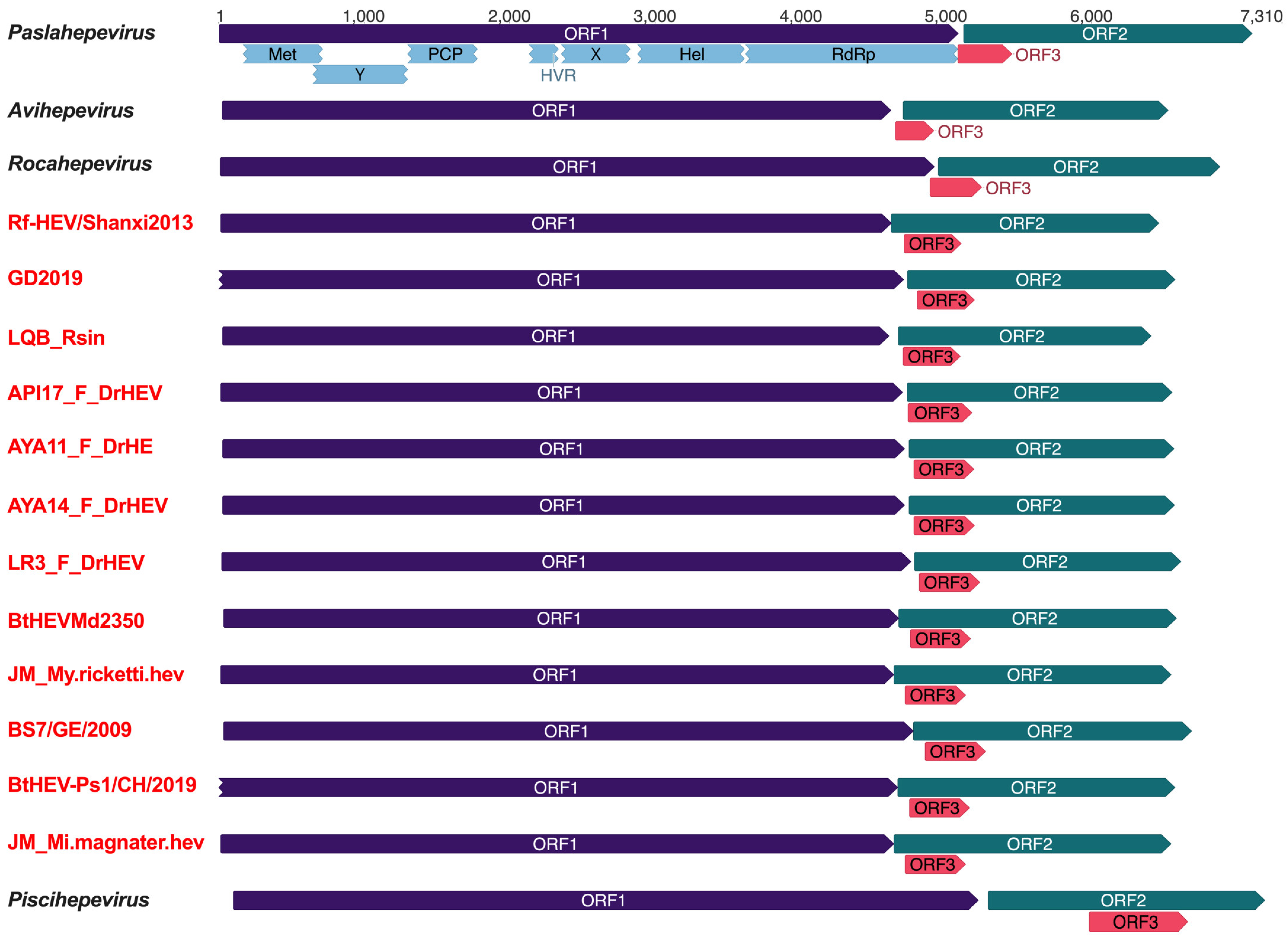
3.2. Genomic Characterization of Bat Chirohepeviruses
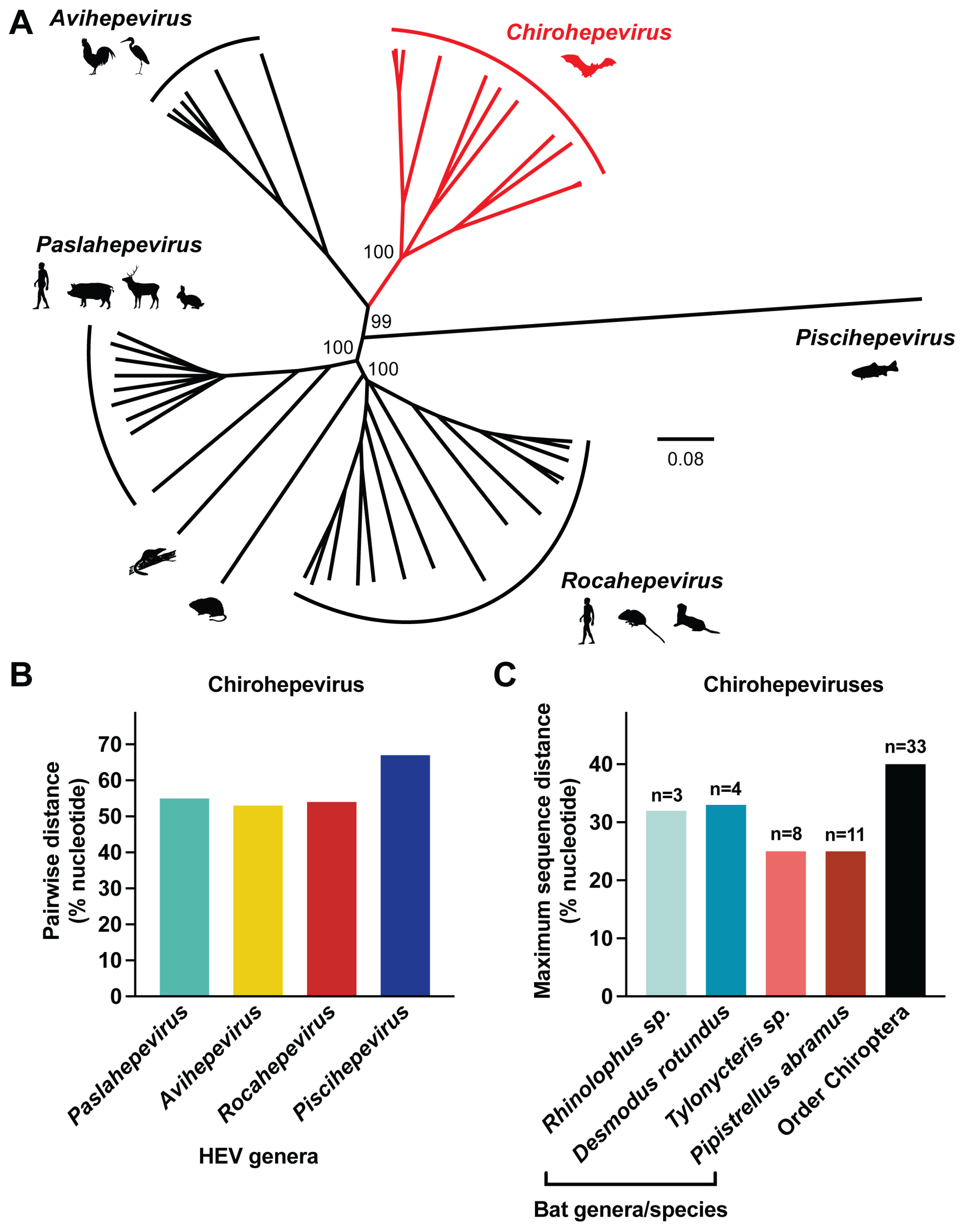
3.3. Genetic Diversity of Chirohepeviruses
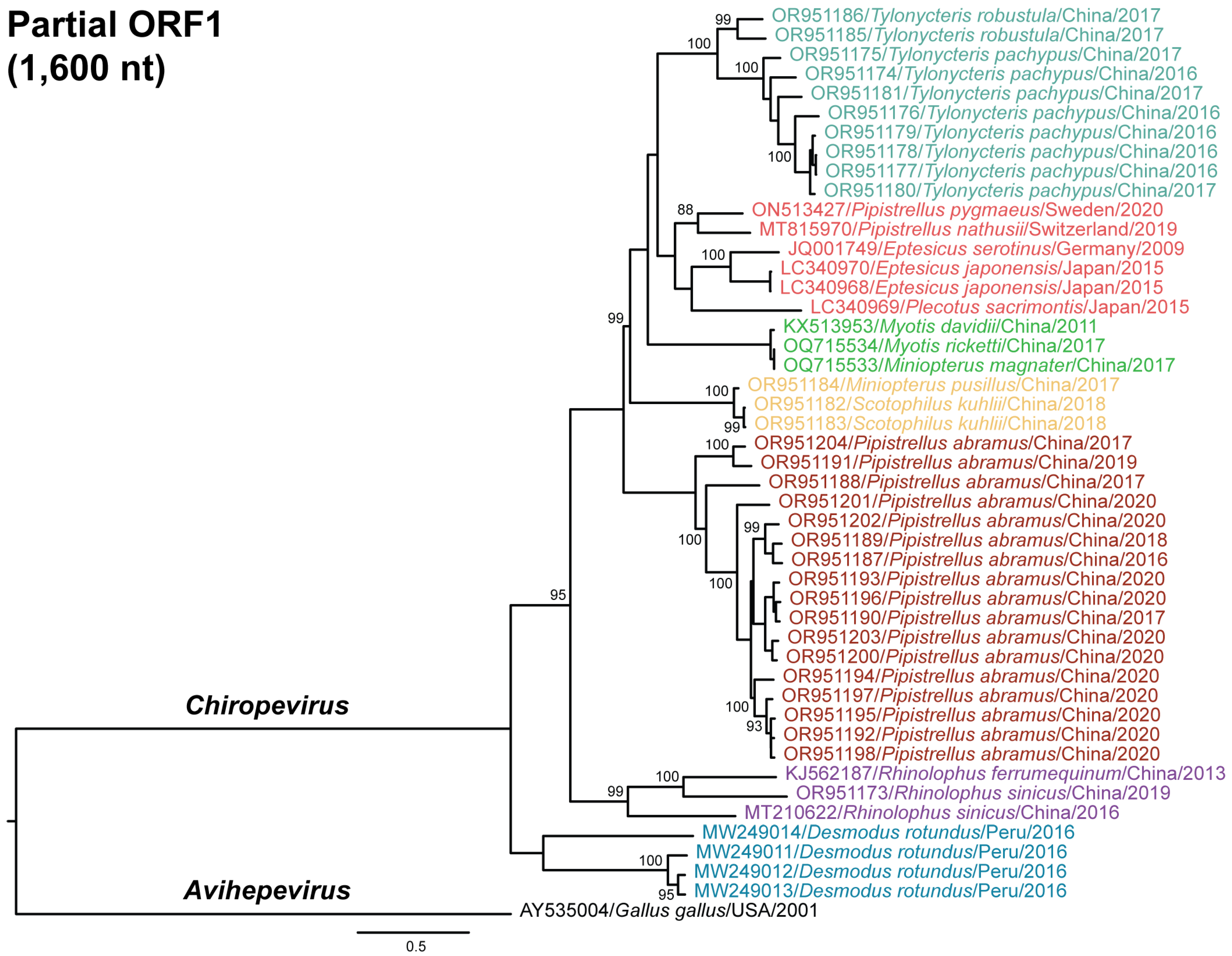
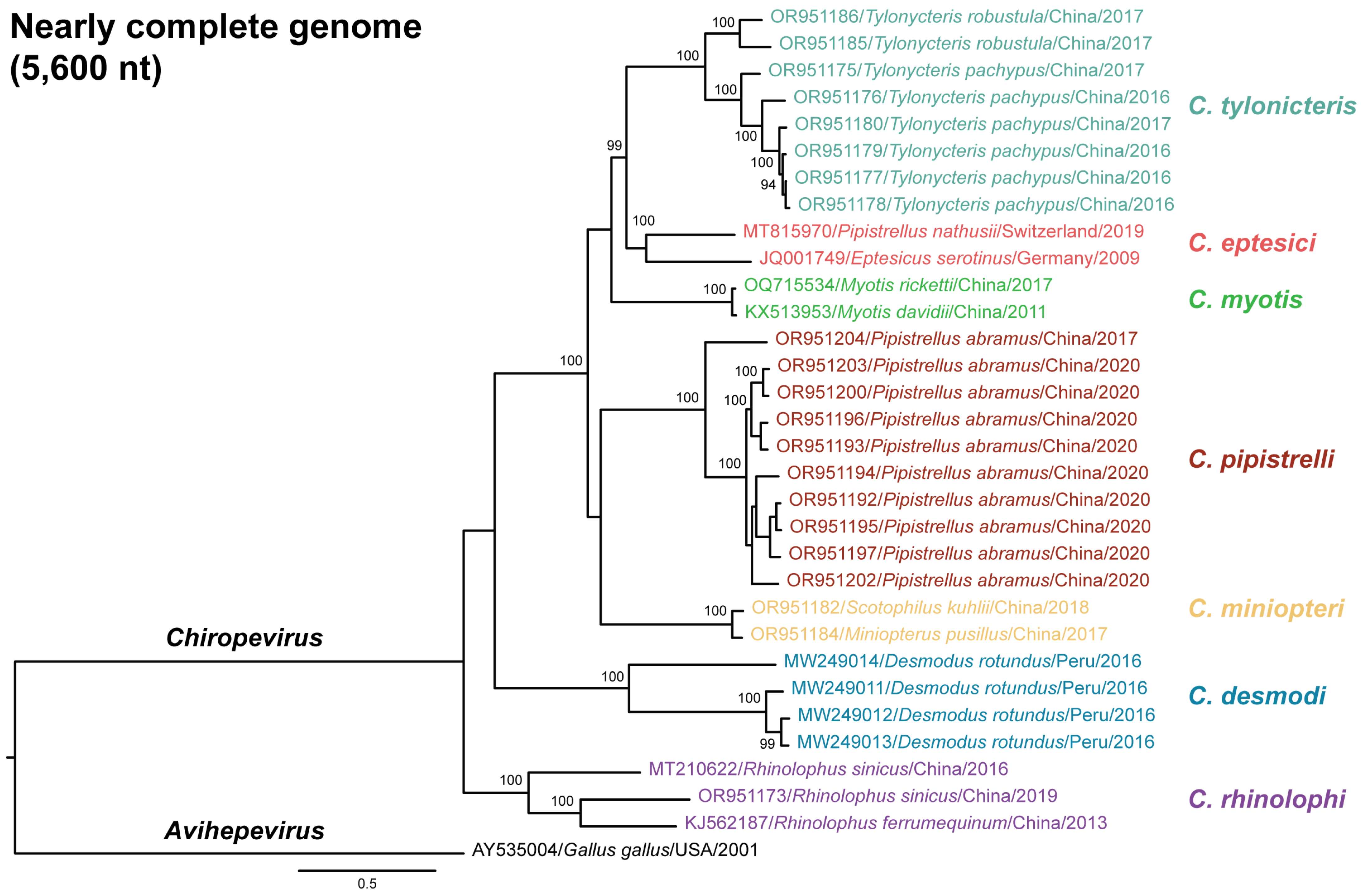
3.4. Phylogenetic Relationships of Chirohepeviruses
3.5. Co-Evolution of Chirohepeviruses and Bat Species
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Rein, D.B.; Stevens, G.A.; Theaker, J.; Wittenborn, J.S.; Wiersma, S.T. The global burden of hepatitis E virus genotypes 1 and 2 in 2005. Hepatology 2012, 55, 988–997. [Google Scholar] [CrossRef]
- Wang, B.; Harms, D.; Papp, C.P.; Niendorf, S.; Jacobsen, S.; Lutgehetmann, M.; Pischke, S.; Wedermeyer, H.; Hofmann, J.; Bock, C.T. Comprehensive Molecular Approach for Characterization of Hepatitis E Virus Genotype 3 Variants. J. Clin. Microbiol. 2018, 56, 1110–1128. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wu, X.; Xia, J. Hepatitis E virus infection during pregnancy. Virol. J. 2020, 17, 73. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; de Man, R.A.; Kamar, N.; Pan, Q. Chronic hepatitis E: Advancing research and patient care. J. Hepatol. 2022, 77, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Meister, T.L.; Steinmann, E. Hepatitis E virus treatment and ribavirin therapy: Viral mechanisms of nonresponse. Curr. Opin. Virol. 2018, 32, 80–87. [Google Scholar] [CrossRef]
- Tian, D.; Li, W.; Heffron, C.L.; Wang, B.; Mahsoub, H.M.; Sooryanarain, H.; Hassebroek, A.M.; Clark-Deener, S.; LeRoith, T.; Meng, X.J. Hepatitis E virus infects brain microvascular endothelial cells, crosses the blood-brain barrier, and invades the central nervous system. Proc. Natl. Acad. Sci. USA 2022, 119, e2201862119. [Google Scholar] [CrossRef]
- Dalton, H.R.; Kamar, N.; Baylis, S.A.; Moradpour, D.; Wedemeyer, H.; Negro, F.; Dalton, H.R.; Kamar, N.; Baylis, S.A.; Moradpour, D.; et al. European Association for the Study of the Liver. EASL Clinical Practice Guidelines on hepatitis E virus infection. J. Hepatol. 2018, 68, 1256–1271. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Meng, X.J. Structural and molecular biology of hepatitis E virus. Comput. Struct. Biotechnol. J. 2021, 19, 1907–1916. [Google Scholar] [CrossRef]
- Purdy, M.A.; Drexler, J.F.; Meng, X.J.; Norder, H.; Okamoto, H.; Van der Poel, W.H.M.; Reuter, G.; de Souza, W.M.; Ulrich, R.G.; Smith, D.B. ICTV Virus Taxonomy Profile: Hepeviridae 2022. J. Gen. Virol. 2022, 103, 001778. [Google Scholar] [CrossRef]
- Wang, B.; Yang, X.L. Chirohepevirus from Bats: Insights into Hepatitis E Virus Diversity and Evolution. Viruses 2022, 14, 905. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Meng, X.J. Hepatitis E virus: Host tropism and zoonotic infection. Curr. Opin. Microbiol. 2021, 59, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Kinast, V.; Klohn, M.; Nocke, M.K.; Todt, D.; Steinmann, E. Hepatitis E virus species barriers: Seeking viral and host determinants. Curr. Opin. Virol. 2022, 56, 101274. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Pavlovich, S.S.; Lovett, S.P.; Koroleva, G.; Guito, J.C.; Arnold, C.E.; Nagle, E.R.; Kulcsar, K.; Lee, A.; Thibaud-Nissen, F.; Hume, A.J.; et al. The Egyptian Rousette Genome Reveals Unexpected Features of Bat Antiviral Immunity. Cell 2018, 173, 1098–1110.e18. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef]
- Ruiz-Aravena, M.; McKee, C.; Gamble, A.; Lunn, T.; Morris, A.; Snedden, C.E.; Yinda, C.K.; Port, J.R.; Buchholz, D.W.; Yeo, Y.Y.; et al. Ecology, evolution and spillover of coronaviruses from bats. Nat. Rev. Microbiol. 2022, 20, 299–314. [Google Scholar] [CrossRef]
- Yang, X.L.; Tan, C.W.; Anderson, D.E.; Jiang, R.D.; Li, B.; Zhang, W.; Zhu, Y.; Lim, X.F.; Zhou, P.; Liu, X.L.; et al. Characterization of a filovirus (Mengla virus) from Rousettus bats in China. Nat. Microbiol. 2019, 4, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Wibbelt, G.; Moore, M.S.; Schountz, T.; Voigt, C.C. Emerging diseases in Chiroptera: Why bats? Biol. Lett. 2010, 6, 438–440. [Google Scholar] [CrossRef]
- Drexler, J.F.; Seelen, A.; Corman, V.M.; Fumie Tateno, A.; Cottontail, V.; Melim Zerbinati, R.; Gloza-Rausch, F.; Klose, S.M.; Adu-Sarkodie, Y.; Oppong, S.K.; et al. Bats worldwide carry hepatitis E virus-related viruses that form a putative novel genus within the family Hepeviridae. J. Virol. 2012, 86, 9134–9147. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Marchand, S.; Sessa, A.; Cappy, P.; Pawlotsky, J.M. Orthohepevirus C hepatitis, an underdiagnosed disease? J. Hepatol. 2023, 79, e39–e41. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, P.; Han, Y.; Zhao, W.; Zhao, L.; Li, R.; Zhang, J.; Zhang, S.; Lu, J.; Daszak, P.; et al. Unveiling bat-borne viruses: A comprehensive classification and analysis of virome evolution. Microbiome 2024, 12, 235. [Google Scholar] [CrossRef] [PubMed]
- Bergner, L.M.; Mollentze, N.; Orton, R.J.; Tello, C.; Broos, A.; Biek, R.; Streicker, D.G. Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats. Viruses 2021, 13, 252. [Google Scholar] [CrossRef]
- Chen, Y.M.; Hu, S.J.; Lin, X.D.; Tian, J.H.; Lv, J.X.; Wang, M.R.; Luo, X.Q.; Pei, Y.Y.; Hu, R.X.; Song, Z.G.; et al. Host traits shape virome composition and virus transmission in wild small mammals. Cell 2023, 186, 4662–4675.e12. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Moore, N.E.; Murray, Z.L.; McInnes, K.; White, D.J.; Tompkins, D.M.; Hall, R.J. Discovery of novel virus sequences in an isolated and threatened bat species, the New Zealand lesser short-tailed bat (Mystacina tuberculata). J. Gen. Virol. 2015, 96, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Waller, S.J.; Tortosa, P.; Thurley, T.; O’Donnell, C.F.J.; Jackson, R.; Dennis, G.; Grimwood, R.M.; Holmes, E.C.; McInnes, K.; Geoghegan, J.L. Virome analysis of New Zealand’s bats reveals cross-species viral transmission among the Coronaviridae. Virus Evol. 2024, 10, veae008. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, X.L.; Li, W.; Zhu, Y.; Ge, X.Y.; Zhang, L.B.; Zhang, Y.Z.; Bock, C.T.; Shi, Z.L. Detection and genome characterization of four novel bat hepadnaviruses and a hepevirus in China. Virol. J. 2017, 14, 40. [Google Scholar] [CrossRef]
- Kobayashi, T.; Murakami, S.; Yamamoto, T.; Mineshita, K.; Sakuyama, M.; Sasaki, R.; Maeda, K.; Horimoto, T. Detection of bat hepatitis E virus RNA in microbats in Japan. Virus Genes 2018, 54, 599–602. [Google Scholar] [CrossRef]
- Hardmeier, I.; Aeberhard, N.; Qi, W.; Schoenbaechler, K.; Kraettli, H.; Hatt, J.M.; Fraefel, C.; Kubacki, J. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PLoS ONE 2021, 16, e0252534. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Simmons, N.; Cirranello, A. Bat Species of the World: A Taxonomic and Geographic Database. 2020. Available online: https://batnames.org/ (accessed on 15 November 2024).
- Rasche, A.; Sander, A.L.; Corman, V.M.; Drexler, J.F. Evolutionary biology of human hepatitis viruses. J. Hepatol. 2019, 70, 501–520. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Walker, P.J.; Poon, L.L. Mass extinctions, biodiversity and mitochondrial function: Are bats ‘special’ as reservoirs for emerging viruses? Curr. Opin. Virol. 2011, 1, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Grange, Z.L.; Goldstein, T.; Johnson, C.K.; Anthony, S.; Gilardi, K.; Daszak, P.; Olival, K.J.; O’Rourke, T.; Murray, S.; Olson, S.H.; et al. Ranking the risk of animal-to-human spillover for newly discovered viruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2002324118. [Google Scholar] [CrossRef]
- Meng, X.J.; Purcell, R.H.; Halbur, P.G.; Lehman, J.R.; Webb, D.M.; Tsareva, T.S.; Haynes, J.S.; Thacker, B.J.; Emerson, S.U. A novel virus in swine is closely related to the human hepatitis E virus. Proc. Natl. Acad. Sci. USA 1997, 94, 9860–9865. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Tan, B.H.; Teo, E.C.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection with Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Harms, D.; Yang, X.L.; Bock, C.T. Orthohepevirus C: An Expanding Species of Emerging Hepatitis E Virus Variants. Pathogens 2020, 9, 154. [Google Scholar] [CrossRef]
- Meng, X.J. Expanding Host Range and Cross-Species Infection of Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005695. [Google Scholar] [CrossRef]
- van Tong, H.; Hoan, N.X.; Wang, B.; Wedemeyer, H.; Bock, C.T.; Velavan, T.P. Hepatitis E Virus Mutations: Functional and Clinical Relevance. EBioMedicine 2016, 11, 31–42. [Google Scholar] [CrossRef]
- Wang, B.; Subramaniam, S.; Tian, D.; Mahsoub, H.M.; Heffron, C.L.; Meng, X.J. Phosphorylation of Ser711 residue in the hypervariable region of zoonotic genotype 3 hepatitis E virus is important for virus replication. mBio 2024, 15, e0263524. [Google Scholar] [CrossRef]
- Wang, B.; Tian, D.; Sooryanarain, H.; Mahsoub, H.M.; Heffron, C.L.; Hassebroek, A.M.; Meng, X.J. Two mutations in the ORF1 of genotype 1 hepatitis E virus enhance virus replication and may associate with fulminant hepatic failure. Proc. Natl. Acad. Sci. USA 2022, 119, e2207503119. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Falkenhagen, A.; Bock, C.T.; Johne, R. Reverse genetics approaches for hepatitis E virus and related viruses. Curr. Opin. Virol. 2020, 44, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Fieulaine, S.; Tubiana, T.; Bressanelli, S. De novo modelling of HEV replication polyprotein: Five-domain breakdown and involvement of flexibility in functional regulation. Virology 2023, 578, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Cambillau, C.; Roussel, A.; Imbert, I. Structure Prediction and Analysis of Hepatitis E Virus Non-Structural Proteins from the Replication and Transcription Machinery by AlphaFold2. Viruses 2022, 14, 1537. [Google Scholar] [CrossRef]
- Jo, W.K.; Anzolini Cassiano, M.H.; de Oliveira-Filho, E.F.; Brunink, S.; Yansanjav, A.; Yihune, M.; Koshkina, A.I.; Lukashev, A.N.; Lavrenchenko, L.A.; Lebedev, V.S.; et al. Ancient evolutionary origins of hepatitis E virus in rodents. Proc. Natl. Acad. Sci. USA 2024, 121, e2413665121. [Google Scholar] [CrossRef] [PubMed]
- Li, F.L.; Wang, B.; Han, P.Y.; Li, B.; Si, H.R.; Zhu, Y.; Yin, H.M.; Zong, L.D.; Tang, Y.; Shi, Z.L.; et al. Identification of novel rodent and shrew orthohepeviruses sheds light on hepatitis E virus evolution. Zool. Res. 2025, 46, 103–121. [Google Scholar] [CrossRef]
- Wang, B.; Li, W.; Zhou, J.H.; Li, B.; Zhang, W.; Yang, W.H.; Pan, H.; Wang, L.X.; Bock, C.T.; Shi, Z.L.; et al. Chevrier’s Field Mouse (Apodemus chevrieri) and Pere David’s Vole (Eothenomys melanogaster) in China Carry Orthohepeviruses that form Two Putative Novel Genotypes Within the Species Orthohepevirus C. Virol. Sin. 2018, 33, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Sander, A.L.; Corman, V.M.; Lukashev, A.N.; Drexler, J.F. Evolutionary Origins of Enteric Hepatitis Viruses. Cold Spring Harb. Perspect. Med. 2018, 8, a031690. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Family | Host Species | Host Common Name a | Sampling Country (Collection Year) | Sample Source | Genomic Sequence (No.) | GenBank Accession No. | Reference |
|---|---|---|---|---|---|---|---|
| Rhinolophidae | Rhinolophus ferrumequinum | greater horseshoe bat | China (2013) | Anal swab | Complete (1) | KJ562187 | [23] |
| Rhinolophidae | Rhinolophus sinicus | Chinese rufous horseshoe bat | China (2016, 2019) | Anal swab | Complete (2) | MT210622, OR951173 | N.A. b, [24] |
| Hipposideridae | Hipposideros abae | Aba roundleaf bat | Ghana (2009) | Feces | Partial (2) | JQ001744, JQ071861 | [21] |
| Phyllostomidae | Vampyrodes caraccioli | great stripe-faced bat | Panama (2009) | Blood | Partial (1) | JQ001745 | [21] |
| Phyllostomidae | Desmodus rotundus | common vampire bat | Peru (2016) | Anal swab | Complete (4) | MW249011–MW249014 | [25] |
| Miniopteridae | Miniopterus magnater | Western long-fingered bat | China (2017) | Pooled animal | Complete (1) | OQ715533 | [26] |
| Miniopteridae | Miniopterus pusillus | small bent-winged bat | China (2017) | Anal swab | Partial (1) | OR951184 | [24] |
| Mystacinidae | Mystacina tuberculata | New Zealand lesser short-tailed bat | New Zealand (2013, 2020, 2021) | Bat guano | Partial (9) | KM204384, KM204385 c, OR248814–OR248820 | [27,28] |
| Vespertilionidae | Chalinolobus tuberculatus | New Zealand long-tailed bat | New Zealand (2020) | Bat guano | Partial (2) | OR248813, OR248821 | [28] |
| Vespertilionidae | Myotis daubentonii | Daubenton’s bat | Germany (2009) | Feces | Partial (2) | JQ001746, JQ001747 | [21] |
| Vespertilionidae | Myotis bechsteinii | Bechstein’s bat | Germany (2008) | Feces | Partial (1) | JQ001748 | [21] |
| Vespertilionidae | Myotis davidii | David’s myotis | China (2011) | Liver | Complete (1) | KX513953 | [29] |
| Vespertilionidae | Myotis ricketti | Rickett’s big-footed myotis | China (2017) | Pooled animal | Complete (1) | OQ715534 | [26] |
| Vespertilionidae | Eptesicus serotinus | common serotine bat | Germany (2009) | Liver | Complete (1) | JQ001749 | [21] |
| Vespertilionidae | Eptesicus japonensis | Japanese short-tailed bat | Japan (2015) | Feces | Partial (2) | LC340968, LC340970 | [30] |
| Vespertilionidae | Pipistrellus nathusii | Nathusius’s pipistrelle | Switzerland (2019) | Feces | Nearly complete (1) d | MT815970 | [31] |
| Vespertilionidae | Pipistrellus pygmaeus | soprano pipistrelle | Sweden (2020) | Feces | Partial (1) | ON513427 | N.A. |
| Vespertilionidae | Pipistrellus abramus | Japanese house bat | China (2016–2020) | Anal swab | Partial (18) | OR951187–OR951204 | [24] |
| Vespertilionidae | Tylonycteris pachypus | lesser bamboo bat | China (2016–2017) | Anal swab | Partial (8) | OR951174–OR951181 | [24] |
| Vespertilionidae | Tylonycteris robustula | greater bamboo bat | China (2017) | Anal swab | Partial (2) | OR951185–OR951186 | [24] |
| Vespertilionidae | Plecotus sacrimontis | Japanese long-eared bat | Japan (2015) | Feces | Partial (1) | LC340969 | [30] |
| Vespertilionidae | Scotophilus kuhlii | lesser Asiatic yellow house bat | China (2018) | Anal swab | Partial (2) | OR951182–OR951183 | [24] |
| Total | 22 species | 64 sequences |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Cronin, P.; Mah, M.G.; Yang, X.-L.; Su, Y.C.F. Genetic Diversity and Molecular Evolution of Hepatitis E Virus Within the Genus Chirohepevirus in Bats. Viruses 2025, 17, 339. https://doi.org/10.3390/v17030339
Wang B, Cronin P, Mah MG, Yang X-L, Su YCF. Genetic Diversity and Molecular Evolution of Hepatitis E Virus Within the Genus Chirohepevirus in Bats. Viruses. 2025; 17(3):339. https://doi.org/10.3390/v17030339
Chicago/Turabian StyleWang, Bo, Peter Cronin, Marcus G. Mah, Xing-Lou Yang, and Yvonne C. F. Su. 2025. "Genetic Diversity and Molecular Evolution of Hepatitis E Virus Within the Genus Chirohepevirus in Bats" Viruses 17, no. 3: 339. https://doi.org/10.3390/v17030339
APA StyleWang, B., Cronin, P., Mah, M. G., Yang, X.-L., & Su, Y. C. F. (2025). Genetic Diversity and Molecular Evolution of Hepatitis E Virus Within the Genus Chirohepevirus in Bats. Viruses, 17(3), 339. https://doi.org/10.3390/v17030339