
Pseudomonas Phage Lydia and the Evolution of the Mesyanzhinovviridae Family

 , , , , , ,
, , , , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage Isolation and Purification
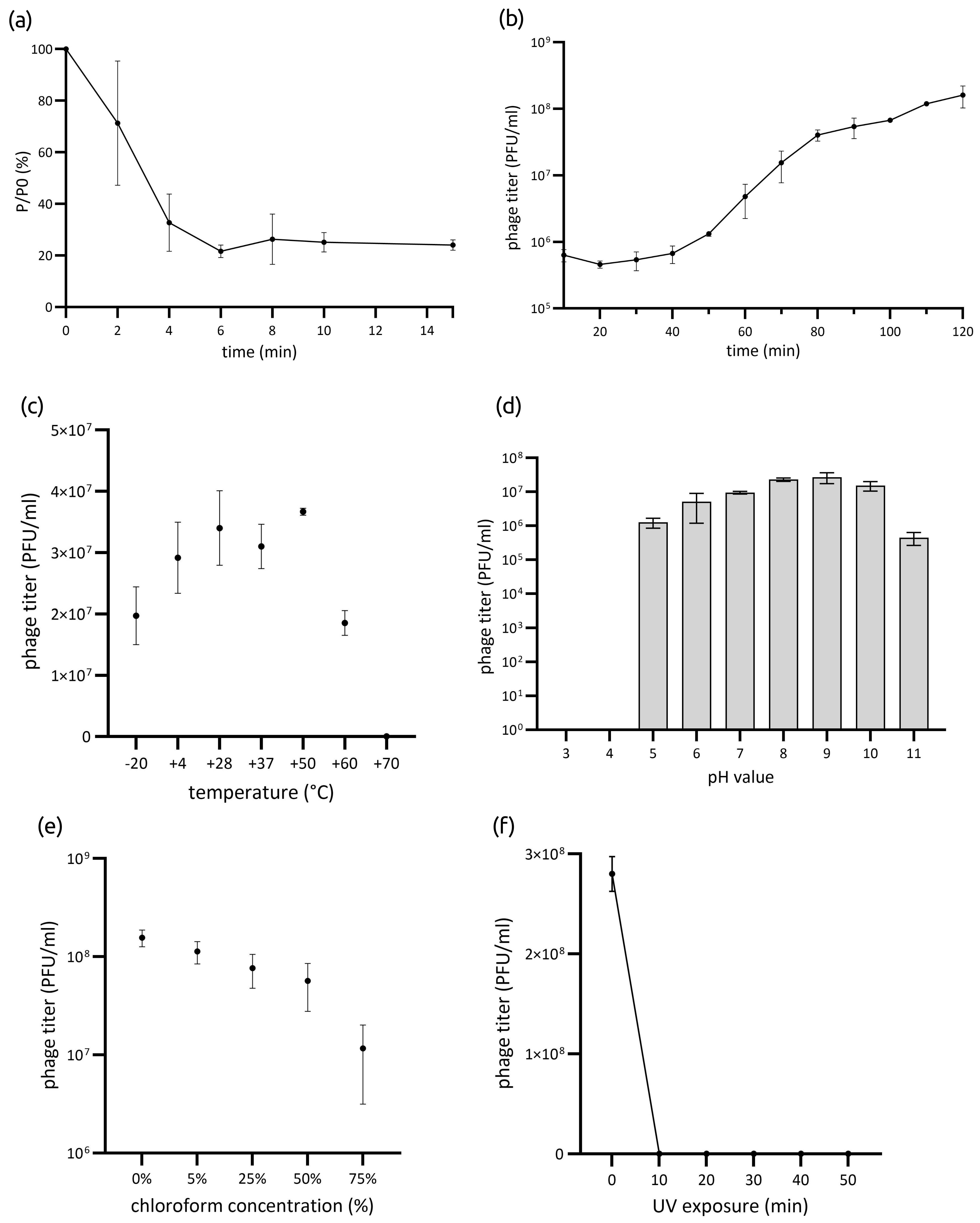
2.2. Phage Stability Under Different Conditions
2.3. Determination of Phage Host Range
2.4. Phage Adsorption and One-Step Growth Experiments
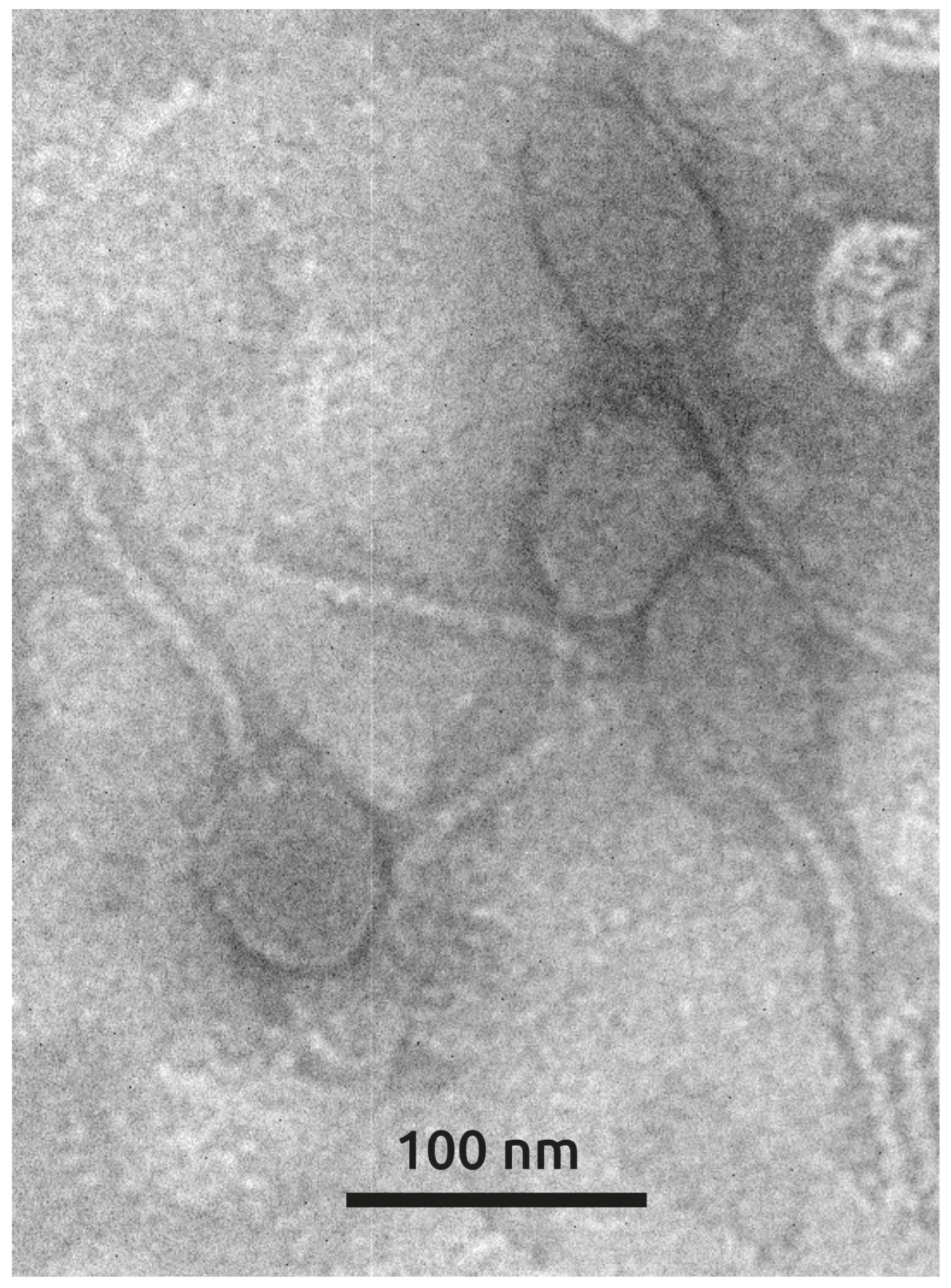
2.5. Transmission Electron Microscopy
2.6. Genome Sequencing and Annotation
2.7. Bioinformatic Analyses
3. Results
3.1. General Biological Features of Phage Lydia
3.1.1. Latency Period and Burst Size Determination
3.1.2. Phage Host Range
3.1.3. Phage Stability
3.1.4. Phage Morphology
3.2. Genome and Proteome Characterization of Phage Lydia
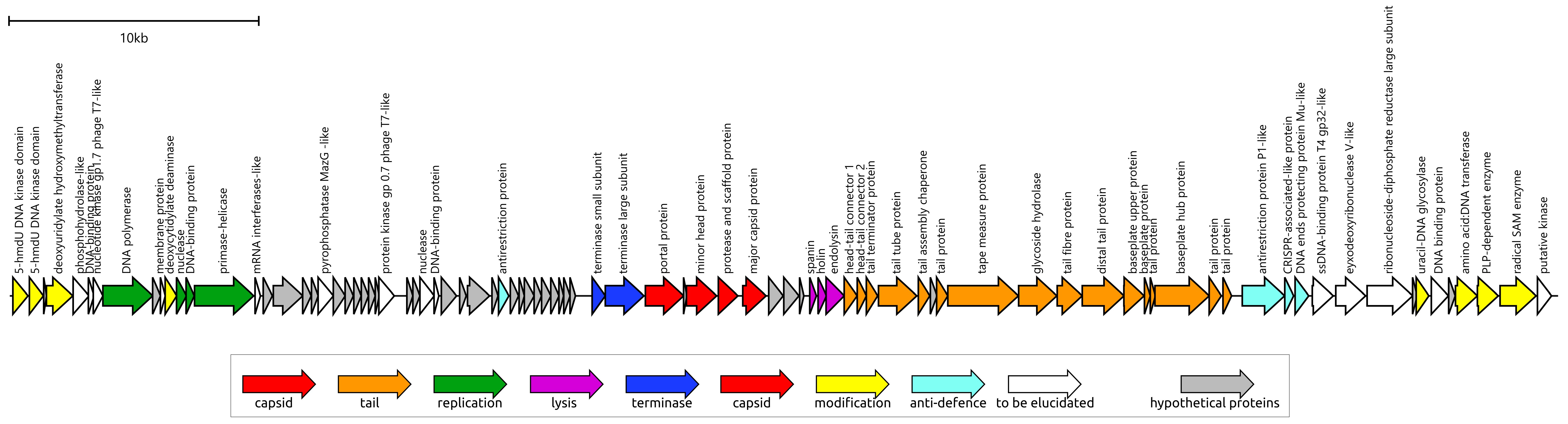
3.2.1. General Features
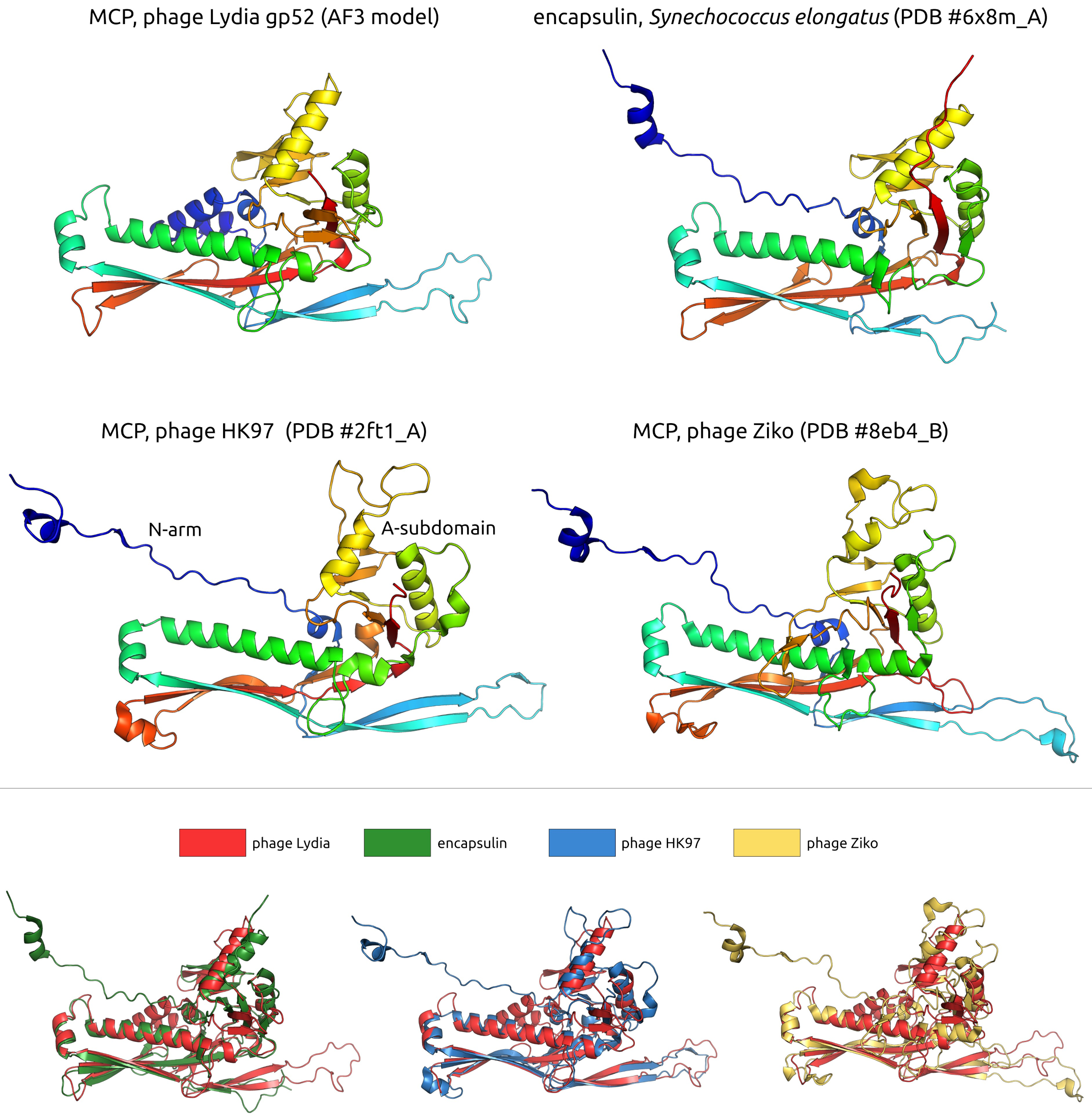
3.2.2. Major Capsid Protein and Terminase Large Subunit
3.2.3. DNA Polymerase
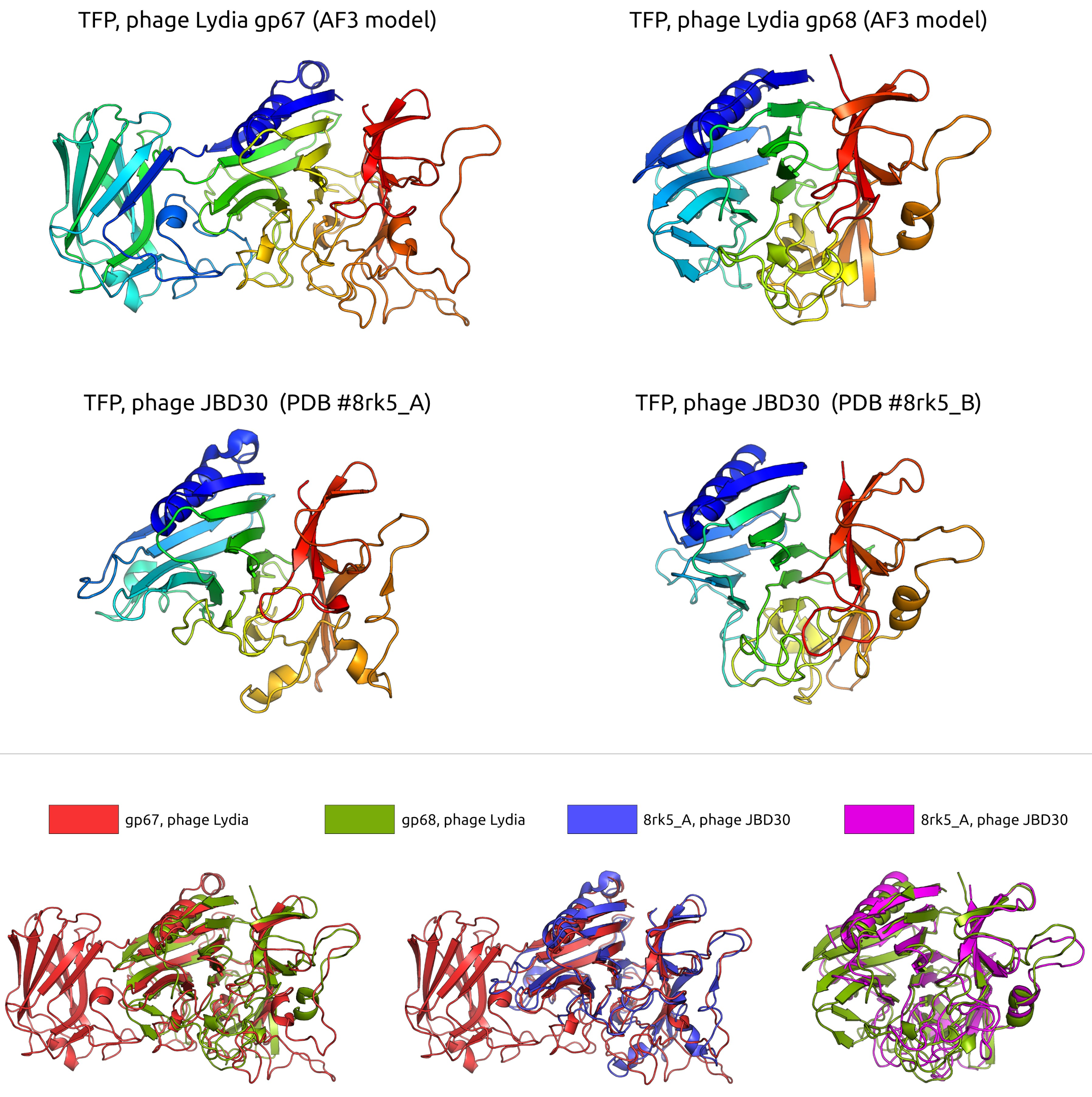
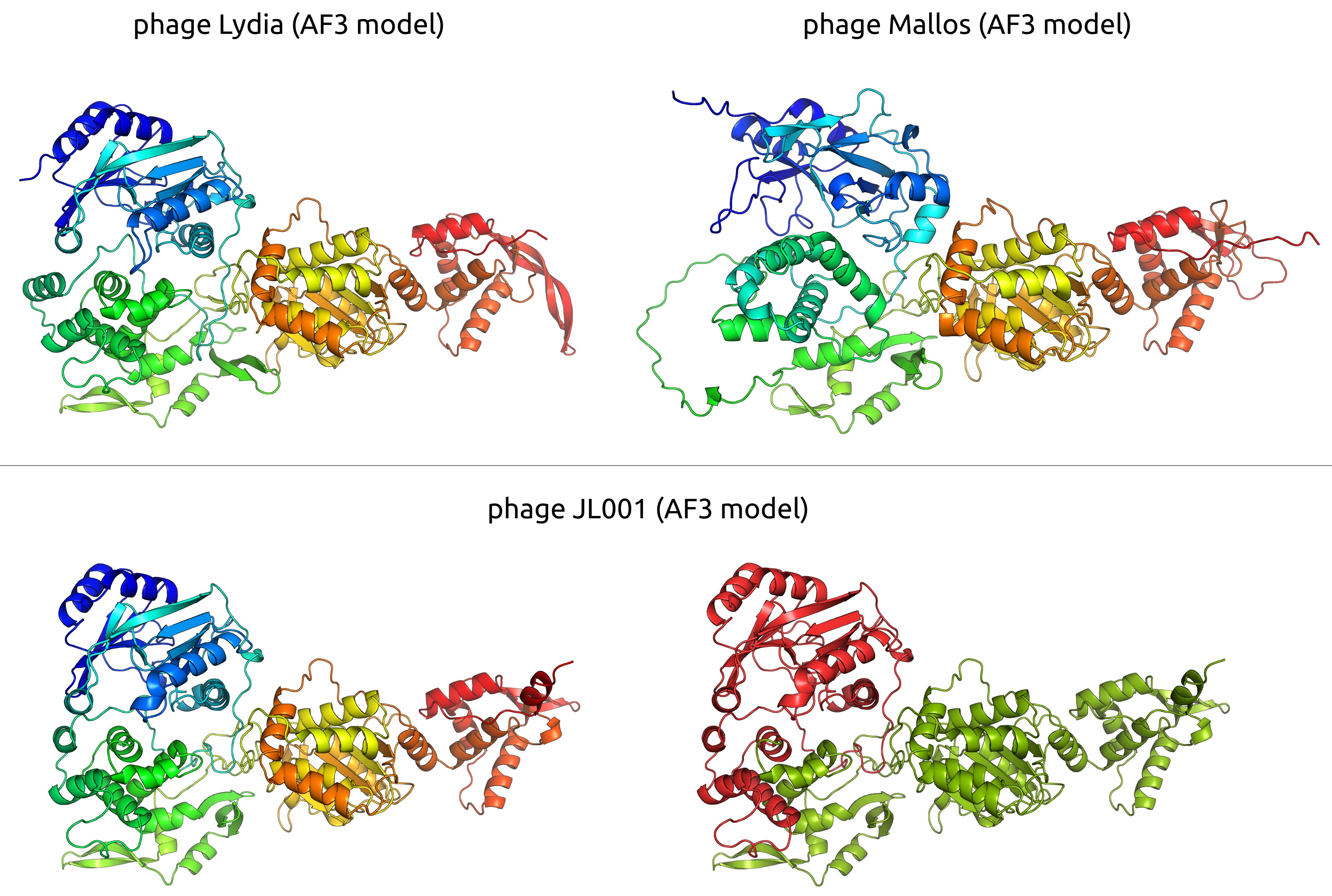
3.2.4. Tail and Adsorption Apparatus
3.2.5. Lysis Module and Endolysin
3.3. Taxonomic Analysis of Phage Lydia and Phylogenomics of Mesyanzhinovviridae Family
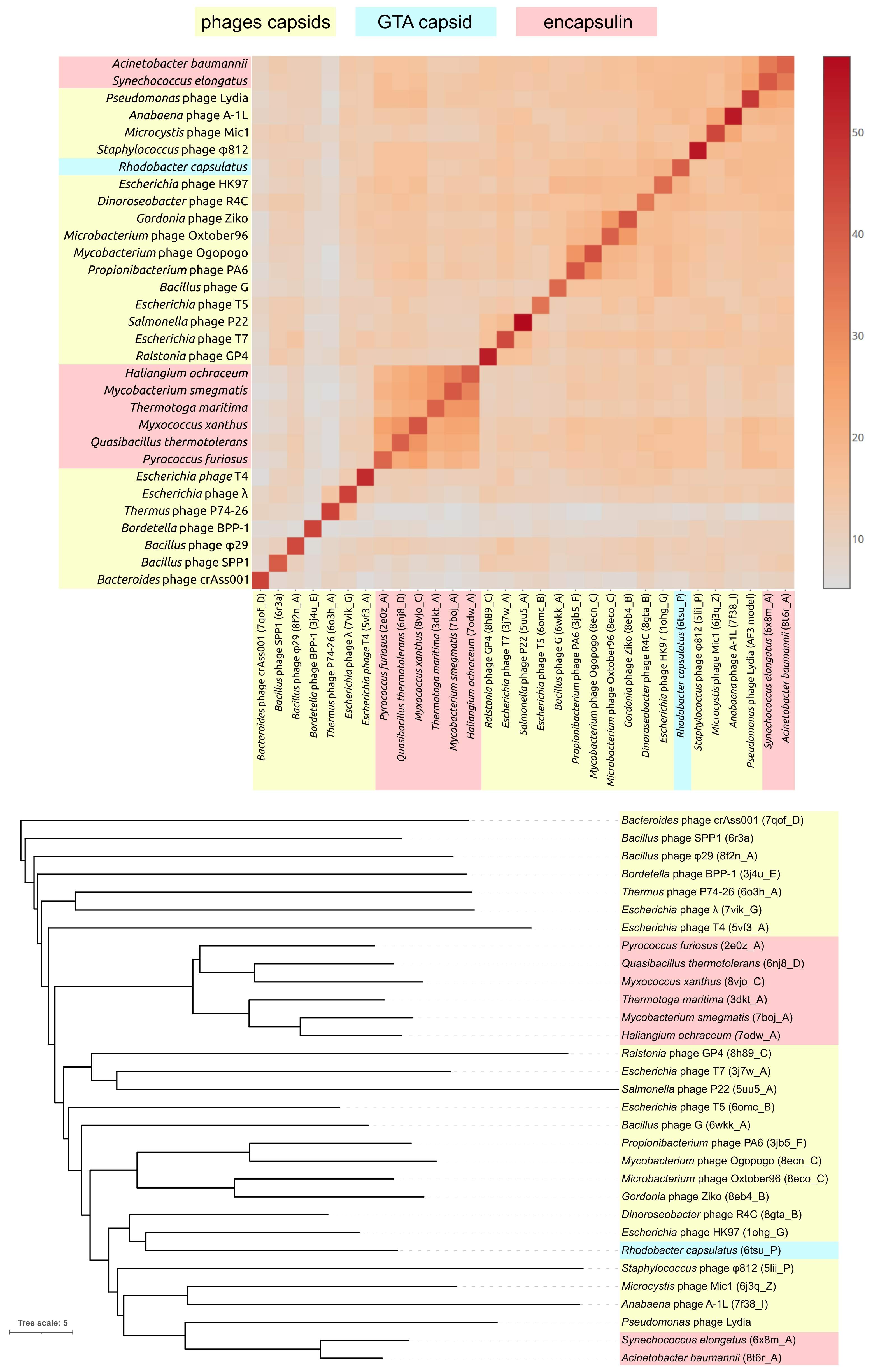
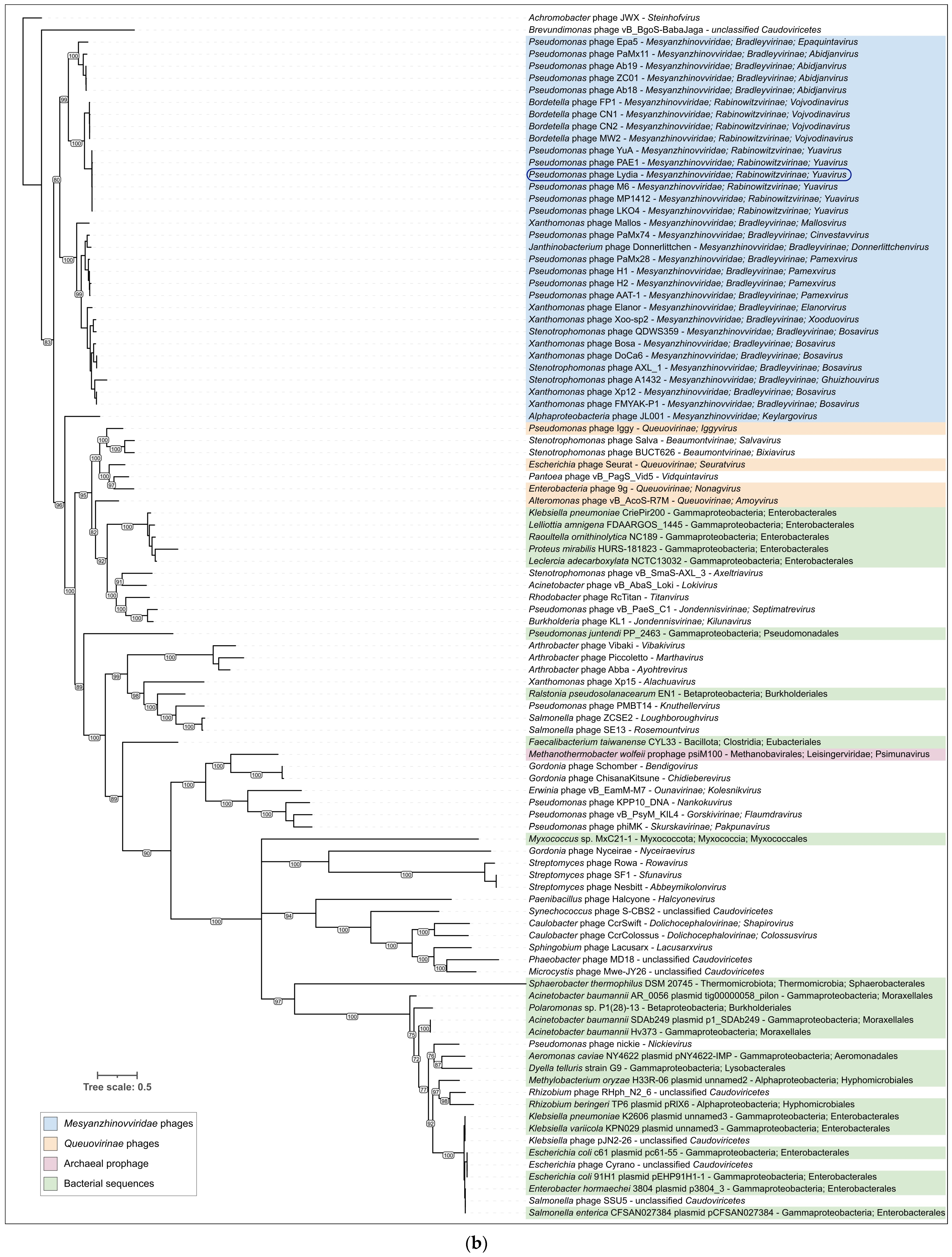
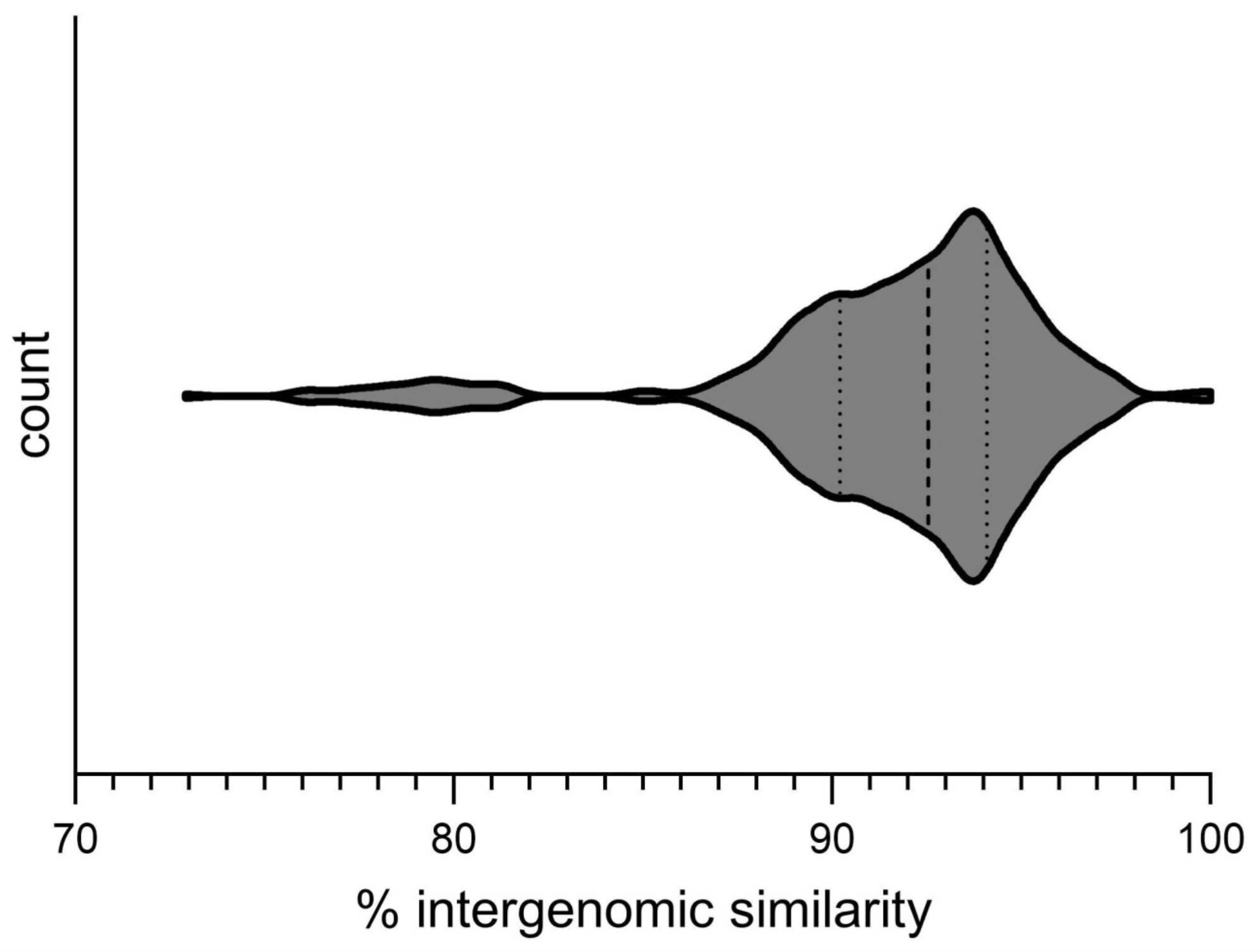
3.3.1. Intergenomic Similarity and Proteome Phylogeny
3.3.2. Phylogeny and Pangenome
3.4. Search for Proteins Associated with the Temperate Lifestyle
4. Discussion
4.1. Phylogenomic Relationships Within the Mesyanzhinovviridae Family
4.2. Classification Issues of Phage Lydia and Related Phages
4.3. Phage Lydia: Host Range, Receptors, and Therapeutic Potential
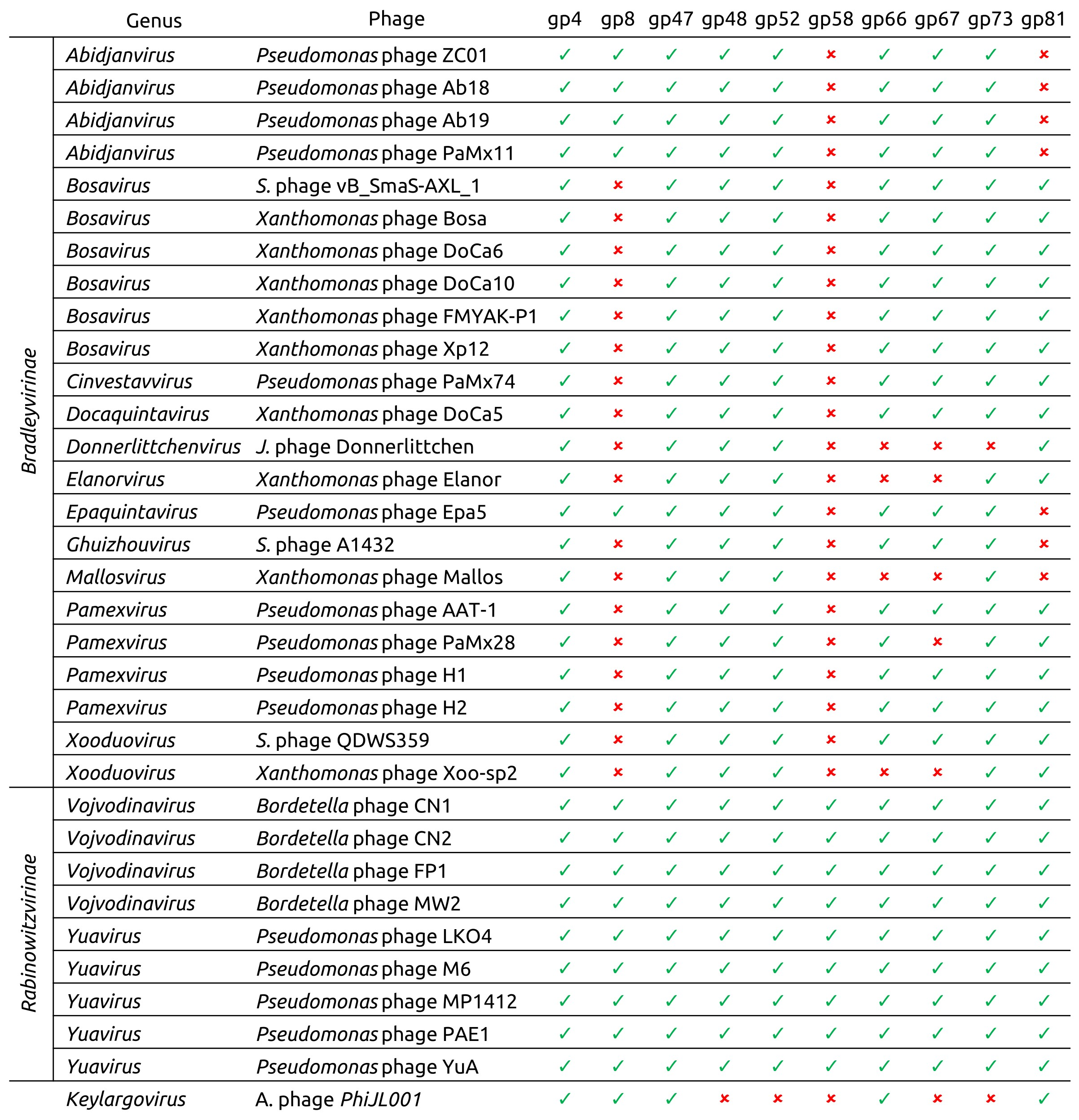
4.4. Evolutionary Dynamics of Mesyanzhinovviridae Phages: Horizontal Gene Transfer and Genomic Architecture
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Naureen, Z.; Dautaj, A.; Anpilogov, K.; Camilleri, G.; Dhuli, K.; Tanzi, B.; Maltese, P.E.; Cristofoli, F.; De Antoni, L.; Beccari, T.; et al. Bacteriophages Presence in Nature and Their Role in the Natural Selection of Bacterial Populations. Acta Bio Medica Atenei Parm. 2020, 91, e2020024. [Google Scholar] [CrossRef]
- Elois, M.A.; da Silva, R.; Pilati, G.V.T.; Rodríguez-Lázaro, D.; Fongaro, G. Bacteriophages as Biotechnological Tools. Viruses 2023, 15, 349. [Google Scholar] [CrossRef] [PubMed]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Phage Evolution and Ecology. Adv. Appl. Microbiol. 2009, 67, 1–45. [Google Scholar] [CrossRef]
- Glazko, G.; Makarenkov, V.; Liu, J.; Mushegian, A. Evolutionary History of Bacteriophages with Double-Stranded DNA Genomes. Biol. Direct 2007, 2, 36. [Google Scholar] [CrossRef]
- Tolstoy, I.; Kropinski, A.M.; Brister, J.R. Bacteriophage Taxonomy: An Evolving Discipline. In Bacteriophage Therapy; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 57–71. [Google Scholar] [CrossRef]
- Kerr, K.G.; Snelling, A.M. Pseudomonas aeruginosa: A Formidable and Ever-Present Adversary. J. Hosp. Infect. 2009, 73, 338–344. [Google Scholar] [CrossRef]
- Qin, S.; Xiao, W.; Zhou, C.; Pu, Q.; Deng, X.; Lan, L.; Liang, H.; Song, X.; Wu, M. Pseudomonas aeruginosa: Pathogenesis, Virulence Factors, Antibiotic Resistance, Interaction with Host, Technology Advances and Emerging Therapeutics. Signal Transduct. Target. Ther. 2022, 7, 1–27. [Google Scholar] [CrossRef]
- Wright, A.; Hawkins, C.H.; Anggård, E.E.; Harper, D.R. A Controlled Clinical Trial of a Therapeutic Bacteriophage Preparation in Chronic Otitis Due to Antibiotic-Resistant Pseudomonas aeruginosa; a Preliminary Report of Efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef]
- Mittal, R.; Aggarwal, S.; Sharma, S.; Chhibber, S.; Harjai, K. Urinary Tract Infections Caused by Pseudomonas aeruginosa: A Minireview. J. Infect. Public Health 2009, 2, 101–111. [Google Scholar] [CrossRef]
- Wei, X.; Li, L.; Li, M.; Liang, H.; He, Y.; Li, S. Risk Factors and Outcomes of Patients with Carbapenem-Resistant Pseudomonas aeruginosa Bloodstream Infection. Infect. Drug Resist. 2023, 16, 337–346. [Google Scholar] [CrossRef]
- Ghasemian, S.; Karami-Zarandi, M.; Heidari, H.; Khoshnood, S.; Kouhsari, E.; Ghafourian, S.; Maleki, A.; Kazemian, H. Molecular Characterizations of Antibiotic Resistance, Biofilm Formation, and Virulence Determinants of Pseudomonas aeruginosa Isolated from Burn Wound Infection. J. Clin. Lab. Anal. 2023, 37, e24850. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.J.; Kuzel, T.M.; Shafikhani, S.H. Pseudomonas aeruginosa: Infections, Animal Modeling, and Therapeutics. Cells 2023, 12, 199. [Google Scholar] [CrossRef] [PubMed]
- Pachori, P.; Gothalwal, R.; Gandhi, P. Emergence of Antibiotic Resistance Pseudomonas aeruginosa in Intensive Care Unit; a Critical Review. Genes Dis. 2019, 6, 109–119. [Google Scholar] [CrossRef]
- Alqahtani, A. Bacteriophage Treatment as an Alternative Therapy for Multidrug-Resistant Bacteria. Saudi Med. J. 2023, 44, 1222–1231. [Google Scholar] [CrossRef]
- Alipour-Khezri, E.; Skurnik, M.; Zarrini, G. Pseudomonas aeruginosa Bacteriophages and Their Clinical Applications. Viruses 2024, 16, 1051. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.E.; Chen, F.; Hill, R.T. Genomic Analysis of Bacteriophage ΦJL001: Insights into Its Interaction with a Sponge-Associated Alpha-Proteobacterium. Appl. Environ. Microbiol. 2005, 71, 1598–1609. [Google Scholar] [CrossRef]
- Peters, D.L.; McCutcheon, J.G.; Stothard, P.; Dennis, J.J. Novel Stenotrophomonas maltophilia Temperate Phage DLP4 Is Capable of Lysogenic Conversion. BMC Genom. 2019, 20, 300. [Google Scholar] [CrossRef]
- Erdrich, S.H.; Sharma, V.; Schurr, U.; Arsova, B.; Frunzke, J. Isolation of Novel Xanthomonas Phages Infecting the Plant Pathogens X. translucens and X. campestris. Viruses 2022, 14, 1449. [Google Scholar] [CrossRef]
- Domingo-Calap, M.L.; Bernabéu-Gimeno, M.; Aure, M.C.; Marco-Noales, E.; Domingo-Calap, P. Comparative Analysis of Novel Lytic Phages for Biological Control of Phytopathogenic Xanthomonas spp. Microbiol. Spectr. 2022, 10, e02960-22. [Google Scholar] [CrossRef]
- Dong, Z.; Xing, S.; Liu, J.; Tang, X.; Ruan, L.; Sun, M.; Tong, Y.; Peng, D. Isolation and Characterization of a Novel Phage Xoo-Sp2 That Infects Xanthomonas oryzae Pv. oryzae. J. Gen. Virol. 2018, 99, 1453–1462. [Google Scholar] [CrossRef]
- Petrovic Fabijan, A.; Aleksic Sabo, V.; Gavric, D.; Doffkay, Z.; Rakhely, G.; Knezevic, P. Are Bordetella Bronchiseptica siphoviruses (Genus Vojvodinavirus) Appropriate for Phage Therapy—Bacterial Allies or Foes? Viruses 2021, 13, 1732. [Google Scholar] [CrossRef] [PubMed]
- Brooke, J.S. Stenotrophomonas maltophilia: An Emerging Global Opportunistic Pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [PubMed]
- Woolfrey, B.F.; Moody, J.A. Human Infections Associated with Bordetella bronchiseptica. Clin. Microbiol. Rev. 1991, 4, 243. [Google Scholar] [CrossRef]
- Zhao, Y.; Laborda, P.; Han, S.-W.; Liu, F. Editorial: Pathogenic Mechanism and Biocontrol of Xanthomonas on Plants. Front. Cell. Infect. Microbiol. 2023, 13, 1270750. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Ehrlich, K.; Mayo, J.A. Unusual Properties of the DNA from Xanthomonas Phage XP-12 in Which 5-Methylcytosine Completely Replaces Cytosine. Biochim. Biophys. Acta 1975, 395, 109–119. [Google Scholar] [CrossRef]
- Wen, Y.; Guo, W.; Meng, C.; Yang, J.; Xu, S.; Chen, H.; Gan, J.; Wu, B. Structural Insights into the Biosynthetic Mechanism of Nα-GlyT and 5-NmdU Hypermodifications of DNA. Nucleic Acids Res. 2024, 52, 11083–11097. [Google Scholar] [CrossRef]
- Ceyssens, P.-J.; Mesyanzhinov, V.; Sykilinda, N.; Briers, Y.; Roucourt, B.; Lavigne, R.; Robben, J.; Domashin, A.; Miroshnikov, K.; Volckaert, G.; et al. The Genome and Structural Proteome of YuA, a New Pseudomonas aeruginosa Phage Resembling M6. J. Bacteriol. 2008, 190, 1429–1435. [Google Scholar] [CrossRef]
- McCutcheon, J.G.; Dennis, J.J. The Potential of Phage Therapy against the Emerging Opportunistic Pathogen Stenotrophomonas Maltophilia. Viruses 2021, 13, 1057. [Google Scholar] [CrossRef]
- Li, S.; Xu, M.; Yang, D.; Yang, M.; Wu, H.; Li, X.; Yang, C.; Fang, Z.; Wu, Q.; Tan, L.; et al. Characterization and Genomic Analysis of a Lytic Stenotrophomonas Maltophilia Short-Tailed Phage A1432 Revealed a New Genus of the Family Mesyanzhinovviridae. Front. Microbiol. 2024, 15, 1400700. [Google Scholar] [CrossRef]
- Wang, R.Y.; Ehrlich, M. 5-Methyl-dCTP Deaminase Induced by Bacteriophage XP-12. J. Virol. 1982, 42, 42–48. [Google Scholar] [CrossRef]
- Farber, M.B.; Ehrlich, M. Bacteriophage XP-12-Induced Exonuclease Which Preferentially Hydrolyzes Nicked DNA. J. Virol. 1980, 33, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Dyson, Z.A.; Seviour, R.J.; Tucci, J.; Petrovski, S. Genome Sequences of Pseudomonas oryzihabitans Phage POR1 and Pseudomonas Aeruginosa Phage PAE1. Genome Announc. 2016, 4, e01515-15. [Google Scholar] [CrossRef]
- Wang, X.; Tang, J.; Dang, W.; Xie, Z.; Zhang, F.; Hao, X.; Sun, S.; Liu, X.; Luo, Y.; Li, M.; et al. Isolation and Characterization of Three Pseudomonas aeruginosa Viruses with Therapeutic Potential. Microbiol. Spectr. 2023, 11, e0463622. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.G.; Lin, A.; Dennis, J.J. Characterization of Stenotrophomonas Maltophilia Phage AXL1 as a Member of the Genus Pamexvirus Encoding Resistance to Trimethoprim-Sulfamethoxazole. Sci. Rep. 2022, 12, 10299. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.V.; Gorshkova, A.S.; Sykilinda, N.N.; Drucker, V.V.; Miroshnikov, K.A. Pseudomonas Bacteriophage AN14—A Baikal-Borne Representative of Yuavirus. Limnol. Freshw. Biol. 2020, 1055–1066. [Google Scholar] [CrossRef]
- Andrade-Domínguez, A.; Kolter, R. Complete Genome Sequence of Pseudomonas Aeruginosa Phage AAT-1. Genome Announc. 2016, 4, e00165-16. [Google Scholar] [CrossRef]
- Martins, L.F.; Dos Santos Junior, A.P.; Nicastro, G.G.; Scheunemann, G.; Angeli, C.B.; Rossi, F.P.N.; Quaggio, R.B.; Palmisano, G.; Sgro, G.G.; Ishida, K.; et al. Phages ZC01 and ZC03 Require Type-IV Pilus for Pseudomonas Aeruginosa Infection and Have a Potential for Therapeutic Applications. Microbiol. Spectr. 2024, 12, e0152724. [Google Scholar] [CrossRef]
- Amgarten, D.; Martins, L.F.; Lombardi, K.C.; Antunes, L.P.; de Souza, A.P.S.; Nicastro, G.G.; Kitajima, E.W.; Quaggio, R.B.; Upton, C.; Setubal, J.C.; et al. Three Novel Pseudomonas Phages Isolated from Composting Provide Insights into the Evolution and Diversity of Tailed Phages. BMC Genom. 2017, 18, 346. [Google Scholar] [CrossRef]
- Farlow, J.; Freyberger, H.R.; He, Y.; Ward, A.M.; Rutvisuttinunt, W.; Li, T.; Campbell, R.; Jacobs, A.C.; Nikolich, M.P.; Filippov, A.A. Complete Genome Sequences of 10 Phages Lytic against Multidrug-Resistant Pseudomonas aeruginosa. Microbiol. Resour. Announc. 2020, 9, e00503-20. [Google Scholar] [CrossRef]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- Wdowiak, M.; Paczesny, J.; Raza, S. Enhancing the Stability of Bacteriophages Using Physical, Chemical, and Nano-Based Approaches: A Review. Pharmaceutics 2022, 14, 1936. [Google Scholar] [CrossRef]
- Guo, M.; Gao, Y.; Xue, Y.; Liu, Y.; Zeng, X.; Cheng, Y.; Ma, J.; Wang, H.; Sun, J.; Wang, Z.; et al. Bacteriophage Cocktails Protect Dairy Cows Against Mastitis Caused by Drug Resistant Escherichia coli Infection. Front. Cell. Infect. Microbiol. 2021, 11, 690377. [Google Scholar] [CrossRef] [PubMed]
- Bocharova, Y.; Chebotar, I.; Savinova, T.; Lyamin, A.; Kondratenko, O.; Polikarpova, S.; Fedorova, N.; Semykin, S.; Korostin, D.; Chaplin, A.; et al. Clonal Diversity, Antimicrobial Resistance, and Genome Features among Non-Fermenting Gram-Negative Bacteria Isolated from Patients with Cystic Fibrosis in Russia. Diagn. Microbiol. Infect. Dis. 2023, 108, 116102. [Google Scholar] [CrossRef]
- Yang, M.; Du, C.; Gong, P.; Xia, F.; Sun, C.; Feng, X.; Lei, L.; Song, J.; Zhang, L.; Wang, B.; et al. Therapeutic Effect of the YH6 Phage in a Murine Hemorrhagic Pneumonia Model. Res. Microbiol. 2015, 166, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Middelboe, M.; Chan, A.; Bertelsen, S.K. Isolation and Life Cycle Characterization of Lytic Viruses Infecting Heterotrophic Bacteria and Cyanobacteria. In Manual of Aquatic Viral Ecology; Wilhelm, S.W., Weinbauer, M.G., Suttle, C.A., Eds.; American Society of Limnology and Oceanography, Inc.: Waco, TX, USA, 2010; pp. 118–133. [Google Scholar]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinform. Oxf. Eng. 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The Viral Proteomic Tree Server. Bioinforma. Oxf. Engl. 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Eren, A.M.; Kiefl, E.; Shaiber, A.; Veseli, I.; Miller, S.E.; Schechter, M.S.; Fink, I.; Pan, J.N.; Yousef, M.; Fogarty, E.C.; et al. Community-Led, Integrated, Reproducible Multi-Omics with Anvi’o. Nat. Microbiol. 2021, 6, 3–6. [Google Scholar] [CrossRef]
- Delmont, T.O.; Eren, A.M. Linking Pangenomes and Metagenomes: The Prochlorococcus Metapangenome. PeerJ 2018, 6, e4320. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-Suite3 for Fast Remote Homology Detection and Deep Protein Annotation. BMC Bioinformatics 2019, 20, 473. [Google Scholar] [CrossRef]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA Packaging Strategy by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. In Bacteriophages: Methods and Protocols, Volume 2: Molecular and Applied Aspects; Humana Press: Totowa, NJ, USA, 2009; Volume 502, pp. 91–111. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Dai, N.; Walsh, S.E.; Müller, S.; Fraser, M.E.; Kauffman, K.M.; Guan, C.; Corrêa, I.R.; Weigele, P.R. Identification and Biosynthesis of Thymidine Hypermodifications in the Genomic DNA of Widespread Bacterial Viruses. Proc. Natl. Acad. Sci. USA 2018, 115, E3116–E3125. [Google Scholar] [CrossRef]
- Flodman, K.; Tsai, R.; Xu, M.Y.; Corrêa, I.R.; Copelas, A.; Lee, Y.-J.; Xu, M.-Q.; Weigele, P.; Xu, S. Type II Restriction of Bacteriophage DNA With 5hmdU-Derived Base Modifications. Front. Microbiol. 2019, 10, 584. [Google Scholar] [CrossRef] [PubMed]
- Serfiotis-Mitsa, D.; Herbert, A.P.; Roberts, G.A.; Soares, D.C.; White, J.H.; Blakely, G.W.; Uhrín, D.; Dryden, D.T.F. The Structure of the KlcA and ArdB Proteins Reveals a Novel Fold and Antirestriction Activity against Type I DNA Restriction Systems in Vivo but Not in Vitro. Nucleic Acids Res. 2010, 38, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Piya, D.; Vara, L.; Russell, W.K.; Young, R.; Gill, J.J. The Multicomponent Antirestriction System of Phage P1 Is Linked to Capsid Morphogenesis. Mol. Microbiol. 2017, 105, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; LaFrance, B.; Phillips, N.R.; Radford, D.R.; Oltrogge, L.M.; Valentin-Alvarado, L.E.; Bischoff, A.J.; Nogales, E.; Savage, D.F. Discovery and Characterization of a Novel Family of Prokaryotic Nanocompartments Involved in Sulfur Metabolism. eLife 2021, 10, e59288. [Google Scholar] [CrossRef]
- Benisch, R.; Andreas, M.P.; Giessen, T.W. A Widespread Bacterial Protein Compartment Sequesters and Stores Elemental Sulfur. Sci. Adv. 2024, 10, eadk9345. [Google Scholar] [CrossRef]
- Gan, L.; Speir, J.A.; Conway, J.F.; Lander, G.; Cheng, N.; Firek, B.A.; Hendrix, R.W.; Duda, R.L.; Liljas, L.; Johnson, J.E. Capsid Conformational Sampling in HK97 Maturation Visualized by X-Ray Crystallography and Cryo-EM. Structure 2006, 14, 1655–1665. [Google Scholar] [CrossRef]
- Cui, N.; Yang, F.; Zhang, J.-T.; Sun, H.; Chen, Y.; Yu, R.-C.; Chen, Z.-P.; Jiang, Y.-L.; Han, S.-J.; Xu, X.; et al. Capsid Structure of Anabaena Cyanophage A-1(L). J. Virol. 2021, 95, e01356-21. [Google Scholar] [CrossRef]
- Podgorski, J.M.; Freeman, K.; Gosselin, S.; Huet, A.; Conway, J.F.; Bird, M.; Grecco, J.; Patel, S.; Jacobs-Sera, D.; Hatfull, G.; et al. A Structural Dendrogram of the Actinobacteriophage Major Capsid Proteins Provides Important Structural Insights into the Evolution of Capsid Stability. Structure 2023, 31, 282–294.e5. [Google Scholar] [CrossRef]
- Bárdy, P.; Füzik, T.; Hrebík, D.; Pantůček, R.; Thomas Beatty, J.; Plevka, P. Structure and Mechanism of DNA Delivery of a Gene Transfer Agent. Nat. Commun. 2020, 11, 3034. [Google Scholar] [CrossRef]
- Xu, R.-G.; Jenkins, H.T.; Antson, A.A.; Greive, S.J. Structure of the Large Terminase from a Hyperthermophilic Virus Reveals a Unique Mechanism for Oligomerization and ATP Hydrolysis. Nucleic Acids Res. 2017, 45, 13029–13042. [Google Scholar] [CrossRef]
- Stockley, M.L.; Ferdinand, A.; Benedetti, G.; Blencowe, P.; Boyd, S.M.; Calder, M.; Charles, M.D.; Edwardes, L.V.; Ekwuru, T.; Finch, H.; et al. Discovery, Characterization, and Structure-Based Optimization of Small-Molecule In Vitro and In Vivo Probes for Human DNA Polymerase Theta. J. Med. Chem. 2022, 65, 13879–13891. [Google Scholar] [CrossRef] [PubMed]
- Czernecki, D.; Hu, H.; Romoli, F.; Delarue, M. Structural Dynamics and Determinants of 2-Aminoadenine Specificity in DNA Polymerase DpoZ of Vibriophage ϕVC8. Nucleic Acids Res. 2021, 49, 11974–11985. [Google Scholar] [CrossRef] [PubMed]
- Desfosses, A.; Venugopal, H.; Joshi, T.; Felix, J.; Jessop, M.; Jeong, H.; Hyun, J.; Heymann, J.B.; Hurst, M.R.H.; Gutsche, I.; et al. Atomic Structures of an Entire Contractile Injection System in Both the Extended and Contracted States. Nat. Microbiol. 2019, 4, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Agnello, E.; Pajak, J.; Liu, X.; Kelch, B.A. Conformational Dynamics Control Assembly of an Extremely Long Bacteriophage Tail Tube. J. Biol. Chem. 2023, 299, 103021. [Google Scholar] [CrossRef]
- Schwemmlein, N.; Pippel, J.; Gazdag, E.-M.; Blankenfeldt, W. Crystal Structures of R-Type Bacteriocin Sheath and Tube Proteins CD1363 and CD1364 From Clostridium Difficile in the Pre-Assembled State. Front. Microbiol. 2018, 9, 1750. [Google Scholar] [CrossRef]
- Valentová, L.; Füzik, T.; Nováček, J.; Hlavenková, Z.; Pospíšil, J.; Plevka, P. Structure and Replication of Pseudomonas aeruginosa Phage JBD30. EMBO J. 2024, 43, 4384–4405. [Google Scholar] [CrossRef]
- Huang, Y.; Sun, H.; Wei, S.; Cai, L.; Liu, L.; Jiang, Y.; Xin, J.; Chen, Z.; Que, Y.; Kong, Z.; et al. Structure and Proposed DNA Delivery Mechanism of a Marine Roseophage. Nat. Commun. 2023, 14, 3609. [Google Scholar] [CrossRef]
- Engilberge, S.; Riobé, F.; Wagner, T.; Di Pietro, S.; Breyton, C.; Franzetti, B.; Shima, S.; Girard, E.; Dumont, E.; Maury, O. Unveiling the Binding Modes of the Crystallophore, a Terbium-Based Nucleating and Phasing Molecular Agent for Protein Crystallography. Chem. Eur. J. 2018, 24, 9739–9746. [Google Scholar] [CrossRef]
- Gadwal, S.; Korotkov, K.V.; Delarosa, J.R.; Hol, W.G.J.; Sandkvist, M. Functional and Structural Characterization of Vibrio Cholerae Extracellular Serine Protease B., VesB. J. Biol. Chem. 2014, 289, 8288–8298. [Google Scholar] [CrossRef]
- Cheng, Y.; Han, J.; Song, M.; Zhang, S.; Cao, Q. Serine Peptidase Vpr Forms Enzymatically Active Fibrils Outside Bacillus Bacteria Revealed by Cryo-EM. Nat. Commun. 2023, 14, 7503. [Google Scholar] [CrossRef]
- Lee, M.; Batuecas, M.T.; Tomoshige, S.; Domínguez-Gil, T.; Mahasenan, K.V.; Dik, D.A.; Hesek, D.; Millán, C.; Usón, I.; Lastochkin, E.; et al. Exolytic and Endolytic Turnover of Peptidoglycan by Lytic Transglycosylase Slt of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2018, 115, 4393–4398. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Gil, T.; Lee, M.; Acebrón-Avalos, I.; Mahasenan, K.V.; Hesek, D.; Dik, D.A.; Byun, B.; Lastochkin, E.; Fisher, J.F.; Mobashery, S.; et al. Activation by Allostery in Cell-Wall Remodeling by a Modular Membrane-Bound Lytic Transglycosylase from Pseudomonas aeruginosa. Structure 2016, 24, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Adouard, V.; Dante, R.; Niveleau, A.; Delain, E.; Revet, B.; Ehrlich, M. The Accessibility of 5-Methylcytosine to Specific Antibodies in Double-Stranded DNA of Xanthomonas Phage XP12. Eur. J. Biochem. 1985, 152, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.T.; Chow, T.Y.; Lin, Y.T. A New Thymidylate Biosynthesis in Xanthomonas Oryzae Infected by Phage Xp 12. Virology 1982, 118, 293–300. [Google Scholar] [CrossRef]
- Flores, V.; Sepúlveda-Robles, O.; Cazares, A.; Kropinski, A.; Adriaenssens, E.; Kuhn, J.; Kameyama, L.; Guarneros, G. To Create One (1) New Genus, Pamx74virus, Including Two (2) New Species in the Family Siphoviridae; ICTV Taxonomy Proposal; International Committee for Taxonomy of Viruses: Stevenage, UK, 2016; p. 57. [Google Scholar]
- Friedrich, I. Structure and Function of Bacterial Viruses and Viral Communities. Ph.D. Thesis, Göttingen State and University, Göttingen, Germany, 2023. [Google Scholar] [CrossRef]
- Bae, H.-W.; Chung, I.-Y.; Sim, N.; Cho, Y.-H. Complete Genome Sequence of Pseudomonas Aeruginosa Siphophage MP1412. J. Virol. 2012, 86, 9537. [Google Scholar] [CrossRef]
- Botstein, D. A Theory of Modular Evolution for Bacteriophages. Ann. N. Y. Acad. Sci. 1980, 354, 484–490. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage Evolution Differs by Host, Lifestyle and Genome. Nat. Microbiol. 2017, 2, 17112. [Google Scholar] [CrossRef]
- Brüssow, H.; Desiere, F. Comparative Phage Genomics and the Evolution of Siphoviridae: Insights from Dairy Phages. Mol. Microbiol. 2001, 39, 213–222. [Google Scholar] [CrossRef]
- Moura de Sousa, J.A.; Pfeifer, E.; Touchon, M.; Rocha, E.P.C. Causes and Consequences of Bacteriophage Diversification via Genetic Exchanges across Lifestyles and Bacterial Taxa. Mol. Biol. Evol. 2021, 38, 2497–2512. [Google Scholar] [CrossRef]
- De Paepe, M.; Hutinet, G.; Son, O.; Amarir-Bouhram, J.; Schbath, S.; Petit, M.-A. Temperate Phages Acquire DNA from Defective Prophages by Relaxed Homologous Recombination: The Role of Rad52-Like Recombinases. PLoS Genet. 2014, 10, e1004181. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in Viruses: Mechanisms, Methods of Study, and Evolutionary Consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.-M.; Rocha, E.P.C.; Touchon, M. The Adaptation of Temperate Bacteriophages to Their Host Genomes. Mol. Biol. Evol. 2013, 30, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Mastrantonio, P.; Spigaglia, P.; van Oirschot, H.; van der Heide, H.G.J.; Heuvelman, K.; Stefanelli, P.; Mooi, F.R. Antigenic Variants in Bordetella Pertussis Strains Isolated from Vaccinated and Unvaccinated Children. Microbiology 1999, 145 Pt 8, 2069–2075. [Google Scholar] [CrossRef] [PubMed]
- Darch, S.E.; McNally, A.; Harrison, F.; Corander, J.; Barr, H.L.; Paszkiewicz, K.; Holden, S.; Fogarty, A.; Crusz, S.A.; Diggle, S.P. Recombination Is a Key Driver of Genomic and Phenotypic Diversity in a Pseudomonas aeruginosa Population during Cystic Fibrosis Infection. Sci. Rep. 2015, 5, 7649. [Google Scholar] [CrossRef]
- Evseev, P.V.; Tarakanov, R.I.; Vo, H.T.N.; Suzina, N.E.; Vasilyeva, A.A.; Ignatov, A.N.; Miroshnikov, K.A.; Dzhalilov, F.S.-U. Characterisation of New Foxunavirus Phage Murka with the Potential of Xanthomonas campestris Pv. Campestris Control. Viruses 2024, 16, 198. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Evseev, P.; Lukianova, A.; Sykilinda, N.; Gorshkova, A.; Bondar, A.; Shneider, M.; Kabilov, M.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages. Int. J. Mol. Sci. 2021, 22, 10350. [Google Scholar] [CrossRef]
- Evseev, P.; Gutnik, D.; Shneider, M.; Miroshnikov, K. Use of an Integrated Approach Involving AlphaFold Predictions for the Evolutionary Taxonomy of Duplodnaviria Viruses. Biomolecules 2023, 13, 110. [Google Scholar] [CrossRef]
- Evseev, P.; Gutnik, D.; Evpak, A.; Kasimova, A.; Miroshnikov, K. Origin, Evolution and Diversity of Φ29-like Phages—Review and Bioinformatic Analysis. Int. J. Mol. Sci. 2024, 25, 10838. [Google Scholar] [CrossRef]
- Evseev, P.; Bocharova, J.; Shagin, D.; Chebotar, I. Analysis of Pseudomonas Aeruginosa Isolates from Patients with Cystic Fibrosis Revealed Novel Groups of Filamentous Bacteriophages. Viruses 2023, 15, 2215. [Google Scholar] [CrossRef]
- Tarakanov, R.I.; Evseev, P.V.; Vo, H.T.N.; Troshin, K.S.; Gutnik, D.I.; Ignatov, A.N.; Toshchakov, S.V.; Miroshnikov, K.A.; Jafarov, I.H.; Dzhalilov, F.S.-U. Xanthomonas Phage PBR31: Classifying the Unclassifiable. Viruses 2024, 16, 406. [Google Scholar] [CrossRef]
- Harvey, H.; Bondy-Denomy, J.; Marquis, H.; Sztanko, K.M.; Davidson, A.R.; Burrows, L.L. Pseudomonas Aeruginosa Defends against Phages through Type IV Pilus Glycosylation. Nat. Microbiol. 2018, 3, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Pas, C.; Latka, A.; Fieseler, L.; Briers, Y. Phage Tailspike Modularity and Horizontal Gene Transfer Reveals Specificity towards E. coli O-Antigen Serogroups. Virol. J. 2023, 20, 174. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Leiman, P.G.; Drulis-Kawa, Z.; Briers, Y. Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, B.; Jiao, N.; Liu, J.; Chen, F. New Evidence Supports the Prophage Origin of RcGTA. Appl. Environ. Microbiol. 2024, 90, e0043424. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Genus | Species | Phage Name | NCBI Accession | Short Phage Name |
|---|---|---|---|---|---|
| Bradleyvirinae | Abidjanvirus | Abidjanvirus ZC01 | Pseudomonas phage ZC01 [38,39] | KU356689 | |
| Bradleyvirinae | Abidjanvirus | Pseudomonas virus Ab18 | Pseudomonas phage vB_PaeS_PAO1_Ab18 | LN610577 | Pseudomonas phage Ab18 |
| Bradleyvirinae | Abidjanvirus | Pseudomonas virus Ab19 | Pseudomonas phage vB_PaeS_PAO1_Ab19 | LN610584 | Pseudomonas phage Ab19 |
| Bradleyvirinae | Abidjanvirus | Pseudomonas virus PaMx11 | Pseudomonas phage PaMx11 [27] | JQ067087 | |
| Bradleyvirinae | Bosavirus | Bosavirus Axl1 | Stenotrophomonas phage vB_SmaS-AXL_1 [35] | OL674541 | |
| Bradleyvirinae | Bosavirus | Bosavirus bosa | Xanthomonas phage Bosa | LR743532 | |
| Bradleyvirinae | Bosavirus | Bosavirus Doca6 | Xanthomonas phage vB_Xar_IVIA-DoCa6 [20] | ON932080 | Xanthomonas phage DoCa6 |
| Bradleyvirinae | Bosavirus | Bosavirus Doca10 | Xanthomonas phage vB_Xar_IVIA-DoCa10 [20] | ON932084 | Xanthomonas phage DoCa10 |
| Bradleyvirinae | Bosavirus | Bosavirus FMYAK | Xanthomonas phage FMYAK-P1 | OK275492 | |
| Bradleyvirinae | Bosavirus | Bosavirus Xp12 | Xanthomonas phage Xp12 [26,32,86,87] | MT664984 | |
| Bradleyvirinae | Cinvestavvirus | Cinvestavvirus PaMx74 | Pseudomonas phage PaMx74 [88] | JQ067093 | |
| Bradleyvirinae | Docaquintavirus | Docaquintavirus doca5 | Xanthomonas phage vB_Xar_IVIA-DoCa5 [20] | ON932079 | Xanthomonas phage DoCa5 |
| Bradleyvirinae | Donnerlittchenvirus | Donnerlittchenvirus donnerlittchenvirus | Janthinobacterium phagevB_JliM-Donnerlittchen [89] | ON529854 | Janthinobacterium phage Donnerlittchen |
| Bradleyvirinae | Elanorvirus | Elanorvirus elanor | Xanthomonas phage Elanor [19] | ON189045 | |
| Bradleyvirinae | Epaquintavirus | Epaquintavirus Epa5 | Pseudomonas phage Epa5 [40] | MT108725 | |
| Bradleyvirinae | Ghuizhouvirus | Ghuizhouvirus A1432 | Stenotrophomonas phage A1432 [30] | ON005621 | |
| Bradleyvirinae | Mallosvirus | Mallosvirus mallos | Xanthomonas phage Mallos [19] | ON189047 | |
| Bradleyvirinae | Pamexvirus | Pamexvirus AAT1 | Pseudomonas phage AAT-1 [37] | KU204984 | |
| Bradleyvirinae | Pamexvirus | Pamexvirus PaMx28 | Pseudomonas phage PaMx28 | JQ067089 | |
| Bradleyvirinae | Pamexvirus | Pamexvirus phiH1 | Pseudomonas phage phiH1 | OP380269 | Pseudomonas phage H1 |
| Bradleyvirinae | Pamexvirus | Pamexvirus phiH2 | Pseudomonas phage phiH2 | OP361299 | Pseudomonas phage H2 |
| Bradleyvirinae | Xooduovirus | Xooduovirus QDWS359 | Stenotrophomonas phage vB_Sm_QDWS359 | ON331942 | Stenotrophomonas phage QDWS359 |
| Bradleyvirinae | Xooduovirus | Xooduovirus Xoosp2 | Xanthomonas phage Xoo-sp2 [21] | KX241618 | |
| Rabinowitzvirinae | Vojvodinavirus | Bordetella virus CN1 | Bordetella phage CN1 [22] | KY000221 | |
| Rabinowitzvirinae | Vojvodinavirus | Bordetella virus CN2 | Bordetella phage CN2 [22] | KY000219 | |
| Rabinowitzvirinae | Vojvodinavirus | Bordetella virus FP1 | Bordetella phage FP1 [22] | KY000220 | |
| Rabinowitzvirinae | Vojvodinavirus | Bordetella virus MW2 | Bordetella phage MW2 [22] | KY000218 | |
| Rabinowitzvirinae | Yuavirus | Pseudomonas virus LKO4 | Pseudomonas phage LKO4 | KC758116 | |
| Rabinowitzvirinae | Yuavirus | Pseudomonas virus M6 | Pseudomonas phage M6 | DQ163916 | |
| Rabinowitzvirinae | Yuavirus | Pseudomonas virus MP1412 | Pseudomonas phage MP1412 [90] | JX131330 | |
| Rabinowitzvirinae | Yuavirus | Pseudomonas virus PAE1 | Pseudomonas phage PAE1 [33] | KT734862 | |
| Rabinowitzvirinae | Yuavirus | Pseudomonas virus Yua | Pseudomonas phage YuA [28] | AM749441 | |
| Keylargovirus | Keylargovirus JL001 | Alphaproteobacteria phage PhiJL001 [17] | AY576273 | Alphaproteobacteria phage JL001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troshin, K.; Sykilinda, N.; Shuraleva, S.; Tokmakova, A.; Tkachenko, N.; Kurochkina, L.; Miroshnikov, K.; Suzina, N.; Brzhozovskaya, E.; Petrova, K.; et al. Pseudomonas Phage Lydia and the Evolution of the Mesyanzhinovviridae Family. Viruses 2025, 17, 369. https://doi.org/10.3390/v17030369
Troshin K, Sykilinda N, Shuraleva S, Tokmakova A, Tkachenko N, Kurochkina L, Miroshnikov K, Suzina N, Brzhozovskaya E, Petrova K, et al. Pseudomonas Phage Lydia and the Evolution of the Mesyanzhinovviridae Family. Viruses. 2025; 17(3):369. https://doi.org/10.3390/v17030369
Chicago/Turabian StyleTroshin, Konstantin, Nina Sykilinda, Sofia Shuraleva, Anna Tokmakova, Nikolay Tkachenko, Lidia Kurochkina, Konstantin Miroshnikov, Natalia Suzina, Ekaterina Brzhozovskaya, Kristina Petrova, and et al. 2025. "Pseudomonas Phage Lydia and the Evolution of the Mesyanzhinovviridae Family" Viruses 17, no. 3: 369. https://doi.org/10.3390/v17030369
APA StyleTroshin, K., Sykilinda, N., Shuraleva, S., Tokmakova, A., Tkachenko, N., Kurochkina, L., Miroshnikov, K., Suzina, N., Brzhozovskaya, E., Petrova, K., Toshchakov, S., & Evseev, P. (2025). Pseudomonas Phage Lydia and the Evolution of the Mesyanzhinovviridae Family. Viruses, 17(3), 369. https://doi.org/10.3390/v17030369