


First Isolation and Characterization of Three Strains of Porcine Sapelovirus in Yunnan Province, China

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, Virus Isolation, and Identification
2.2. Virus Purification and TCID50
2.3. Replication Kinetics Analysis
2.4. Observation of the Virus Using Transmission Electron Microscopy
2.5. Virus Genome Sequencing
2.6. Sequence and Phylogenetic Analysis
3. Results
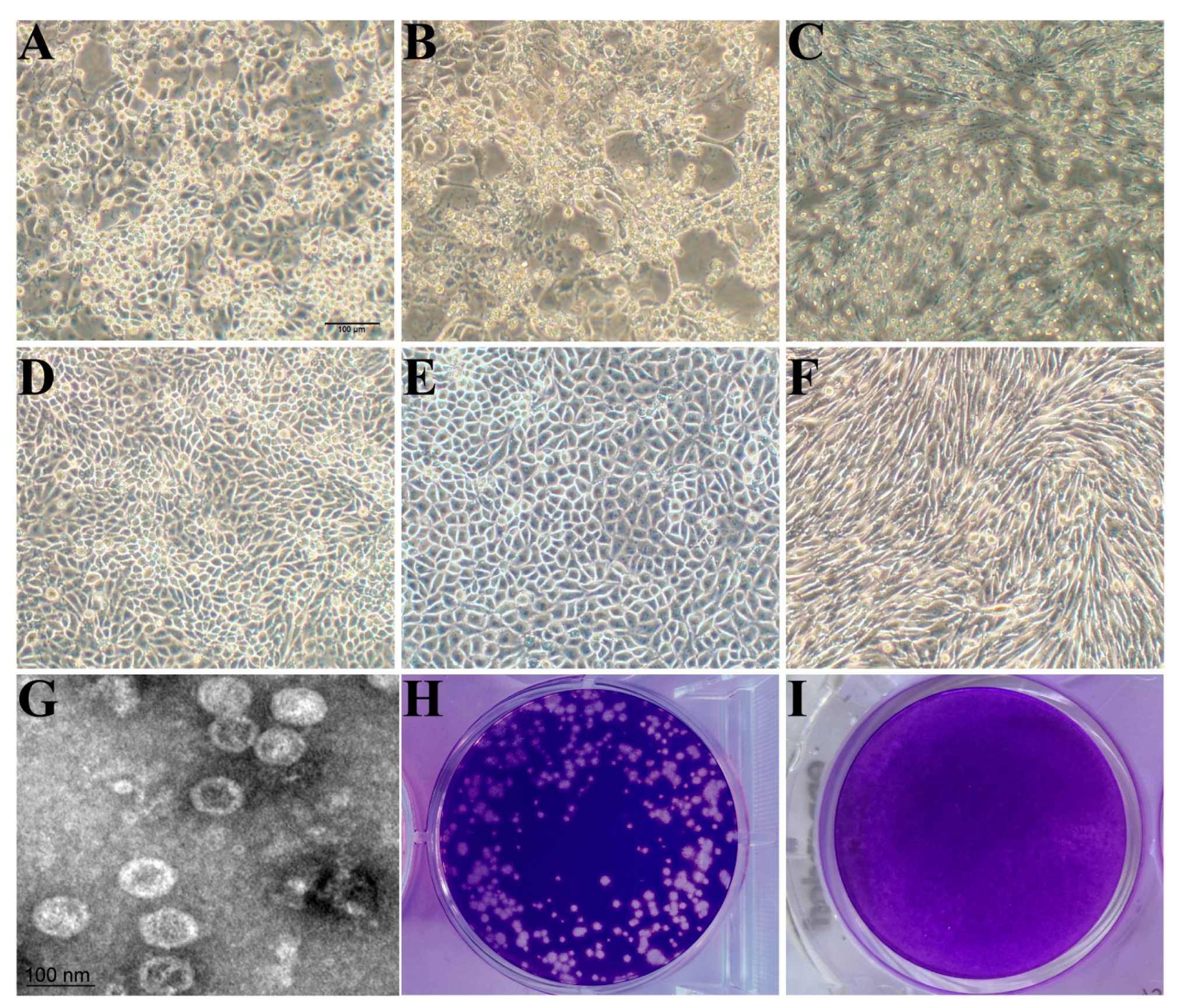
3.1. Isolation, Identification, and Biological Characteristics of PSV
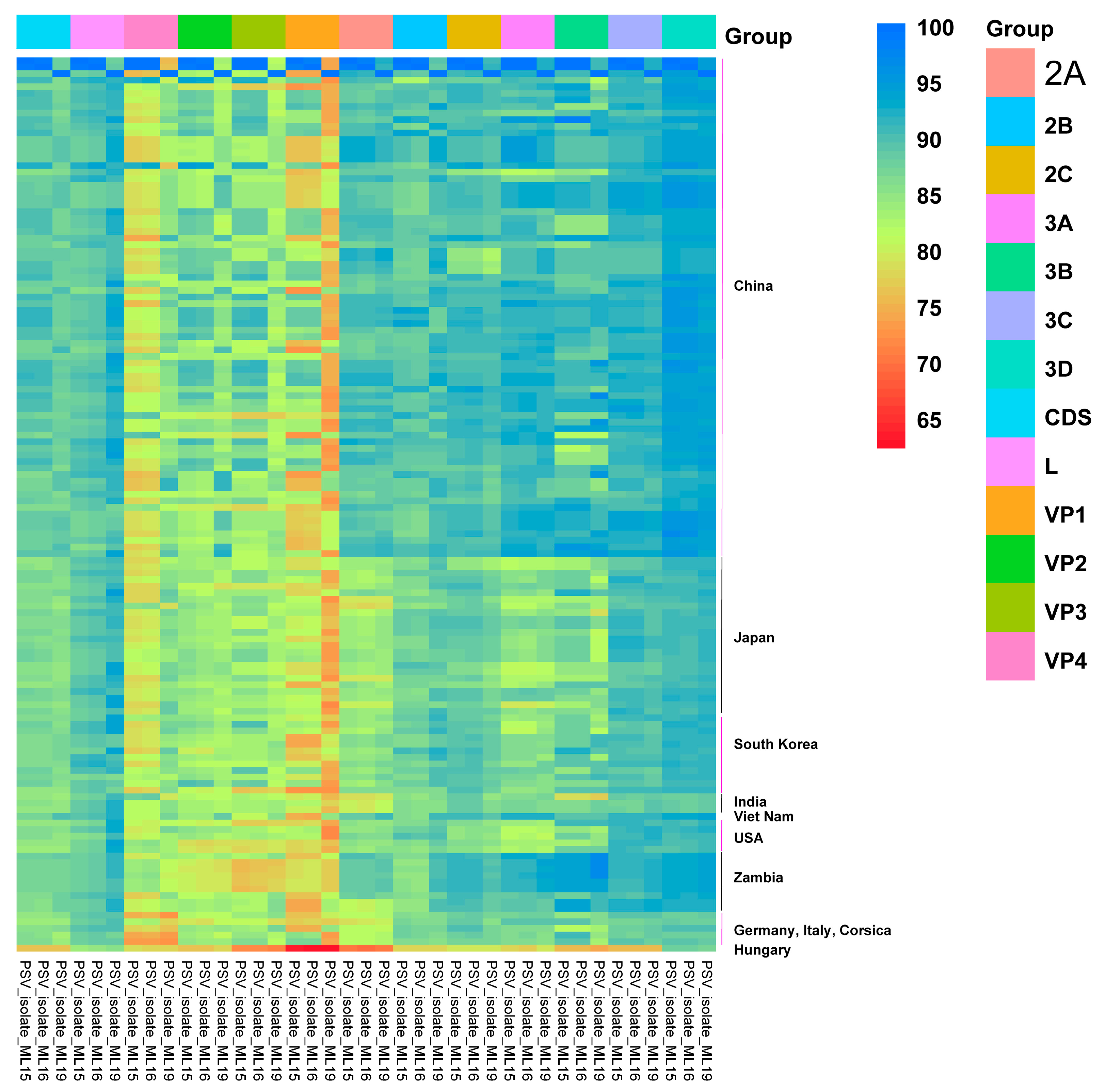
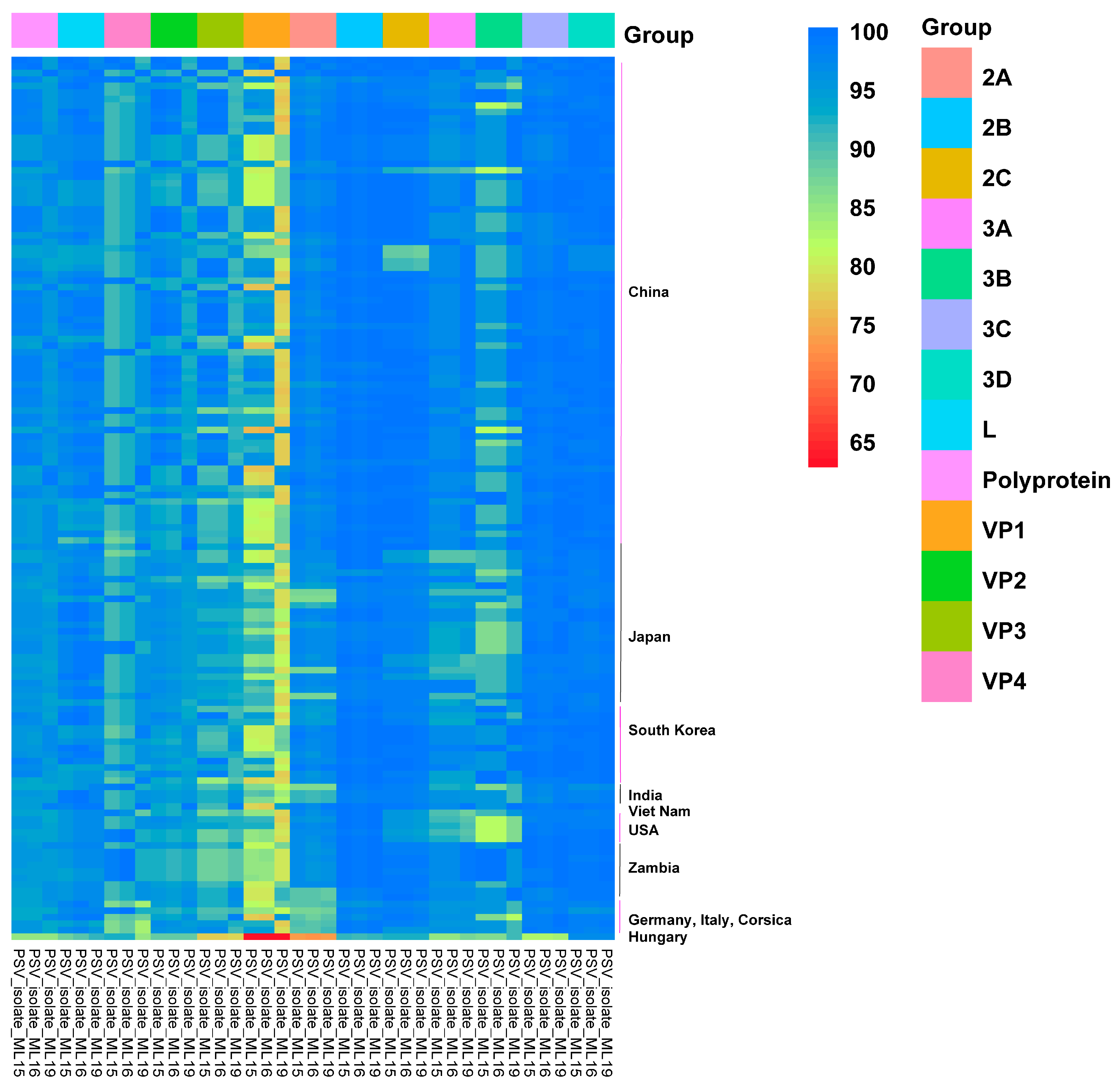
3.2. Genome Organization and Homology Analyses
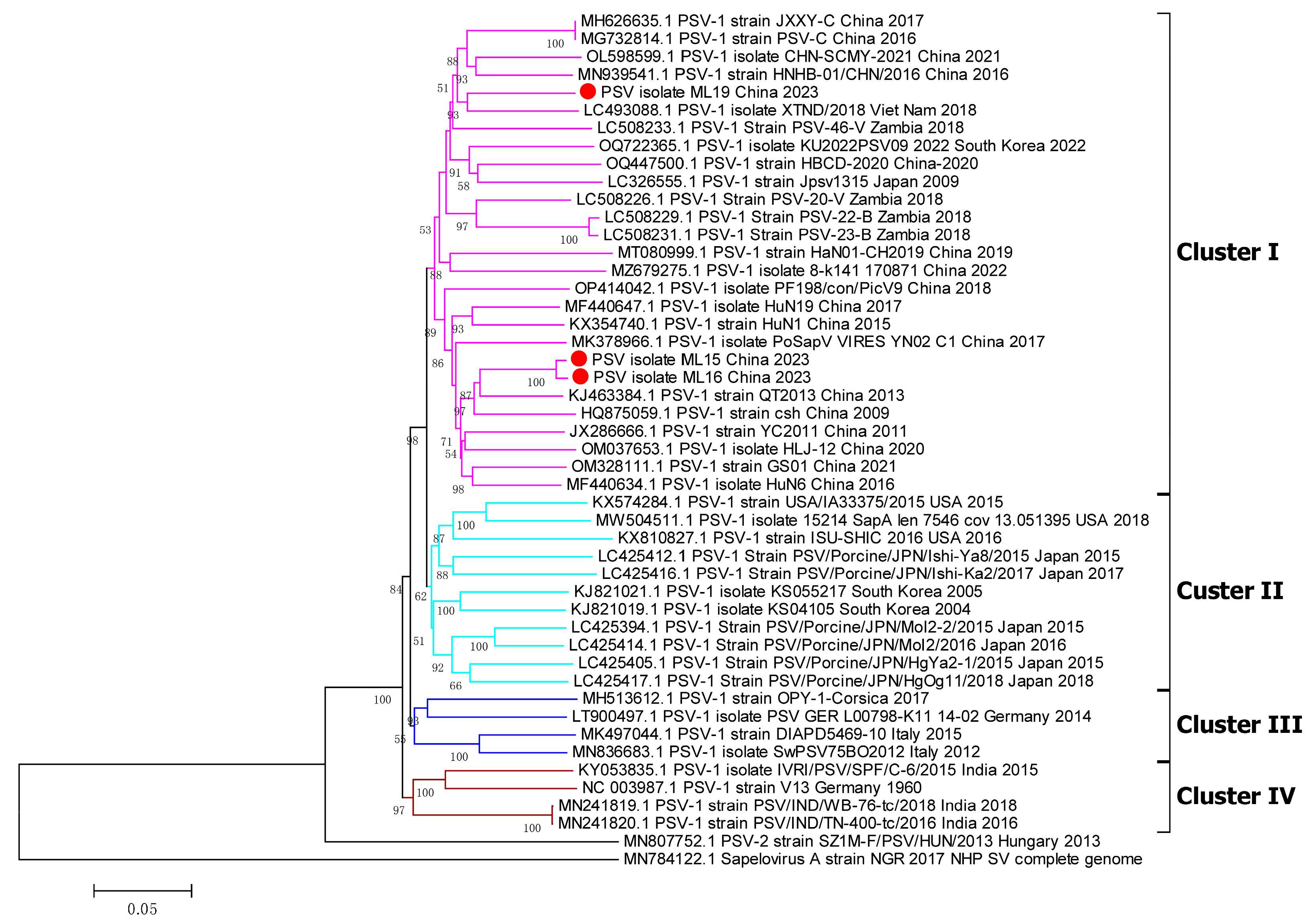
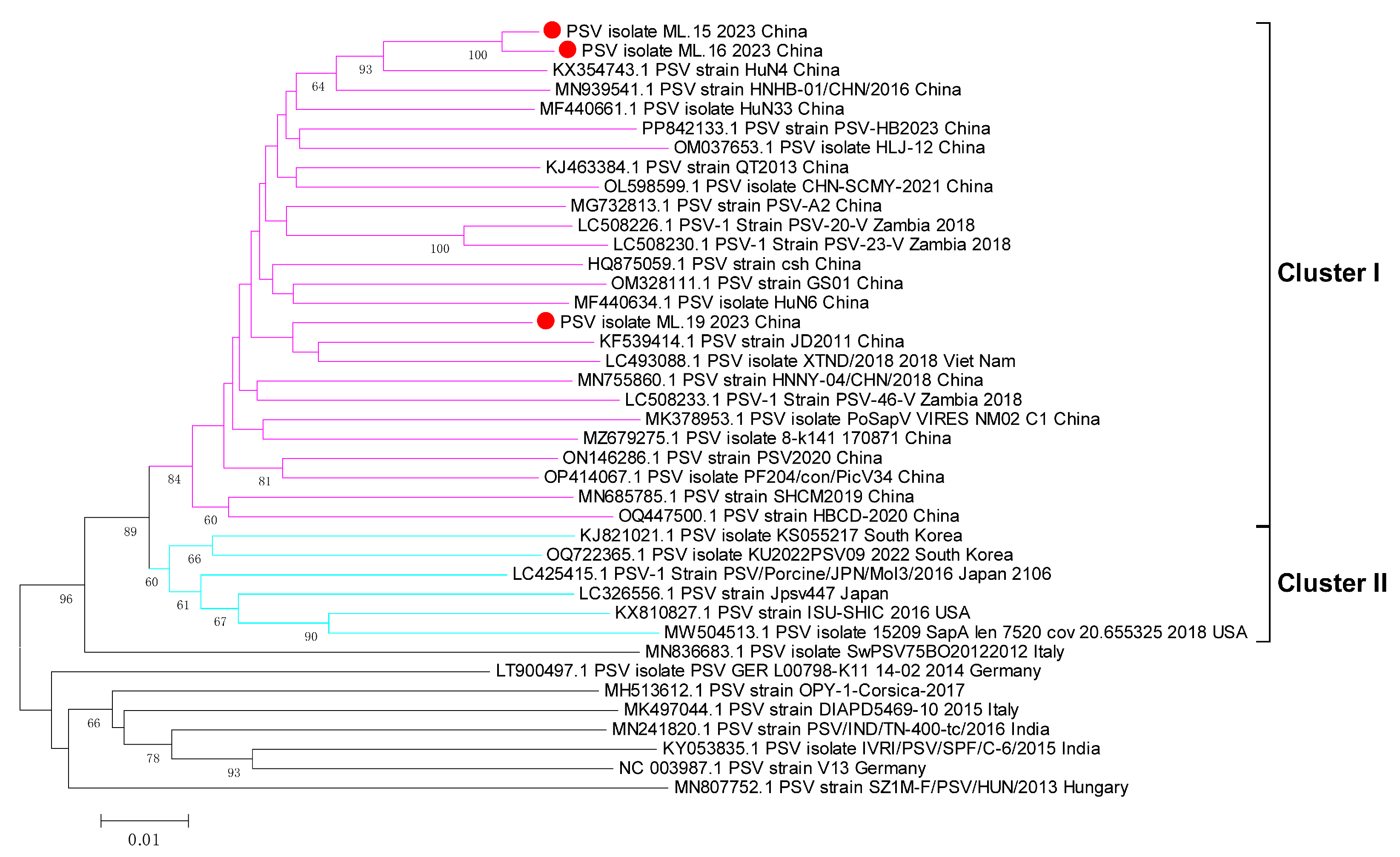
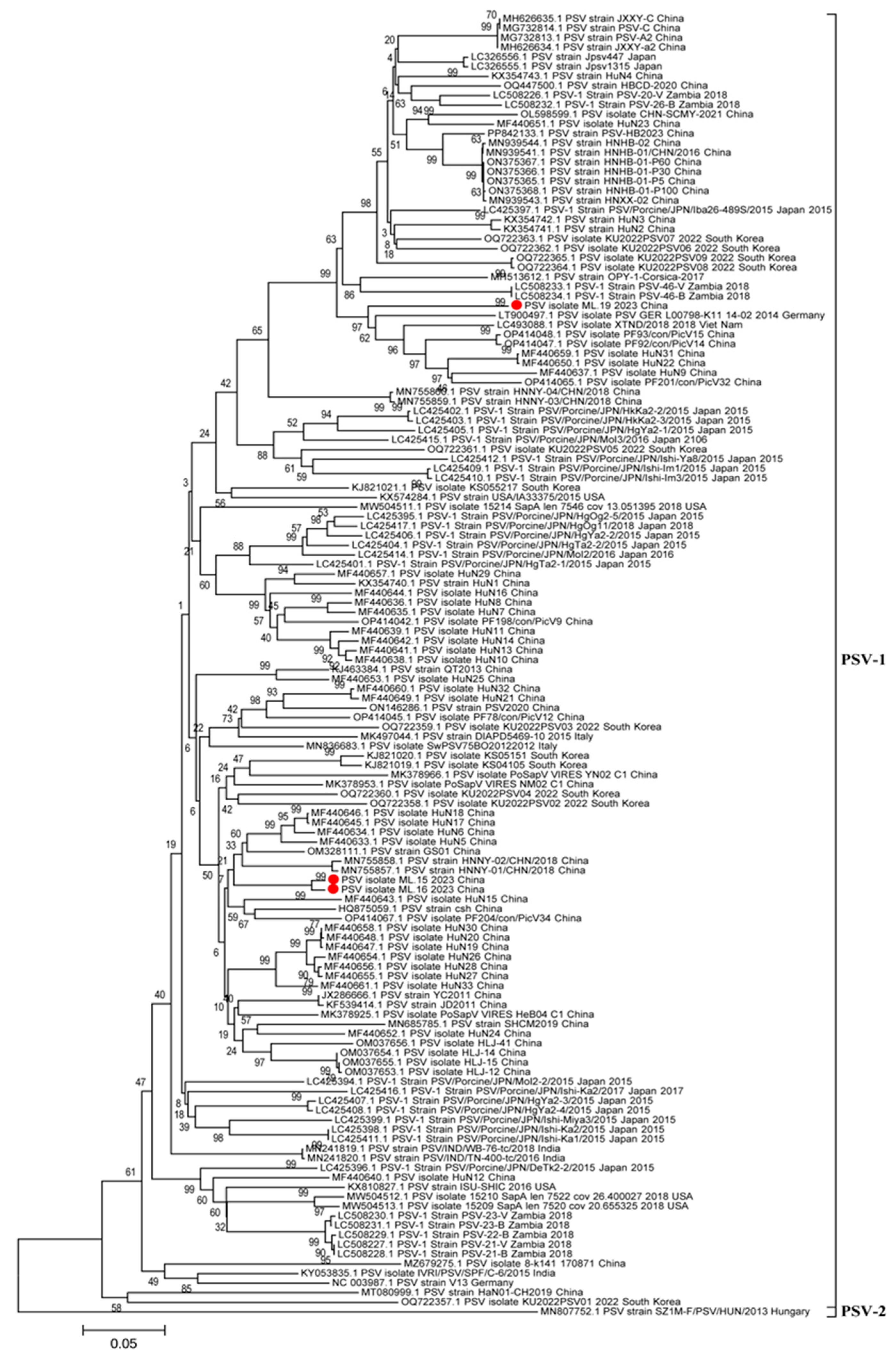
3.3. Phylogenetic Analyses of Whole Genome, 3D Gene, and VP1 Gene
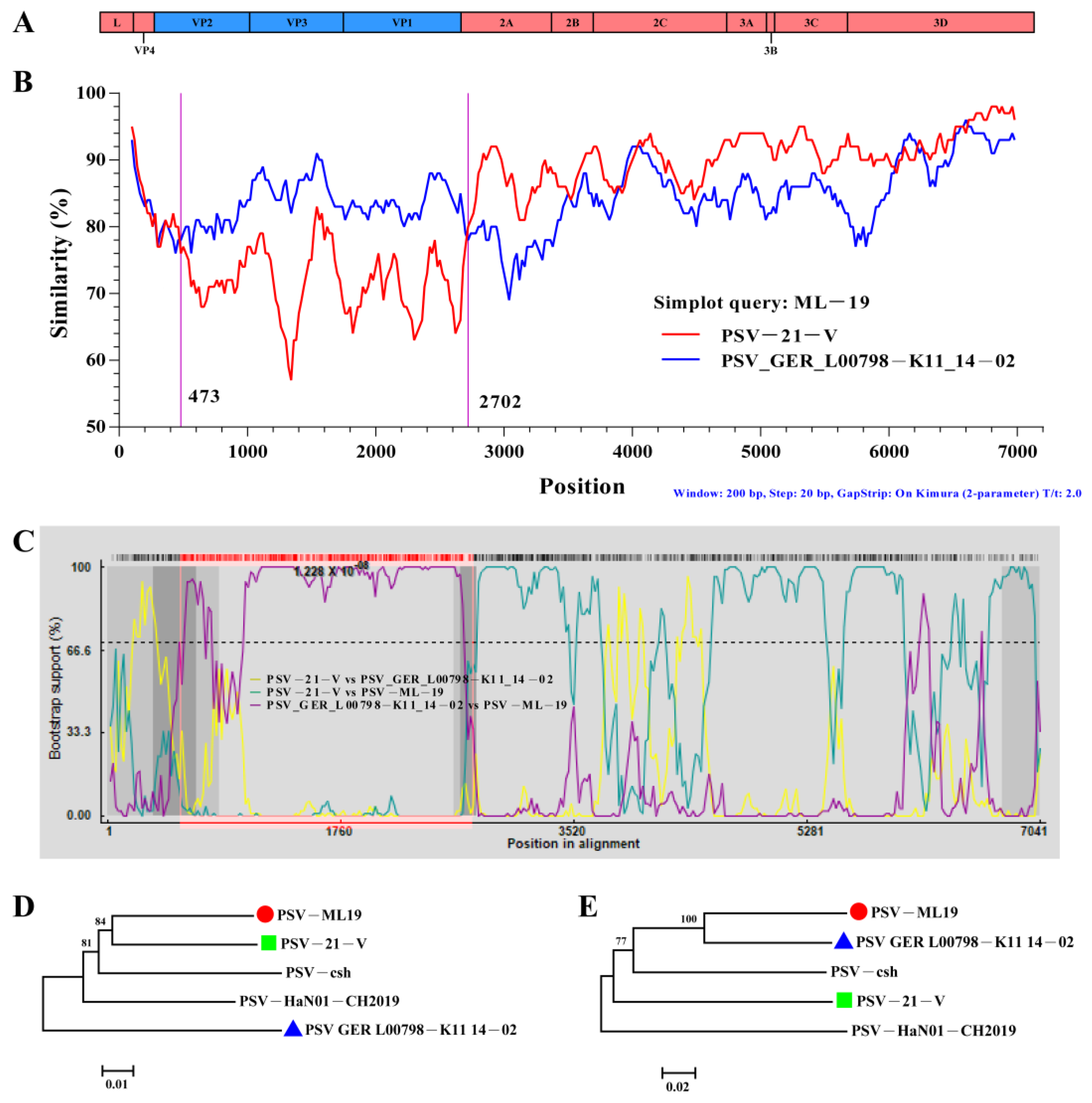
3.4. Recombination Analyses of the PSV Strains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lamont, P.H.; Betts, A.O. Enteroviruses in the pig. Nature 1958, 182, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Ji, W.; Yang, S.; Cui, L.; Yang, Z.; Yuan, C.; Hua, X. Isolation and characterization of the first Chinese porcine sapelovirus strain. Arch. Virol. 2011, 156, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, L.; Jin, T.; Cheng, Y.; Zhang, X.; Jiao, S.; Huang, T.; Zhang, Y.; Yan, Y.; Gu, J.; et al. Characterization and epidemiological survey of porcine sapelovirus in China. Vet Microbiol. 2019, 232, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Capai, L.; Piorkowski, G.; Maestrini, O.; Casabianca, F.; Masse, S.; de Lamballerie, X.; Charrel, R.N.; Falchi, A. Detection of porcine enteric viruses (Kobuvirus, Mamastrovirus and Sapelovirus) in domestic pigs in Corsica, France. PLoS ONE 2022, 17, e0260161. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tseng, C.H.; Tsai, H.J. Sequence analysis of a duck picornavirus isolate indicates that it together with porcine enterovirus type 8 and simian picornavirus type 2 should be assigned to a new picornavirus genus. Virus Res. 2007, 129, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.M.; Zhang, W.; Werid, G.M.; Zhang, H.; Feng, Y.; Pan, Y.; Zhang, L.; Li, C.; Lin, H.; Chen, H.; et al. Isolation, Characterization, and Molecular Detection of Porcine Sapelovirus. Viruses 2022, 14, 349. [Google Scholar] [CrossRef]
- Kaku, Y.; Sarai, A.; Murakami, Y. Genetic reclassification of porcine enteroviruses. J. Gen. Virol. 2001, 82 Pt 2, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Sun, Z.H.; Che, Y.L.; Chen, R.J.; Wu, X.M.; Wu, R.J.; Wang, L.B.; Zhou, L.J. High Prevalence, Genetic Diversity, and Recombination of Porcine Sapelovirus in Pig Farms in Fujian, Southern China. Viruses 2023, 15, 1751. [Google Scholar] [CrossRef]
- Boros, Á.; László, Z.; Pankovics, P.; Marosi, A.; Albert, M.; Cságola, A.; Bíró, H.; Fahsbender, E.; Delwart, E.; Reuter, G. High prevalence, genetic diversity and a potentially novel genotype of Sapelovirus A (Picornaviridae) in enteric and respiratory samples in Hungarian swine farms. J. Gen. Virol. 2020, 101, 609–621. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Oberste, M.S.; Maher, K.; Pallansch, M.A. Molecular phylogeny and proposed classification of the simian picornaviruses. J. Virol. 2002, 76, 1244–1251. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lamont, P.H.; Betts, A.O. Studies on Enteroviruses of the Pig—IV: The Isolation in Tissue Culture of a Possible Enteric Cytopathogenic Swine Orphan (ECSO) Virus (V 13) from the Faeces of a Pig. Res. Vet. Sci. 1960, 1, 152–161. [Google Scholar] [CrossRef]
- Bai, H.; Liu, J.; Fang, L.; Kataoka, M.; Takeda, N.; Wakita, T.; Li, T.C. Characterization of porcine sapelovirus isolated from Japanese swine with PLC/PRF/5 cells. Transbound. Emerg. Dis. 2018, 65, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Ito, N.; Sakai, K.; Kaku, Y.; Oba, M.; Nishimura, M.; Kurane, I.; Saijo, M.; Morikawa, S.; Sugiyama, M.; et al. A novel sapelovirus-like virus isolation from wild boar. Virus Genes 2011, 43, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Ji, W.; Wang, C.; Sun, H.; Chen, M.; Cui, L.; Tong, G.; Hua, X. Molecular epidemiologic investigation of porcine Sapro virus in swine herds in some areas of East China. Prog. Vet. Med. 2012, 33, 116–121. [Google Scholar]
- Schock, A.; Gurrala, R.; Fuller, H.; Foyle, L.; Dauber, M.; Martelli, F.; Scholes, S.; Roberts, L.; Steinbach, F.; Dastjerdi, A. Investigation into an outbreak of encephalomyelitis caused by a neuroinvasive porcine sapelovirus in the United Kingdom. Vet. Microbiol. 2014, 172, 381–389. [Google Scholar] [CrossRef]
- Arruda, P.; Arruda, B.L.; Schwartz, K.J.; Vannucci, F.; Resende, T.; Rovira, A.; Sundberg, P.; Nietfeld, J.; Hause, B.M. Detection of a novel sapelovirus in central nervous tissue of pigs with polioencephalomyelitis in the USA. Transbound. Emerg. Dis. 2017, 64, 311–315. [Google Scholar] [CrossRef]
- Harima, H.; Kajihara, M.; Simulundu, E.; Bwalya, E.; Qiu, Y.; Isono, M.; Okuya, K.; Gonzalez, G.; Yamagishi, J.; Hang’ombe, B.M.; et al. Genetic and Biological Diversity of Porcine Sapeloviruses Prevailing in Zambia. Viruses 2020, 12, 180. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/nt. Nuclc. Acids. Symp. 1999, 41, 95–98. [Google Scholar]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE. 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar]
- Kim, D.S.; Kang, M.I.; Son, K.Y.; Bak, G.Y.; Park, J.G.; Hosmillo, M.; Seo, J.Y.; Kim, J.Y.; Alfajaro, M.M.; Soliman, M.; et al. Pathogenesis of Korean SapelovirusA in piglets and chicks. J. Gen. Virol. 2016, 97, 2566–2574. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Su, M.; Li, C.; Qi, S.; Yang, D.; Jiang, N.; Yin, B.; Guo, D.; Kong, F.; Yuan, D.; Feng, L.; et al. A molecular epidemiological investigation of PEDV in China: Characterization of co-infection and genetic diversity of S1-based genes. Transbound. Emerg. Dis. 2020, 67, 1129–1140, Erratum in Transbound. Emerg. Dis. 2021, 68, 2634–2635. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vilar, M.J.; Peralta, B.; García-Bocanegra, I.; Simon-Grifé, M.; Bensaid, A.; Casal, J.; Segalés, J.; Pina-Pedrero, S. Distribution and genetic characterization of Enterovirus G and Sapelovirus A in six Spanish swine herds. Virus Res. 2016, 215, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Stäubli, T.; Rickli, C.I.; Torgerson, P.R.; Fraefel, C.; Lechmann, J. Porcine teschovirus, sapelovirus, and enterovirus in Swiss pigs: Multiplex RT-PCR investigation of viral frequencies and disease association. J. Vet. Diagn. Investig. 2021, 33, 864–874. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hammerschmitt, M.E.; de Almeida, P.R.; de Cecco, B.S.; Lorenzett, M.P.; Schwertz, C.I.; da Cruz, R.A.S.; Caprioli, R.A.; Schuh, D.T.; Demoliner, M.; Eisen, A.K.A.; et al. Swine polioencephalomyelitis in Brazil: Identification of Teschovirus A, Sapelovirus A, and Enterovirus G in a farm from Southern Brazil. Braz. J. Microbiol. 2021, 52, 1617–1622. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, T.; Yu, X.; Yan, M.; Luo, B.; Li, R.; Qu, T.; Luo, Z.; Ge, M.; Zhao, D. Molecular characterization of Porcine sapelovirus in Hunan, China. J. Gen. Virol. 2017, 98, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, M.; Mimouli, K.; Kyriakopoulou, Z.; Tsimpidis, M.; Tsakogiannis, D.; Markoulatos, P.; Amoutzias, G.D. Large-scale genomic analysis reveals recurrent patterns of intertypic recombination in human enteroviruses. Virology 2019, 526, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhou, Y.S.; Wang, H.N.; Zhang, Y.; Wei, K.; Wang, T. Isolation, identification and complete genome sequence analysis of a strain of foot-and-mouth disease virus serotype Asia1 from pigs in southwest of China. Virol. J. 2011, 8, 175. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Z.H.; Li, Z.R.; Zhu, P.; Zhang, Z.X.; Song, J.L. First Identification and Pathogenicity Evaluation of an EV-G17 Strain Carrying a Torovirus Papain-like Cysteine Protease (PLCP) Gene in China. Viruses 2023, 15, 1747. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, T.; Li, R.; Peng, W.; Ge, M.; Luo, B.; Qu, T.; Yu, X. First isolation and genetic characteristics of porcine sapeloviruses in Hunan, China. Arch. Virol. 2017, 162, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Sunaga, F.; Masuda, T.; Ito, M.; Akagami, M.; Naoi, Y.; Sano, K.; Katayama, Y.; Omatsu, T.; Oba, M.; Sakaguchi, S.; et al. Complete genomic analysis and molecular characterization of Japanese porcine sapeloviruses. Virus Genes 2019, 55, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Golender, N.; Hoffmann, B. The Molecular Epidemiology of Epizootic Hemorrhagic Disease Viruses Identified in Israel between 2015 and 2023. Epidemiologia 2024, 5, 90–105. [Google Scholar] [CrossRef]
- Primadharsini, P.P.; Nagashima, S.; Okamoto, H. Genetic Variability and Evolution of Hepatitis E Virus. Viruses 2019, 11, 456. [Google Scholar] [CrossRef]
- Lan, D.; Tang, C.; Yue, H.; Sun, H.; Cui, L.; Hua, X.; Li, J. Microarray analysis of differentially expressed transcripts in porcine intestinal epithelial cells (IPEC-J2) infected with porcine sapelovirus as a model to study innate immune responses to enteric viruses. Arch. Virol. 2013, 158, 1467–1475. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Gene/Protein a | Asia b | America b | Africa b | Europe b |
|---|---|---|---|---|---|
| PSV15 | CDS (6996/2332) | 87.76 ± 1.85/96.05 ± 1.58 | 84.92 ± 0.97/94.46 ± 0.79 | 86.38 ± 0.46/94.58 ± 0.42 | 84.98 ± 0.98/94.10 ± 1.29 |
| L (252/84) | 88.37 ± 1.29/96.42 ± 1.87 | 86.48 ± 1.06/95.20 ± 0 | 87.63 ± 0.46/95.07 ± 0.72 | 87.94 ± 0.63/96.40 ± 1.47 | |
| VP4 (159/53) | 79.59 ± 2.97/91.32 ± 2.58 | 81.46 ± 2.28/95.06 ± 3.80 | 81.57 ± 2.72/96.62 ± 4.01 | 76.26 ± 3.83/90.74 ± 5.21 | |
| VP2 (714/238) | 85.39 ± 3.09/96.12 ± 2.46 | 81.50 ± 3.19/94.14 ± 1.86 | 81.11 ± 1.87/93.33 ± 1.50 | 82.66 ± 2.30/94.66 ± 1.76 | |
| VP3 (702/234) | 85.07 ± 3.62/94.15 ± 3.90 | 79.78 ± 2.93/89.72 ± 3.19 | 78.72 ± 3.45/89.48 ± 2.03 | 83.56 ± 1.90/92.94 ± 2.85 | |
| VP1 (879/293) | 81.27 ± 4.83/88.32 ± 6.40 | 79.52 ± 2.46/87.52 ± 3.88 | 77.03 ± 2.28/82.78 ± 2.58 | 79.78 ± 5.67/85.82 ± 7.16 | |
| 2A(678/226) | 87.57 ± 3.54/97.00 ± 2.64 | 85.14 ± 0.96/96.88 ± 0.53 | 86.74 ± 3.46/94.99 ± 3.23 | 81.00 ± 1.29/88.88 ± 1.04 | |
| 2B (315/105) | 87.93 ± 1.82/99.41 ± 0.84 | 86.76 ± 1.75/99.80 ± 0.45 | 84.96 ± 0.65/99.56 ± 0.88 | 86.76 ± 0.59/97.84 ± 1.25 | |
| 2C (996/332) | 89.24 ± 1.80/98.25 ± 2.09 | 85.26 ± 0.45/94.74 ± 0.13 | 91.07 ± 0.56/98.70 ± 0.30 | 87.90 ± 1.00/97.38 ± 1.51 | |
| 3A (300/100) | 89.30 ± 4.04/96.00 ± 2.09 | 86.16 ± 1.36/91.20 ± 0.84 | 86.04 ± 2.92/96.89 ± 0.33 | 86.24 ± 5.46/95.40 ± 0.55 | |
| 3B (66/22) | 88.87 ± 3.26/92.97 ± 3.94 | 86.92 ± 2.96/83.62 ± 4.07 | 93.22 ± 2.03/97.46 ± 3.32 | 87.22 ± 3.79/93.58 ± 4.07 | |
| 3C (546/182) | 90.81 ± 1.52/98.61 ± 0.72 | 91.08 ± 0.31/98.54 ± 0.84 | 91.10 ± 0.74/98.67 ± 0.95 | 87.90 ± 1.15/98.20 ± 0.62 | |
| 3D (1389/463) | 92.66 ± 1.93/98.72 ± 0.75 | 90.04 ± 0.64/98.36 ± 0.68 | 92.81 ± 0.40/98.69 ± 0.13 | 88.48 ± 0.94/97.44 ± 1.31 | |
| PSV16 | CDS (6996/2332) | 87.69 ± 1.86/95.81 ± 1.57 | 84.92 ± 1.02/94.46 ± 0.83 | 86.34 ± 0.54/94.41 ± 0.45 | 84.92 ± 0.93/93.94 ± 1.30 |
| L (252/84) | 89.13 ± 1.30/97.54 ± 1.81 | 86.48 ± 1.05/95.20 ± 0 | 88.40 ± 0.49/96.27 ± 0.72 | 88.72 ± 0.66/97.60 ± 1.47 | |
| VP4 (159/53) | 80.21 ± 2.97/93.19 ± 2.59 | 81.46 ± 2.50/95.06 ± 4.16 | 82.20 ± 2.71/94.72 ± 4.01 | 76.86 ± 3.89/89.00 ± 5.12 | |
| VP2 (714/238) | 84.75 ± 3.07/95.31 ± 2.46 | 81.50 ± 3.26/94.14 ± 1.83 | 80.66 ± 1.79/92.47 ± 1.55 | 81.92 ± 2.35/93.78 ± 1.75 | |
| VP3 (702/234) | 85.10 ± 3.71/94.15 ± 3.90 | 79.78 ± 2.90/89.72 ± 3.19 | 78.99 ± 3.13/89.48 ± 2.03 | 83.52 ± 1.93/92.94 ± 2.85 | |
| VP1 (879/293) | 81.31 ± 4.72/88.20 ± 6.50 | 79.52 ± 2.36/87.52 ± 3.67 | 77.31 ± 2.28/82.67 ± 2.81 | 79.88 ± 5.25/85.56 ± 7.35 | |
| 2A(678/226) | 87.12 ± 3.56/95.96 ± 2.40 | 85.14 ± 0.95/96.88 ± 0.53 | 86.31 ± 3.81/94.19 ± 3.01 | 80.68 ± 1.00/88.36 ± 1.04 | |
| 2B (315/105) | 87.62 ± 1.81/98.42 ± 0.81 | 86.76 ± 1.83/99.80 ± 0.45 | 84.78 ± 0.47/98.58 ± 0.84 | 86.22 ± 0.72/96.88 ± 1.22 | |
| 2C (996/332) | 89.39 ± 1.81/98.25 ± 2.09 | 85.26 ± 0.51/94.74 ± 0.13 | 90.92 ± 0.64/98.70 ± 0.30 | 88.08 ± 0.83/97.38 ± 1.51 | |
| 3A (300/100) | 89.48 ± 4.15/96.00 ± 2.09 | 86.16 ± 1.09/91.20 ± 0.84 | 86.12 ± 2.92/96.89 ± 0.33 | 86.50 ± 5.21/95.40 ± 0.55 | |
| 3B (66/22) | 88.87 ± 3.26/92.97 ± 3.94 | 86.92 ± 2.96/83.62 ± 4.07 | 93.22 ± 2.03/97.46 ± 3.32 | 87.22 ± 3.79/93.58 ± 4.07 | |
| 3C (546/182) | 90.84 ± 1.55/98.08 ± 0.71 | 91.08 ± 0.29/98.54 ± 0.45 | 90.90 ± 0.74/98.17 ± 0.95 | 87.80 ± 1.06/97.68 ± 0.59 | |
| 3D (1389/463) | 92.51 ± 1.92/98.53 ± 0.75 | 90.04 ± 0.64/98.36 ± 0.75 | 92.76 ± 0.43/98.69 ± 0.13 | 88.38 ± 1.03/97.40 ± 1.28 | |
| PSV19 | CDS (6996/2332) | 86.92 ± 1.46/95.14 ± 1.24 | 84.50 ± 0.42/93.36 ± 0.21 | 87.07 ± 0.72/95.16 ± 0.92 | 84.26 ± 1.20/94.00 ± 1.52 |
| L (252/84) | 91.18 ± 1.79/97.14 ± 1.65 | 92.48 ± 0.95/96.40 ± 0 | 91.07 ± 0.72/96.27 ± 0.72 | 88.66 ± 1.18/96.16 ± 2.31 | |
| VP4 (159/53) | 84.96 ± 2.36/96.00 ± 1.77 | 83.48 ± 1.89/93.16 ± 3.18 | 81.88 ± 1.95/94.09 ± 2.00 | 74.1 ± 2.35/85.82 ± 4.20 | |
| VP2 (714/238) | 84.26 ± 2.82/94.45 ± 2.14 | 80.96 ± 2.54/93.56 ± 0.76 | 80.80 ± 2.49/93.71 ± 2.05 | 82.22 ± 1.69/94.98 ± 1.43 | |
| VP3 (702/234) | 82.84 ± 1.74/92.73 ± 2.08 | 79.88 ± 1.87/91.04 ± 0.95 | 80.81 ± 4.64/91.83 ± 3.37 | 82.66 ± 3.31/93.62 ± 3.37 | |
| VP1 (855/285) | 76.30 ± 3.58/81.65 ± 5.32 | 73.64 ± 2.02/78.20 ± 0.79 | 79.01 ± 2.74/83.72 ± 5.37 | 77.72 ± 4.75/83.96 ± 7.55 | |
| 2A(678/226) | 87.67 ± 3.34/96.99 ± 2.67 | 85.06 ± 0.73/97.12 ± 0.71 | 86.99 ± 2.87/94.50 ± 3.47 | 81.50 ± 1.07/89.52 ± 1.08 | |
| 2B (315/105) | 89.54 ± 1.94/99.41 ± 0.84 | 88.52 ± 1.10/99.80 ± 0.45 | 89.59 ± 0.86/99.56 ± 0.88 | 85.92 ± 0.68/97.84 ± 1.25 | |
| 2C (996/332) | 88.16 ± 2.06/97.87 ± 2.16 | 84.62 ± 0.56/94.14 ± 0.13/ | 89.09 ± 0.64/98.60 ± 0.21 | 86.42 ± 0.61/96.90 ± 1.25 | |
| 3A (300/100) | 89.46 ± 3.60/96.88 ± 2.55 | 85.88 ± 1.63/89.20 ± 0.84 | 85.82 ± 2.91/98.22 ± 1.20 | 86.24 ± 6.03/95.60 ± 0.55 | |
| 3B (66/22) | 88.23 ± 4.01/96.23 ± 3.57 | 86.32 ± 3.04/88.12 ± 4.07 | 94.23 ± 2.78/96.93 ± 2.30 | 84.80 ± 1.50/89.08 ± 4.07 | |
| 3C (546/182) | 90.51 ± 1.37/98.94 ± 0.56 | 90.70 ± 1.20/98.66 ± 0.62 | 91.74 ± 0.80/99.16 ± 0.49 | 87.88 ± 0.58/98.86 ± 0.55 | |
| 3D (1389/463) | 92.54 ± 1.36/99.06 ± 0.68 | 90.88 ± 0.56/98.60 ± 0.66 | 93.16 ± 0.49/99.17 ± 0.22 | 88.34 ± 0.95/97.82 ± 1.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, P.; Li, Z.; Li, Z.; Meng, L.; Liu, P.; Sun, X.; Yang, Q.; Song, J. First Isolation and Characterization of Three Strains of Porcine Sapelovirus in Yunnan Province, China. Viruses 2025, 17, 505. https://doi.org/10.3390/v17040505
Zhu P, Li Z, Li Z, Meng L, Liu P, Sun X, Yang Q, Song J. First Isolation and Characterization of Three Strains of Porcine Sapelovirus in Yunnan Province, China. Viruses. 2025; 17(4):505. https://doi.org/10.3390/v17040505
Chicago/Turabian StyleZhu, Pei, Zhanhong Li, Zhuoran Li, Li Meng, Peng Liu, Xiutao Sun, Qi Yang, and Jianling Song. 2025. "First Isolation and Characterization of Three Strains of Porcine Sapelovirus in Yunnan Province, China" Viruses 17, no. 4: 505. https://doi.org/10.3390/v17040505
APA StyleZhu, P., Li, Z., Li, Z., Meng, L., Liu, P., Sun, X., Yang, Q., & Song, J. (2025). First Isolation and Characterization of Three Strains of Porcine Sapelovirus in Yunnan Province, China. Viruses, 17(4), 505. https://doi.org/10.3390/v17040505