

Chikungunya Replication and Infection Is Dependent upon and Alters Cellular Hexosylceramide Levels in Vero Cells

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Viruses
2.3. Viral Infection for Lipidomic Analysis
2.4. Lipid Extraction
2.5. HILIC-IM-MS
2.6. Data Analysis and Processing
2.7. Data Availability
2.8. Drug Inhibition Assays
2.9. Virus Entry Assays
2.10. Effect of Drugs on CHIKV Multi-Step Infection Titers
2.11. Virucidal Assay
2.12. Replicon Generation and Assay
2.13. Statistics
3. Results
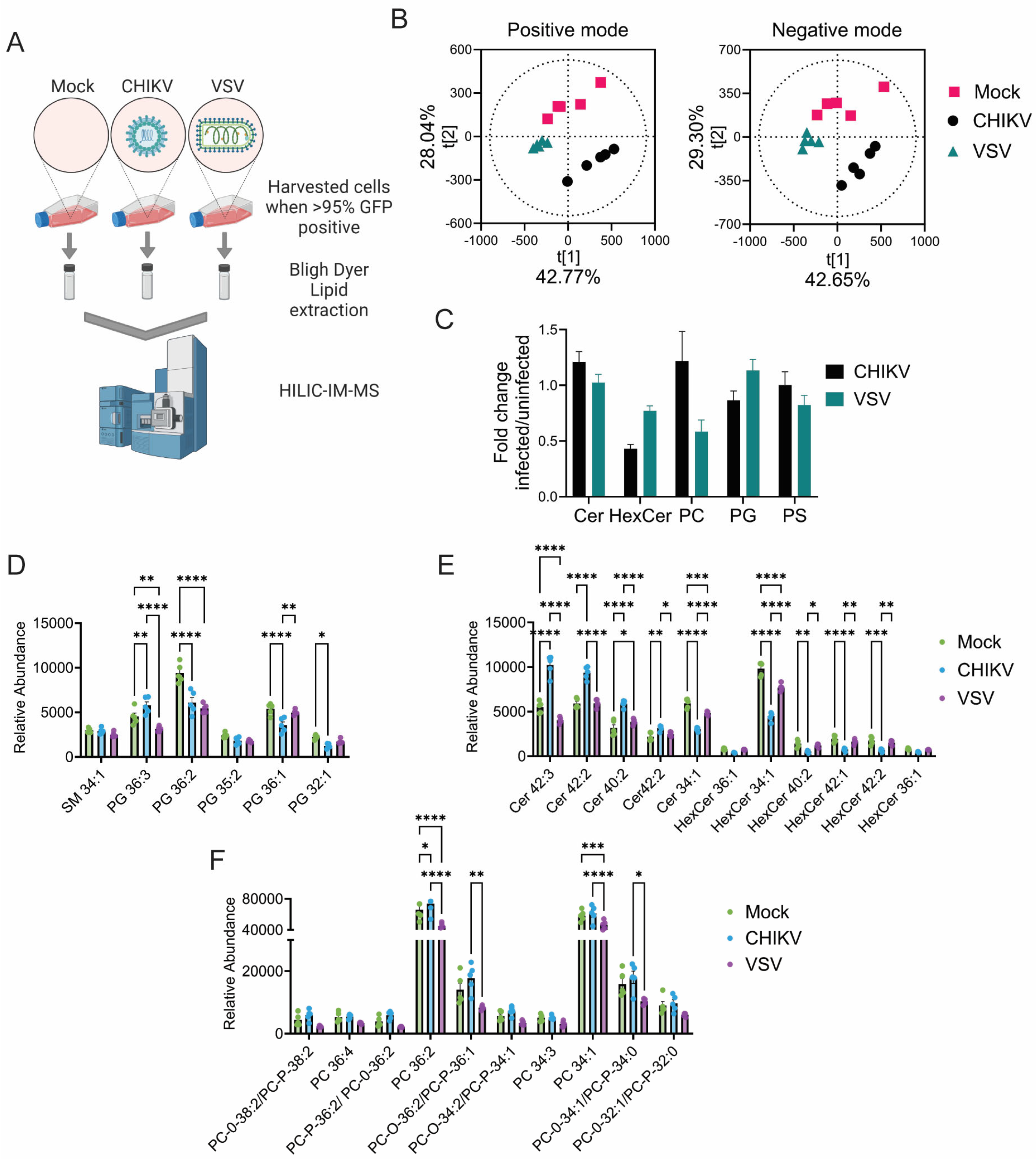
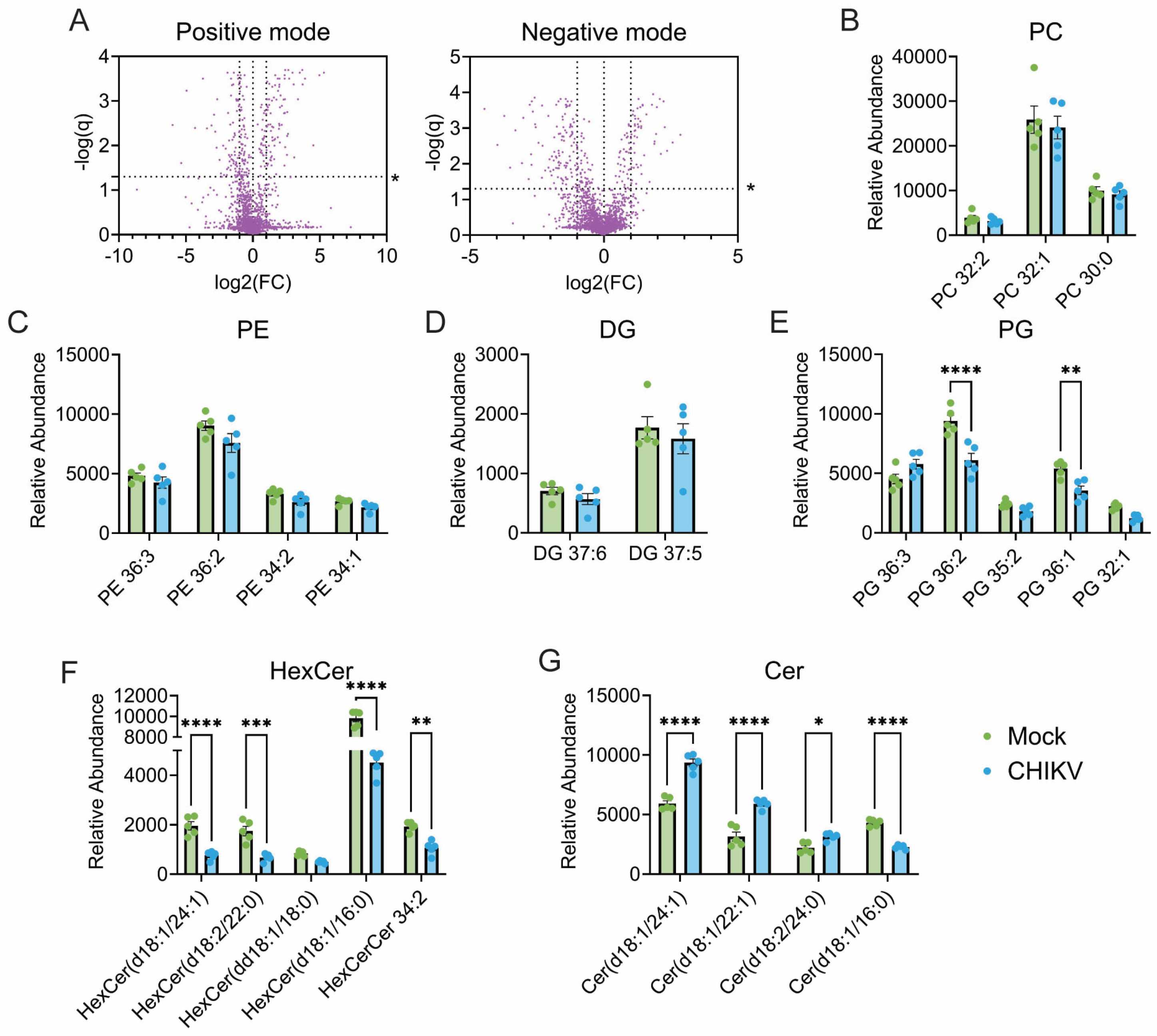
3.1. CHIKV Infection Alters Cellular Lipid Levels
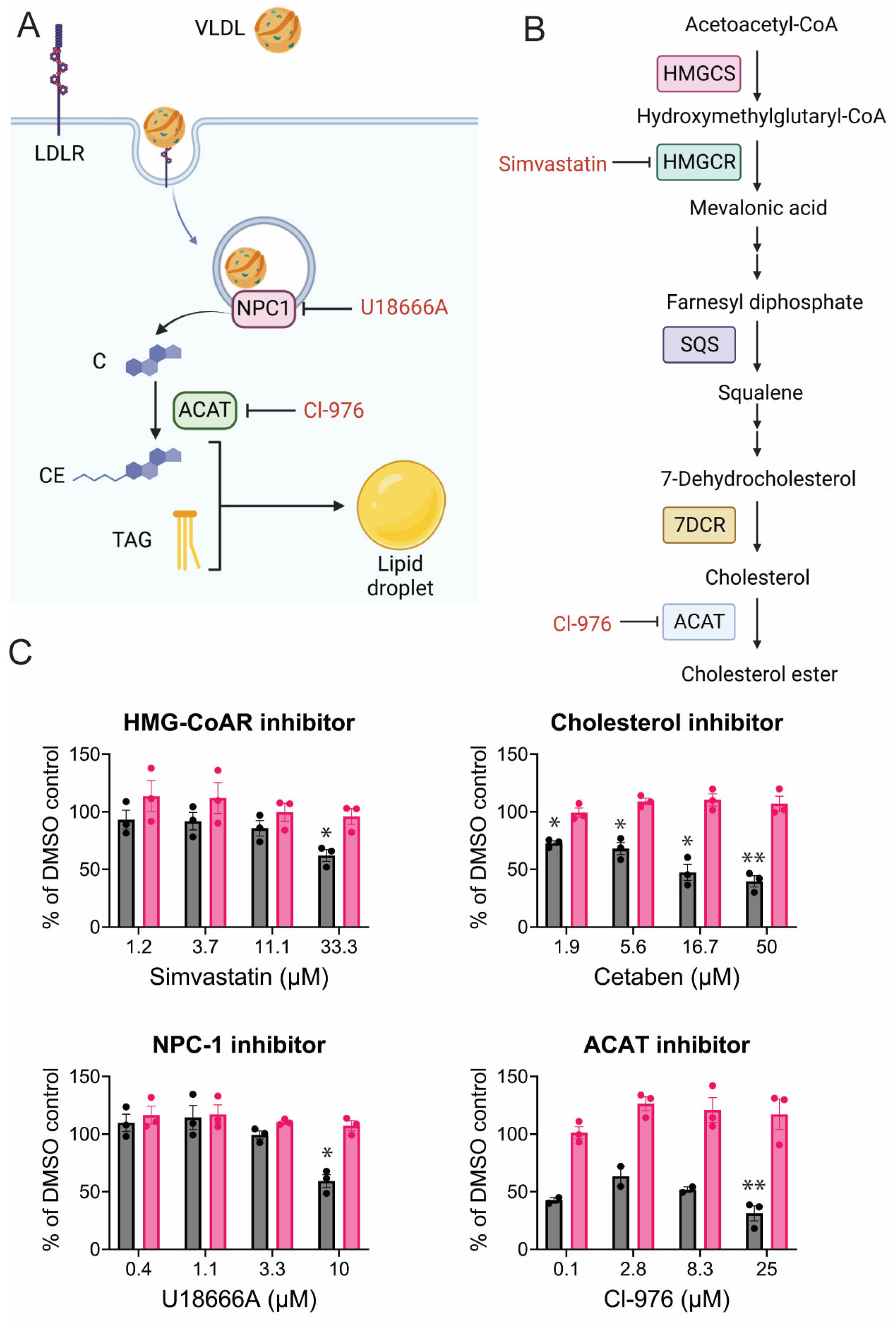
3.2. Alteration of Lipid Regulatory Elements Disturbs CHIKV Replication
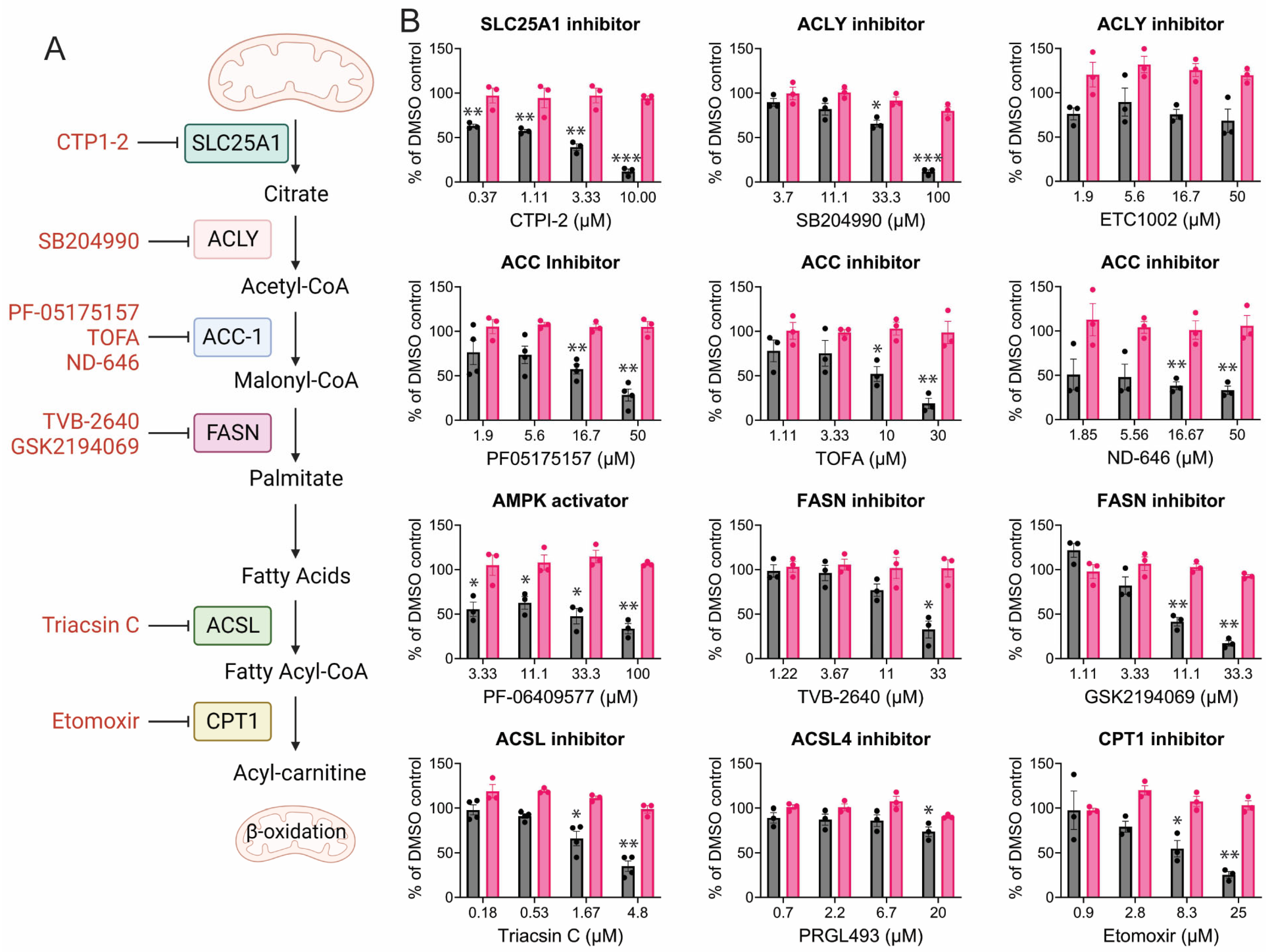
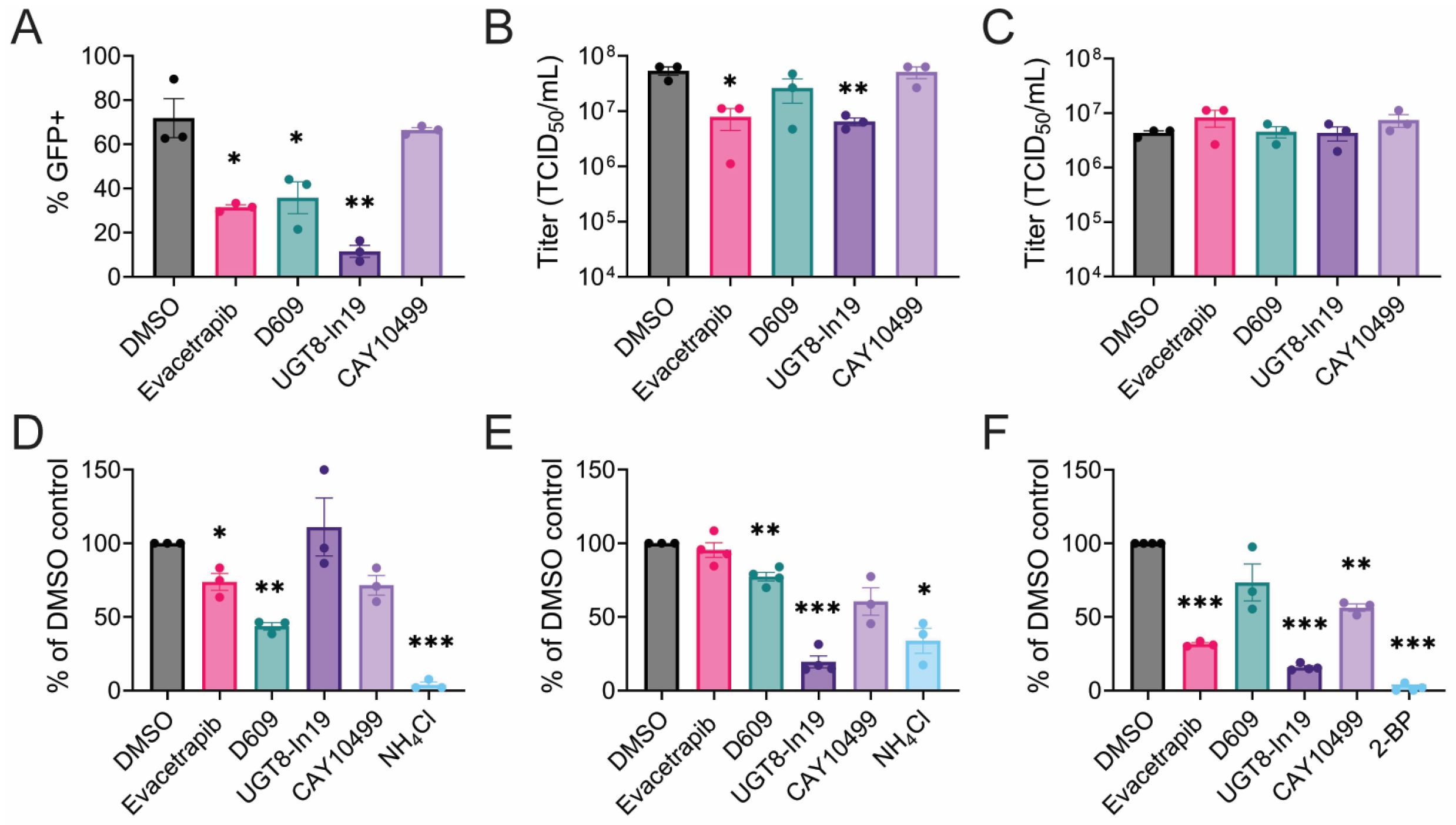
3.3. CHIKV Replication Requires Active Fatty Acid Synthesis and β-Oxidation
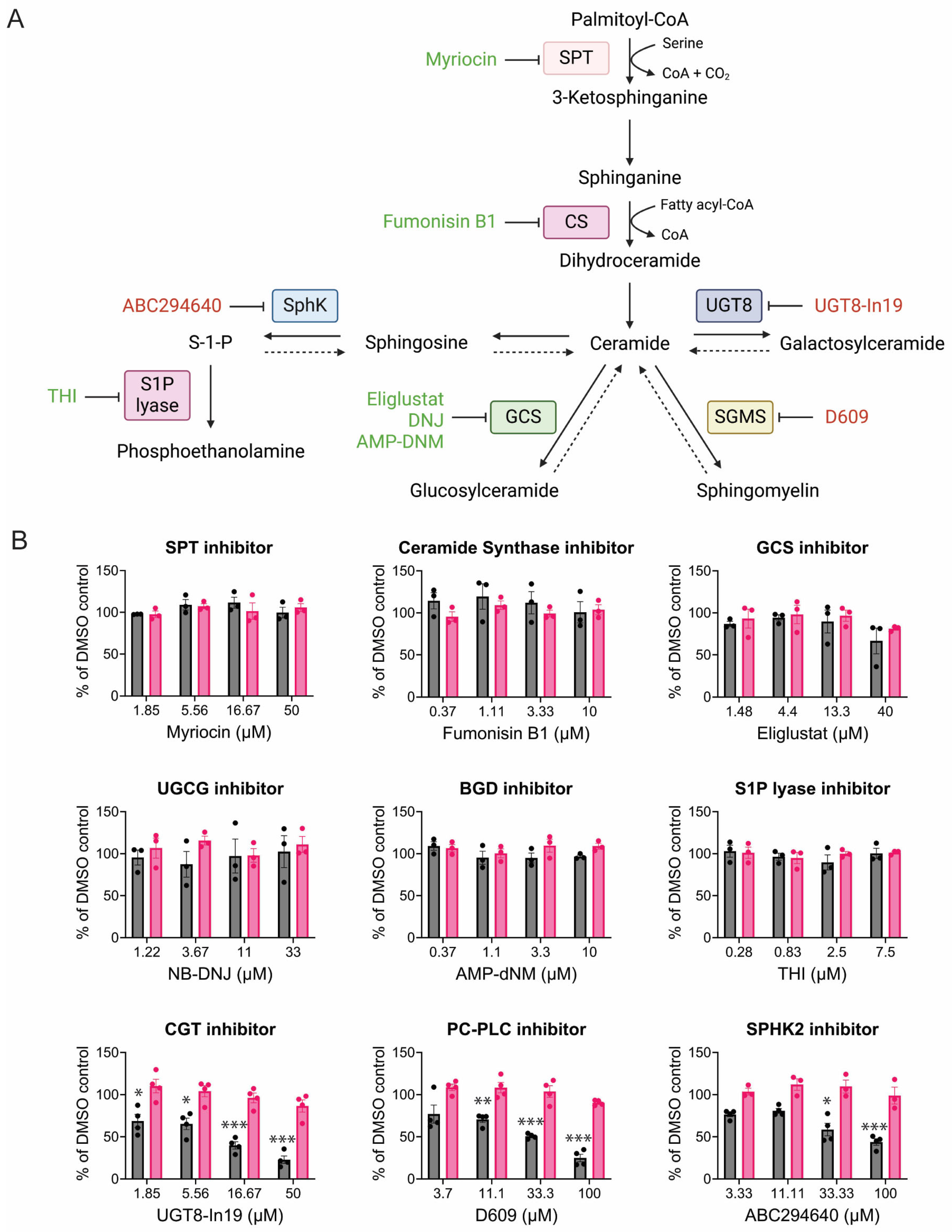
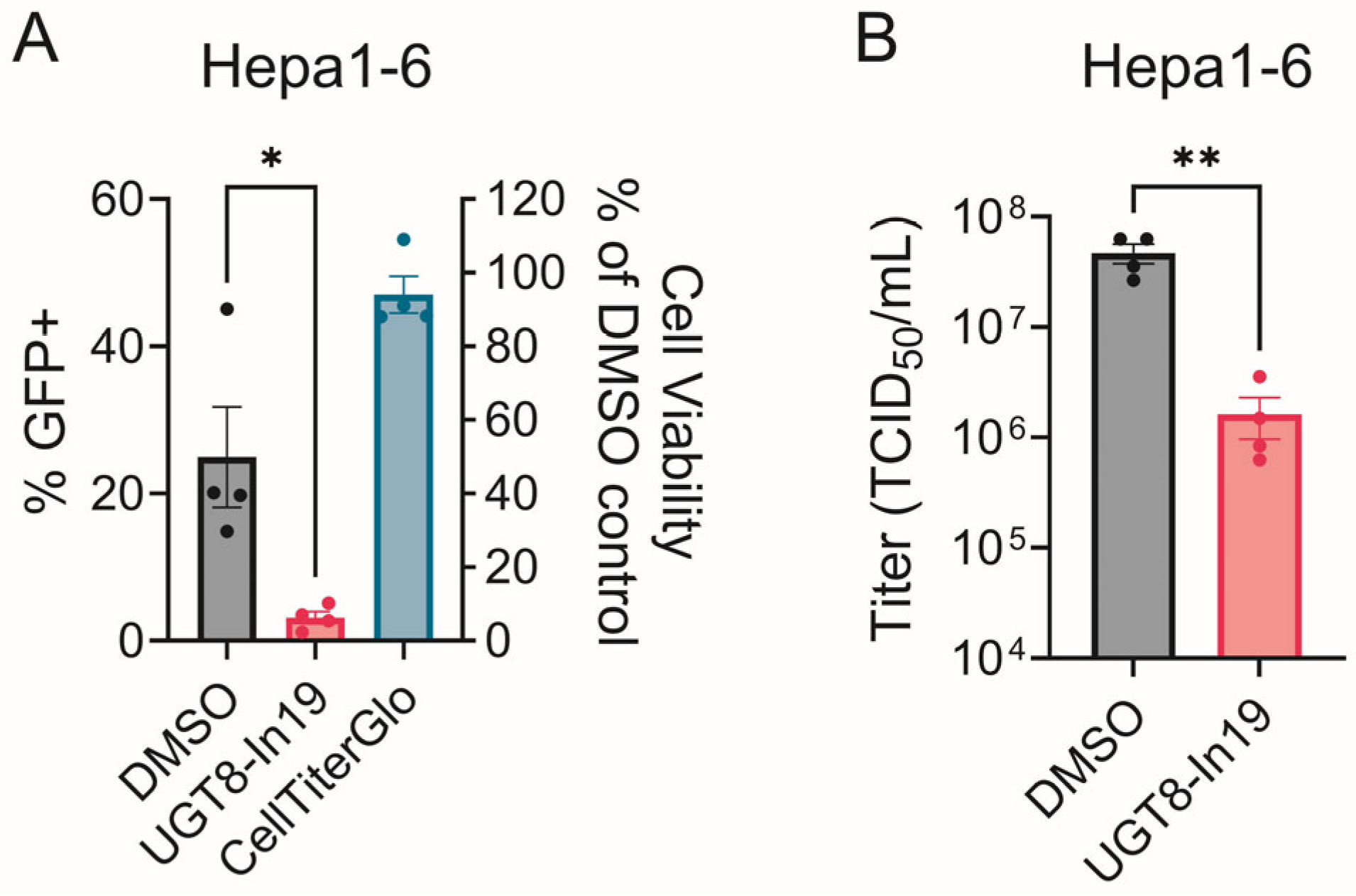
3.4. CHIKV Sensitivity to Galactosylceramide Modulation
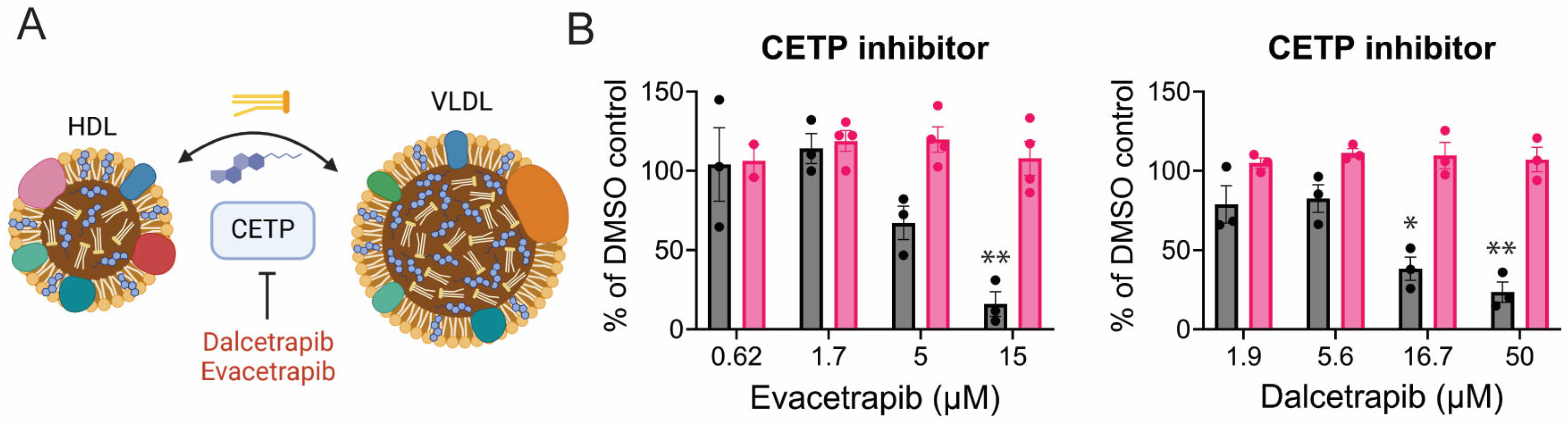
3.5. CHIKV Relies on Cholesterol Synthesis, Modification, and Transport
3.6. Mechanisms of Inhibition
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Weaver, S.C.; Lecuit, M. Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [PubMed]
- Rodriguez-Morales, A.J.; Cardona-Ospina, J.A.; Fernanda Urbano-Garzon, S.; Sebastian Hurtado-Zapata, J. Prevalence of Post-Chikungunya Infection Chronic Inflammatory Arthritis: A Systematic Review and Meta-Analysis. Arthritis Care Res. 2016, 68, 1849–1858. [Google Scholar]
- Serfaty, A.; Mendonça, S.; Canella, C.; Marchiori, E. Detection of musculoskeletal inflammatory lesions in patients with chronic chikungunya infection using 3T whole-body magnetic resonance imaging. Rev. Da Soc. Bras. De Med. Trop. 2024, 57, e004062024. [Google Scholar]
- Kee, A.C.; Yang, S.; Tambyah, P. Atypical chikungunya virus infections in immunocompromised patients. Emerg. Infect. Dis. 2010, 16, 1038–1040. [Google Scholar]
- Gogia, A.; B, S.; Kakar, A. CHIKUNGUNYA: A Mortality Report. Open Forum Infect. Dis. 2017, 4, S518. [Google Scholar]
- Leparc-Goffart, I.; Nougairede, A.; Cassadou, S.; Prat, C.; de Lamballerie, X. Chikungunya in the Americas. Lancet 2014, 383, 514. [Google Scholar] [CrossRef]
- Morens, D.M.; Fauci, A.S. Chikungunya at the door—Deja vu all over again? N. Engl. J. Med. 2014, 371, 885–887. [Google Scholar] [CrossRef]
- Feldstein, L.R.; Ellis, E.M.; Rowhani-Rahbar, A.; Hennessey, M.J.; Staples, J.E.; Halloran, M.E.; Weaver, M.R. Estimating the cost of illness and burden of disease associated with the 2014–2015 chikungunya outbreak in the U.S. Virgin Islands. PLoS Neglected Trop. Dis. 2019, 13, e0007563. [Google Scholar]
- Liu-Helmersson, J.; Brannstrom, A.; Sewe, M.O.; Semenza, J.C.; Rocklov, J. Estimating Past, Present, and Future Trends in the Global Distribution and Abundance of the Arbovirus Vector Aedes aegypti Under Climate Change Scenarios. Front. Public Health 2019, 7, 148. [Google Scholar]
- Ryan, S.J.; Carlson, C.J.; Mordecai, E.A.; Johnson, L.R. Global expansion and redistribution of Aedes-borne virus transmission risk with climate change. PLoS Neglected Trop. Dis. 2019, 13, e0007213. [Google Scholar]
- Chen, L.H.; Fritzer, A.; Hochreiter, R.; Dubischar, K.; Meyer, S. From bench to clinic: The development of VLA1553/IXCHIQ, a live-attenuated chikungunya vaccine. J. Travel Med. 2024, 31, taae123. [Google Scholar] [CrossRef] [PubMed]
- Balgoma, D.; Gil-de-Gomez, L.; Montero, O. Lipidomics Issues on Human Positive ssRNA Virus Infection: An Update. Metabolites 2020, 10, 356. [Google Scholar] [CrossRef] [PubMed]
- Harak, C.; Lohmann, V. Ultrastructure of the replication sites of positive-strand RNA viruses. Virology 2015, 479–480, 418–433. [Google Scholar] [CrossRef]
- Smit, J.M.; Bittman, R.; Wilschut, J. Low-pH-dependent fusion of Sindbis virus with receptor-free cholesterol- and sphingolipid-containing liposomes. J. Virol. 1999, 73, 8476–8484. [Google Scholar] [CrossRef]
- Kielian, M.; Chanel-Vos, C.; Liao, M. Alphavirus Entry and Membrane Fusion. Viruses 2010, 2, 796–825. [Google Scholar] [CrossRef]
- Wichit, S.; Hamel, R.; Bernard, E.; Talignani, L.; Diop, F.; Ferraris, P.; Liegeois, F.; Ekchariyawat, P.; Luplertlop, N.; Surasombatpattana, P.; et al. Imipramine Inhibits Chikungunya Virus Replication in Human Skin Fibroblasts through Interference with Intracellular Cholesterol Trafficking. Sci. Rep. 2017, 7, 3145. [Google Scholar] [CrossRef]
- Hoornweg, T.E.; van Duijl-Richter, M.K.S.; Ayala Nunez, N.V.; Albulescu, I.C.; van Hemert, M.J.; Smit, J.M. Dynamics of Chikungunya Virus Cell Entry Unraveled by Single-Virus Tracking in Living Cells. J. Virol. 2016, 90, 4745–4756. [Google Scholar] [CrossRef]
- Lorizate, M.; Krausslich, H.G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef]
- Angelini, M.M.; Neuman, B.W.; Buchmeier, M.J. Untangling membrane rearrangement in the nidovirales. DNA Cell Biol. 2014, 33, 122–127. [Google Scholar] [CrossRef]
- Strating, J.R.; van Kuppeveld, F.J. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr. Opin. Cell Biol. 2017, 47, 24–33. [Google Scholar] [CrossRef]
- Bakhache, W.; Neyret, A.; Bernard, E.; Merits, A.; Briant, L. Palmitoylated Cysteines in Chikungunya Virus nsP1 Are Critical for Targeting to Cholesterol-Rich Plasma Membrane Microdomains with Functional Consequences for Viral Genome Replication. J. Virol. 2020, 94, e02183-19. [Google Scholar] [PubMed]
- Frolova, E.I.; Gorchakov, R.; Pereboeva, L.; Atasheva, S.; Frolov, I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 2010, 84, 11679–11695. [Google Scholar] [PubMed]
- Gottipati, K.; Woodson, M.; Choi, K.H. Membrane binding and rearrangement by chikungunya virus capping enzyme nsP1. Virology 2020, 544, 31–41. [Google Scholar] [PubMed]
- Froshauer, S.; Kartenbeck, J.; Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol. 1988, 107, 2075–2086. [Google Scholar]
- Elmasri, Z.; Nasal, B.L.; Jose, J. Alphavirus-Induced Membrane Rearrangements during Replication, Assembly, and Budding. Pathogens 2021, 10, 984. [Google Scholar] [CrossRef]
- Jose, J.; Taylor, A.B.; Kuhn, R.J. Spatial and Temporal Analysis of Alphavirus Replication and Assembly in Mammalian and Mosquito Cells. mBio 2017, 8, e02294-16. [Google Scholar]
- Pietila, M.K.; Hellstrom, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar]
- Frolov, I.; Frolova, E.I. Molecular Virology of Chikungunya Virus. Curr. Top. Microbiol. Immunol. 2019, 435, 1–31. [Google Scholar]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef]
- Ketter, E.; Randall, G. Virus Impact on Lipids and Membranes. Annu. Rev. Virol. 2019, 6, 319–340. [Google Scholar] [CrossRef]
- Leier, H.C.; Weinstein, J.B.; Kyle, J.E.; Lee, J.Y.; Bramer, L.M.; Stratton, K.G.; Kempthorne, D.; Navratil, A.R.; Tafesse, E.G.; Hornemann, T.; et al. A global lipid map defines a network essential for Zika virus replication. Nat. Commun. 2020, 11, 3652. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.F.; de Oliveira, D.N.; Lima, E.O.; Guerreiro, T.M.; Esteves, C.Z.; Beck, R.M.; Padilla, M.A.; Milanez, G.P.; Arns, C.W.; Proenca-Modena, J.L.; et al. A Lipidomics Approach in the Characterization of Zika-Infected Mosquito Cells: Potential Targets for Breaking the Transmission Cycle. PLoS ONE 2016, 11, e0164377. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.A.; Vazquez, A.; Furiya, K.; Splattstoesser, M.K.; Bashmail, A.K.; Schwartz, H.; Russell, M.; Bhark, S.J.; Moreno, O.K.; McGovern, M.; et al. Rewiring of the Host Cell Metabolome and Lipidome during Lytic Gammaherpesvirus Infection Is Essential for Infectious-Virus Production. J. Virol. 2023, 97, e0050623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Chukkapalli, V.; Nchoutmboube, J.A.; Li, J.; Randall, G.; Belov, G.A.; Wang, X. Positive-strand RNA viruses stimulate host phosphatidylcholine synthesis at viral replication sites. Proc. Natl. Acad. Sci. USA 2016, 113, E1064–E1073. [Google Scholar] [CrossRef]
- Xu, K.; Nagy, P.D. RNA virus replication depends on enrichment of phosphatidylethanolamine at replication sites in subcellular membranes. Proc. Natl. Acad. Sci. USA 2015, 112, E1782–E1091. [Google Scholar] [CrossRef]
- Nguyen, A.; Guedan, A.; Mousnier, A.; Swieboda, D.; Zhang, Q.; Horkai, D.; Le Novere, N.; Solari, R.; Wakelam, M.J.O. Host lipidome analysis during rhinovirus replication in HBECs identifies potential therapeutic targets. J. Lipid Res. 2018, 59, 1671–1684. [Google Scholar] [CrossRef]
- Perera, R.; Riley, C.; Isaac, G.; Hopf-Jannasch, A.S.; Moore, R.J.; Weitz, K.W.; Pasa-Tolic, L.; Metz, T.O.; Adamec, J.; Kuhn, R.J. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 2012, 8, e1002584. [Google Scholar] [CrossRef]
- Hofmann, S.; Krajewski, M.; Scherer, C.; Scholz, V.; Mordhorst, V.; Truschow, P.; Schobel, A.; Reimer, R.; Schwudke, D.; Herker, E. Complex lipid metabolic remodeling is required for efficient hepatitis C virus replication. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1041–1056. [Google Scholar] [CrossRef]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef]
- Yan, B.; Chu, H.; Yang, D.; Sze, K.H.; Lai, P.M.; Yuan, S.; Shuai, H.; Wang, Y.; Kao, R.Y.; Chan, J.F.; et al. Characterization of the Lipidomic Profile of Human Coronavirus-Infected Cells: Implications for Lipid Metabolism Remodeling upon Coronavirus Replication. Viruses 2019, 11, 73. [Google Scholar] [CrossRef]
- Morita, M.; Kuba, K.; Ichikawa, A.; Nakayama, M.; Katahira, J.; Iwamoto, R.; Watanebe, T.; Sakabe, S.; Daidoji, T.; Nakamura, S.; et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell 2013, 153, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Woods, P.S.; Doolittle, L.M.; Rosas, L.E.; Joseph, L.M.; Calomeni, E.P.; Davis, I.C. Lethal H1N1 influenza A virus infection alters the murine alveolar type II cell surfactant lipidome. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L1160–L1169. [Google Scholar] [CrossRef] [PubMed]
- Havranek, K.E.; Reyes Ballista, J.M.; Hines, K.M.; Brindley, M.A. Untargeted Lipidomics of Vesicular Stomatitis Virus-Infected Cells and Viral Particles. Viruses 2021, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Brugger, B.; Glass, B.; Haberkant, P.; Leibrecht, I.; Wieland, F.T.; Krausslich, H.G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646. [Google Scholar] [CrossRef]
- Hines, K.M.; Herron, J.; Xu, L. Assessment of altered lipid homeostasis by HILIC-ion mobility-mass spectrometry-based lipidomics. J. Lipid Res. 2017, 58, 809–819. [Google Scholar]
- Ono, N.; Tatsuo, H.; Hidaka, Y.; Aoki, T.; Minagawa, H.; Yanagi, Y. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J. Virol. 2001, 75, 4399–4401. [Google Scholar]
- Acciani, M.D.; Lay Mendoza, M.F.; Havranek, K.E.; Duncan, A.M.; Iyer, H.; Linn, O.L.; Brindley, M.A. Ebola virus requires phosphatidylserine scrambling activity for efficient budding and optimal infectivity. J. Virol. 2021, 95, JVI0116521. [Google Scholar]
- Reyes Ballista, J.M.; Miazgowicz, K.L.; Acciani, M.D.; Jimenez, A.R.; Belloli, R.S.; Havranek, K.E.; Brindley, M.A. Chikungunya virus entry and infectivity is primarily facilitated through cell line dependent attachment factors in mammalian and mosquito cells. Front. Cell Dev. Biol. 2023, 11, 1085913. [Google Scholar]
- Bligh, E.G. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 39, 911–917. [Google Scholar]
- Hines, K.M.; Waalkes, A.; Penewit, K.; Holmes, E.A.; Salipante, S.J.; Werth, B.J.; Xu, L. Characterization of the Mechanisms of Daptomycin Resistance among Gram-Positive Bacterial Pathogens by Multidimensional Lipidomics. mSphere 2017, 2, e00492-17. [Google Scholar]
- Bimpeh, K.; Hines, K.M. A rapid single-phase extraction for polar staphylococcal lipids. Anal. Bioanal. Chem. 2023, 415, 4591–4602. [Google Scholar] [PubMed]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.Y.; Terada, T. Enzymatic measurement of phosphatidylglycerol and cardiolipin in cultured cells and mitochondria. Sci. Rep. 2015, 5, 11737. [Google Scholar]
- Moyano, A.L.; Li, G.; Lopez-Rosas, A.; Mansson, J.E.; van Breemen, R.B.; Givogri, M.I. Distribution of C16:0, C18:0, C24:1, and C24:0 sulfatides in central nervous system lipid rafts by quantitative ultra-high-pressure liquid chromatography tandem mass spectrometry. Anal. Biochem. 2014, 467, 31–39. [Google Scholar]
- Lunghi, G.; Fazzari, M.; Di Biase, E.; Mauri, L.; Chiricozzi, E.; Sonnino, S. The structure of gangliosides hides a code for determining neuronal functions. FEBS Open Biol. 2021, 11, 3193–3200. [Google Scholar]
- Sonnino, S.; Mauri, L.; Chigorno, V.; Prinetti, A. Gangliosides as components of lipid membrane domains. Glycobiology 2007, 17, 1R–13R. [Google Scholar]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar]
- Roingeard, P.; Eymieux, S.; Burlaud-Gaillard, J.; Hourioux, C.; Patient, R.; Blanchard, E. The double-membrane vesicle (DMV): A virus-induced organelle dedicated to the replication of SARS-CoV-2 and other positive-sense single-stranded RNA viruses. Cell Mol. Life Sci. 2022, 79, 425. [Google Scholar]
- Laurent, T.; Kumar, P.; Liese, S.; Zare, F.; Jonasson, M.; Carlson, A.; Carlson, L.A. Architecture of the chikungunya virus replication organelle. elife 2022, 11, e83042. [Google Scholar]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.-S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Q.; Li, Z.W.; Feng, Y.F.; Yang, H.Q.; Hou, C.L.; Geng, C.; Yang, P.R.; Zhao, H.M.; Wang, J. MK2206 attenuates atherosclerosis by inhibiting lipid accumulation, cell migration, proliferation, and inflammation. Acta Pharmacol. Sin. 2022, 43, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Bjune, K.; Sundvold, H.; Leren, T.P.; Naderi, S. MK-2206, an allosteric inhibitor of AKT, stimulates LDLR expression and LDL uptake: A potential hypocholesterolemic agent. Atherosclerosis 2018, 276, 28–38. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11, 584303. [Google Scholar] [CrossRef]
- Collins, J.L.; Fivush, A.M.; Watson, M.A.; Galardi, C.M.; Lewis, M.C.; Moore, L.B.; Parks, D.J.; Wilson, J.G.; Tippin, T.K.; Binz, J.G.; et al. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J. Med. Chem. 2002, 45, 1963–1966. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, H.; Zhang, L. Fatty Acid Synthase Promotes the Palmitoylation of Chikungunya Virus nsP1. J. Virol. 2019, 93, e01747-18. [Google Scholar] [CrossRef]
- Bakhache, W.; Neyret, A.; McKellar, J.; Clop, C.; Bernard, E.; Weger-Lucarelli, J.; Briant, L. Fatty acid synthase and stearoyl-CoA desaturase-1 are conserved druggable cofactors of Old World Alphavirus genome replication. Antivir. Res. 2019, 172, 104642. [Google Scholar] [CrossRef]
- Tan, M.; Mosaoa, R.; Graham, G.T.; Kasprzyk-Pawelec, A.; Gadre, S.; Parasido, E.; Catalina-Rodriguez, O.; Foley, P.; Giaccone, G.; Cheema, A.; et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. 2020, 27, 2143–2157. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Filippov, S.; Srivastava, R.A.K.; Hanselman, J.C.; Bradshaw, C.D.; Hurley, T.R.; Cramer, C.T.; Spahr, M.A.; Brant, A.F.; Houghton, J.L.; et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J. Lipid Res. 2013, 54, 134–151. [Google Scholar] [CrossRef]
- Pearce, N.J.; Yates, J.W.; Berkhout, T.A.; Jackson, B.; Tew, D.; Boyd, H.; Camilleri, P.; Sweeney, P.; Gribble, A.D.; Shaw, A.; et al. The role of ATP citrate-lyase in the metabolic regulation of plasma lipids. Biochem. J. 1998, 334, 113–119. [Google Scholar] [PubMed]
- Angin, Y.; Beauloye, C.; Horman, S.; Bertrand, L. Regulation of Carbohydrate Metabolism, Lipid Metabolism, and Protein Metabolism by AMPK. In AMP-Activated Protein Kinase; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; pp. 23–43. [Google Scholar]
- Oh, J.E.; Jung, B.H.; Park, J.; Kang, S.; Lee, H. Deciphering Fatty Acid Synthase Inhibition-Triggered Metabolic Flexibility in Prostate Cancer Cells through Untargeted Metabolomics. Cells 2020, 9, 2447. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Mohseni, R.; Lucas, K.J.; Gutierrez, J.A.; Perry, R.G.; Trotter, J.F.; Rahimi, R.S.; Harrison, S.A.; Ajmera, V.; Wayne, J.D.; et al. TVB-2640 (FASN Inhibitor) for the Treatment of Nonalcoholic Steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial. Gastroenterology 2021, 161, 1475–1486. [Google Scholar] [CrossRef]
- Mishra, S.; Shelke, V.; Gaikwad, A.B. Acyl-CoA Synthetase Long-Chain Isoenzymes in Kidney Diseases: Mechanistic Insights and Therapeutic Implications. Cell Biochem. Funct. 2024, 42, e4114. [Google Scholar]
- Castillo, A.F.; Orlando, U.D.; Maloberti, P.M.; Prada, J.G.; Dattilo, M.A.; Solano, A.R.; Bigi, M.M.; Medrano, M.A.R.; Torres, M.T.; Indo, S.; et al. New inhibitor targeting Acyl-CoA synthetase 4 reduces breast and prostate tumor growth, therapeutic resistance and steroidogenesis. Cell. Mol. Life Sci. 2020, 78, 2893–2910. [Google Scholar] [CrossRef]
- Bu, S.Y.; Mashek, M.T.; Mashek, D.G. Suppression of long chain acyl-CoA synthetase 3 decreases hepatic de novo fatty acid synthesis through decreased transcriptional activity. J. Biol. Chem. 2009, 284, 30474–30483. [Google Scholar]
- Kan, C.F.K.; Singh, A.B.; Stafforini, D.M.; Azhar, S.; Liu, J. Arachidonic acid downregulates acyl-CoA synthetase 4 expression by promoting its ubiquitination and proteasomal degradation. J. Lipid Res. 2014, 55, 1657–1667. [Google Scholar]
- Kuwata, H.; Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019, 144, 106363. [Google Scholar] [CrossRef]
- Lee, H.; Gan, B. Ferroptosis execution: Is it all about ACSL4? Cell Chem. Biol. 2022, 29, 1363–1365. [Google Scholar] [CrossRef]
- Sauer, G.; Amtmann, E.; Melber, K.; Knapp, A.; Müller, K.; Hummel, K.; Scherm, A. DNA and RNA virus species are inhibited by xanthates, a class of antiviral compounds with unique properties. Proc. Natl. Acad. Sci. USA 1984, 81, 3263–3267. [Google Scholar]
- Amtmann, E.; Hummel, K.; Sauer, G. The Therapeutic Efficacy of a Xanthate Compound on Herpes Simplex Virus in Skin Lesions of Mice and Guinea-pigs. J. Gen. Virol. 1985, 66, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Tritsch, S.R.; Kota, K.; Chiang, C.Y.; Dong, L.; Kenny, T.; Brueggemann, E.E.; Ward, M.D.; Cazares, L.H.; Bavari, S. Sphingosine kinase 2 is a chikungunya virus host factor co-localized with the viral replication complex. Emerg. Microbes Infect. 2015, 4, e61. [Google Scholar] [CrossRef] [PubMed]
- Ershov, P.; Kaluzhskiy, L.; Mezentsev, Y.; Yablokov, E.; Gnedenko, O.; Ivanov, A. Enzymes in the Cholesterol Synthesis Pathway: Interactomics in the Cancer Context. Biomedicines 2021, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Chandoga, J.; Hampl, L.; Turecky, L.; Rojekova, I.; Uhlikova, E.; Hocman, G. Cetaben is an exceptional type of peroxisome proliferator. Int. J. Biochem. 1994, 26, 679–696. [Google Scholar]
- Kovacs, W.J.; Schrader, M.; Walter, I.; Stangl, H. The hypolipidemic compound cetaben induces changes in Golgi morphology and vesicle movement. Histochem. Cell Biol. 2004, 122, 95–109. [Google Scholar]
- Nicholls, S.J.; Bubb, K. The mystery of evacetrapib—Why are CETP inhibitors failing? Expert Rev. Cardiovasc. Ther. 2020, 18, 127–130. [Google Scholar]
- Ribeiro, Y.P.; Falcão, L.F.M.; Smith, V.C.; de Sousa, J.R.; Pagliari, C.; Franco, E.C.S.; Cruz, A.C.R.; Chiang, J.O.; Martins, L.C.; Nunes, J.A.L.; et al. Comparative Analysis of Human Hepatic Lesions in Dengue, Yellow Fever, and Chikungunya: Revisiting Histopathological Changes in the Light of Modern Knowledge of Cell Pathology. Pathogens 2023, 12, 680. [Google Scholar] [CrossRef]
- Schwartz, O.; Albert, M.L. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 2010, 8, 491–500. [Google Scholar]
- Couderc, T.; Chrétien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A Mouse Model for Chikungunya: Young Age and Inefficient Type-I Interferon Signaling Are Risk Factors for Severe Disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef]
- Tam, V.C.; Quehenberger, O.; Oshansky, C.M.; Suen, R.; Armando, A.M.; Treuting, P.M.; Thomas, P.G.; Dennis, E.A.; Aderem, A. Lipidomic Profiling of Influenza Infection Identifies Mediators that Induce and Resolve Inflammation. Cell 2013, 154, 213–227. [Google Scholar] [CrossRef]
- Girard, J.; Le Bihan, O.; Lai-Kee-Him, J.; Girleanu, M.; Bernard, E.; Castellarin, C.; Chee, M.; Neyret, A.; Spehner, D.; Holy, X.; et al. In situ fate of Chikungunya virus replication organelles. J. Virol. 2024, 98, e0036824. [Google Scholar] [CrossRef] [PubMed]
- Aiqui-Reboul-Paviet, O.; Bakhache, W.; Bernard, E.; Holsteyn, L.; Neyret, A.; Briant, L. The Rac1-PAK1-Arp2/3 signaling axis regulates CHIKV nsP1-induced filopodia and optimal viral genome replication. J. Virol. 2024, 98, e0061224. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef]
- Shaimardanova, A.A.; Solovyeva, V.V.; Issa, S.S.; Rizvanov, A.A. Gene Therapy of Sphingolipid Metabolic Disorders. Int. J. Mol. Sci. 2023, 24, 3627. [Google Scholar] [CrossRef]
- Braunstein, H.; Maor, G.; Chicco, G.; Filocamo, M.; Zimran, A.; Horowitz, M. UPR activation and CHOP mediated induction of GBA1 transcription in Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 21–29. [Google Scholar] [CrossRef]
- Reich, S.; Nguyen, C.D.L.; Has, C.; Steltgens, S.; Soni, H.; Coman, C.; Freyberg, M.; Bichler, A.; Seifert, N.; Conrad, D.; et al. A multi-omics analysis reveals the unfolded protein response regulon and stress-induced resistance to folate-based antimetabolites. Nat. Commun. 2020, 11, 2936. [Google Scholar] [CrossRef]
- Zhou, X.; Lee, Y.-K.; Li, X.; Kim, H.; Sanchez-Priego, C.; Han, X.; Tan, H.; Zhou, S.; Fu, Y.; Purtell, K.; et al. Integrated proteomics reveals autophagy landscape and an autophagy receptor controlling PKA-RI complex homeostasis in neurons. Nat. Commun. 2024, 15, 3113. [Google Scholar] [CrossRef]
- Krejbich-Trotot, P.; Gay, B.; Li-Pat-Yuen, G.; Hoarau, J.-J.; Jaffar-Bandjee, M.-C.; Briant, L.; Gasque, P.; Denizot, M. Chikungunya triggers an autophagic process which promotes viral replication. Virol. J. 2011, 8, 432. [Google Scholar] [CrossRef]
- Joubert, P.-E.; Werneke, S.W.; De La Calle, C.; Guivel-Benhassine, F.; Giodini, A.; Peduto, L.; Levine, B.; Schwartz, O.; Lenschow, D.J.; Albert, M.L. Chikungunya virus–induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012, 209, 1029–1047. [Google Scholar] [CrossRef]
- Khongwichit, S.; Wikan, N.; Abere, B.; Thepparit, C.; Kuadkitkan, A.; Ubol, S.; Smith, D.R. Cell-type specific variation in the induction of ER stress and downstream events in chikungunya virus infection. Microb. Pathog. 2016, 101, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.P.S.; Ng, M.-L.; Vasudevan, S.G. Differential unfolded protein response during Chikungunya and Sindbis virus infection: CHIKV nsP4 suppresses eIF2α phosphorylation. Virol. J. 2013, 10, 36. [Google Scholar] [PubMed]
- Fros, J.J.; Major, L.D.; Scholte, F.E.M.; Gardner, J.; Van Hemert, M.J.; Suhrbier, A.; Pijlman, G.P. Chikungunya virus non-structural protein 2-mediated host shut-off disables the unfolded protein response. J. Gen. Virol. 2015, 96, 580–589. [Google Scholar]
- Oyewole, O.O.; Dunnavant, K.; Bhattarai, S.; Kharel, Y.; Lynch, K.R.; Santos, W.L.; Reid, S.P. A Novel Sphingosine Kinase Inhibitor Suppresses Chikungunya Virus Infection. Viruses 2022, 14, 1123. [Google Scholar] [CrossRef]
- Prell, A.; Wigger, D.; Huwiler, A.; Schumacher, F.; Kleuser, B. The sphingosine kinase 2 inhibitors ABC294640 and K145 elevate (dihydro)sphingosine 1-phosphate levels in various cells. J. Lipid Res. 2024, 65, 100631. [Google Scholar]
- Takahashi, T.; Suzuki, T. Role of sulfatide in normal and pathological cells and tissues. J. Lipid Res. 2012, 53, 1437–1450. [Google Scholar]
- Honke, K. Enzyme assay of cerebroside sulfotransferase. In Glycoscience Protocols (GlycoPODv2); Nishihara, S., Angata, K., Aoki-Kinoshita, K.F., Hirabayashi, J., Eds.; Japan Consortium for Glycobiology and Glycotechnology: Saitama, Japan, 2021. [Google Scholar]
- Ohba, Y.; Motohashi, M.; Arita, M. Characterization of UGT8 as a monogalactosyl diacylglycerol synthase in mammals. J. Biochem. 2024, 177, 141–152. [Google Scholar]
- Hall, A.; Róg, T.; Karttunen, M.; Vattulainen, I. Role of Glycolipids in Lipid Rafts: A View through Atomistic Molecular Dynamics Simulations with Galactosylceramide. J. Phys. Chem. B 2010, 114, 7797–7807. [Google Scholar]
- Forte, T.M.; Bell-Quint, J.J.; Cheng, F. Lipoproteins of fetal and newborn calves and adult steer: A study of developmental changes. Lipids 1981, 16, 240–245. [Google Scholar]
- Jose, J.; Hoque, M.; Engel, J.; Beevi, S.S.; Wahba, M.; Georgieva, M.I.; Murphy, K.J.; Hughes, W.E.; Cochran, B.J.; Lu, A.; et al. Annexin A6 and NPC1 regulate LDL-inducible cell migration and distribution of focal adhesions. Sci. Rep. 2022, 12, 596. [Google Scholar]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Beyer, T.P.; Zhang, Y.; Schmidt, R.J.; Chen, Y.Q.; Cockerham, S.L.; Zimmerman, K.M.; Karathanasis, S.K.; Cannady, E.A.; Fields, T.; et al. Evacetrapib is a novel, potent, and selective inhibitor of cholesteryl ester transfer protein that elevates HDL cholesterol without inducing aldosterone or increasing blood pressure. J. Lipid Res. 2011, 52, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Funnell, S.G.P.; Afrough, B.; Baczenas, J.J.; Berry, N.; Bewley, K.R.; Bradford, R.; Florence, C.; Le Duff, Y.; Lewis, M.; Moriarty, R.V.; et al. A cautionary perspective regarding the isolation and serial propagation of SARS-CoV-2 in Vero cells. npj Vaccines 2021, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Konishi, K.; Yamaji, T.; Sakuma, C.; Kasai, F.; Endo, T.; Kohara, A.; Hanada, K.; Osada, N. Whole-Genome Sequencing of Vero E6 (VERO C1008) and Comparative Analysis of Four Vero Cell Sublines. Front. Genet. 2022, 13, 801382. [Google Scholar] [CrossRef]
- Mercado, M.; Acosta-Reyes, J.; Parra, E.; Guzman, L.; Beltran, M.; Gasque, P.; Mejia-Garcia, C.; Viasus, D. Renal involvement in fatal cases of chikungunya virus infection. J. Clin. Virol. 2018, 103, 16–18. [Google Scholar] [CrossRef]
- Mosca, J.D.; Pitha, P.M. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell Biol. 1986, 6, 2279–2283. [Google Scholar]
- Her, Z.; Malleret, B.; Chan, M.; Ong, E.K.; Wong, S.C.; Kwek, D.J.; Tolou, H.; Lin, R.T.; Tambyah, P.A.; Renia, L.; et al. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J. Immunol. 2010, 184, 5903–5913. [Google Scholar]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef]
- Elliott, K.; Caicedo, P.A.; Haunerland, N.H.; Lowenberger, C. Profiling lipidomic changes in dengue-resistant and dengue-susceptible strains of Colombian Aedes aegypti after dengue virus challenge. PLoS Neglected Trop. Dis. 2023, 17, e0011676. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; McGee, C.E.; Higgs, S. Chikungunya virus adaptation to Aedes albopictus mosquitoes does not correlate with acquisition of cholesterol dependence or decreased pH threshold for fusion reaction. Virol. J. 2011, 8, 376. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Lupien, L.E.; Dunkley, E.M.; Maloy, M.J.; Lehner, I.B.; Foisey, M.G.; Ouellette, M.E.; Lewis, L.D.; Pooler, D.B.; Kinlaw, W.B.; Baures, P.W. An Inhibitor of Fatty Acid Synthase Thioesterase Domain with Improved Cytotoxicity against Breast Cancer Cells and Stability in Plasma. J. Pharmacol. Exp. Ther. 2019, 371, 171–185. [Google Scholar] [PubMed]
- Choi, J.; Smith, D.M.; Lee, Y.J.; Cai, D.; Hossain, M.J.; O’Connor, T.J.; Deme, P.; Haughey, N.J.; Scafidi, S.; Riddle, R.C.; et al. Etomoxir repurposed as a promiscuous fatty acid mimetic chemoproteomic probe. iScience 2024, 27, 110642. [Google Scholar] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noble, J.T.; Bimpeh, K.; Pisciotta, M.A.; Reyes Ballista, J.M.; Hines, K.M.; Brindley, M.A. Chikungunya Replication and Infection Is Dependent upon and Alters Cellular Hexosylceramide Levels in Vero Cells. Viruses 2025, 17, 509. https://doi.org/10.3390/v17040509
Noble JT, Bimpeh K, Pisciotta MA, Reyes Ballista JM, Hines KM, Brindley MA. Chikungunya Replication and Infection Is Dependent upon and Alters Cellular Hexosylceramide Levels in Vero Cells. Viruses. 2025; 17(4):509. https://doi.org/10.3390/v17040509
Chicago/Turabian StyleNoble, Joseph Thomas, Kingsley Bimpeh, Michael Anthony Pisciotta, Judith Mary Reyes Ballista, Kelly Marie Hines, and Melinda Ann Brindley. 2025. "Chikungunya Replication and Infection Is Dependent upon and Alters Cellular Hexosylceramide Levels in Vero Cells" Viruses 17, no. 4: 509. https://doi.org/10.3390/v17040509
APA StyleNoble, J. T., Bimpeh, K., Pisciotta, M. A., Reyes Ballista, J. M., Hines, K. M., & Brindley, M. A. (2025). Chikungunya Replication and Infection Is Dependent upon and Alters Cellular Hexosylceramide Levels in Vero Cells. Viruses, 17(4), 509. https://doi.org/10.3390/v17040509