
Phylodynamic of Tomato Brown Rugose Fruit Virus and Tomato Chlorosis Virus, Two Emergent Viruses in Mixed Infections in Argentina

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Collection and High-Throughput Sequencing
2.2. ToBRFV and ToCV Complete Genome Reconstruction from Leaves Samples
2.3. Environmental Water Sample Collection, Processing, and Viral RNA Extraction
2.4. ToBRFV Genome Reconstruction from Raw Sewage Samples
2.5. Phylodynamic Analysis of ToBRFV
2.6. Phylogenetic and Phylodynamic Analysis of ToCV
2.7. Intra-Sample Diversity in Argentine ToBRFV Sequences
3. Results
3.1. Viral Complete Genome Sequences Through Metagenomics Analysis
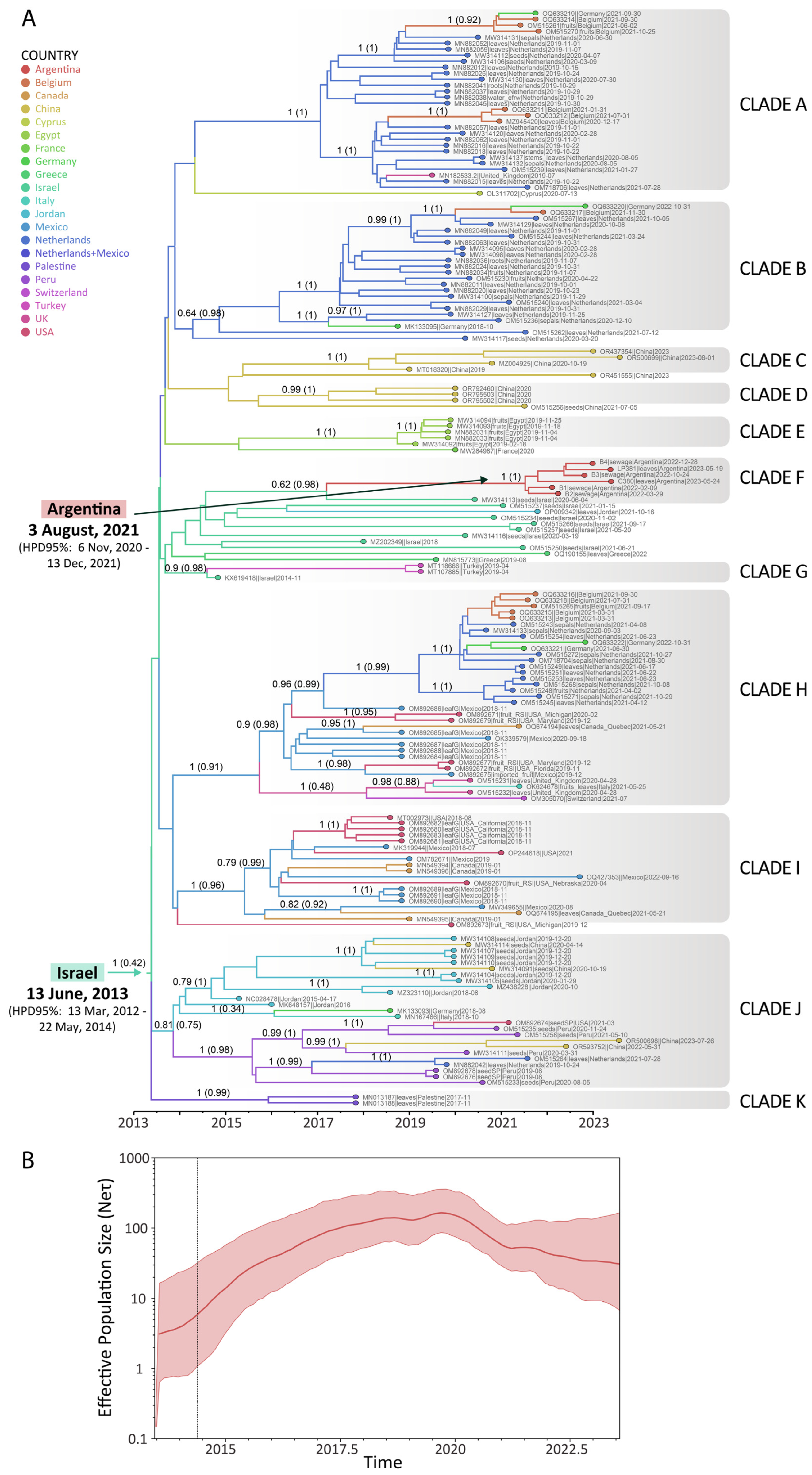
3.2. Spatial and Temporal Dynamics of ToBRFV
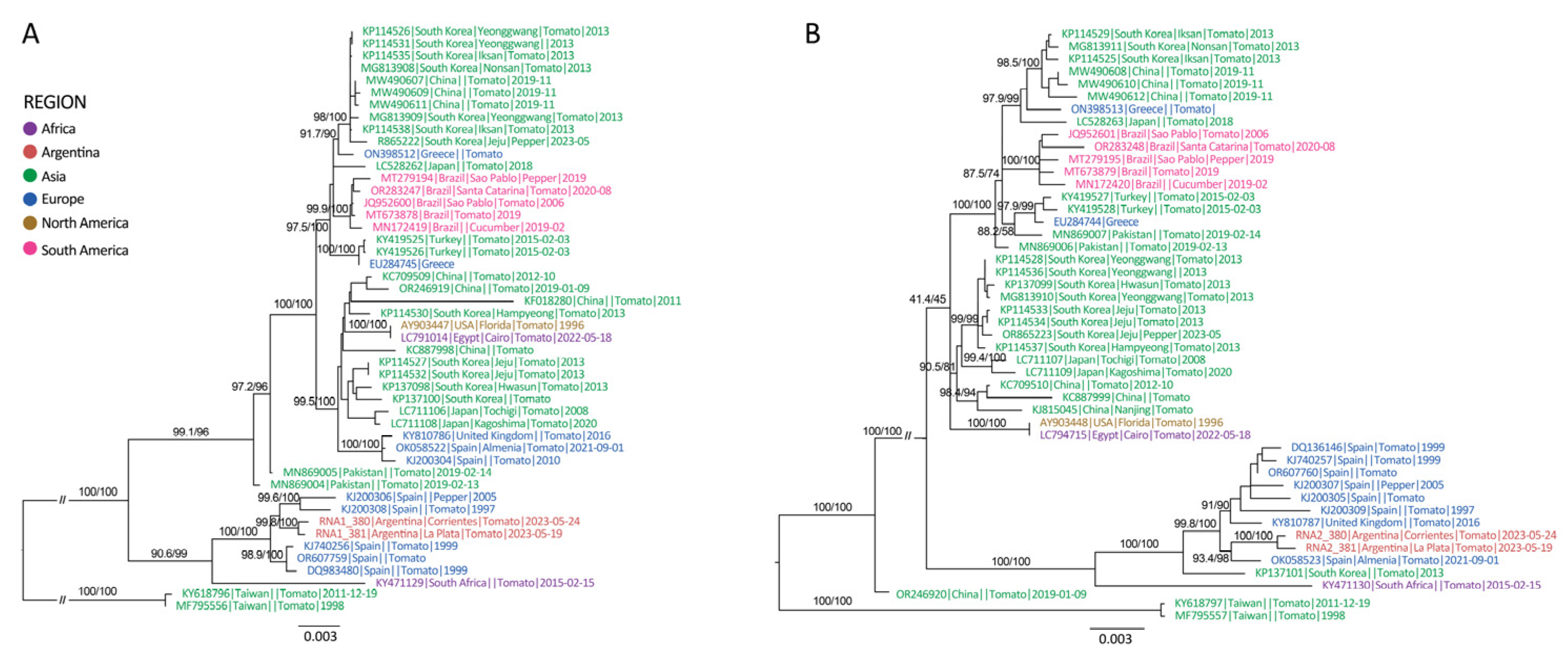
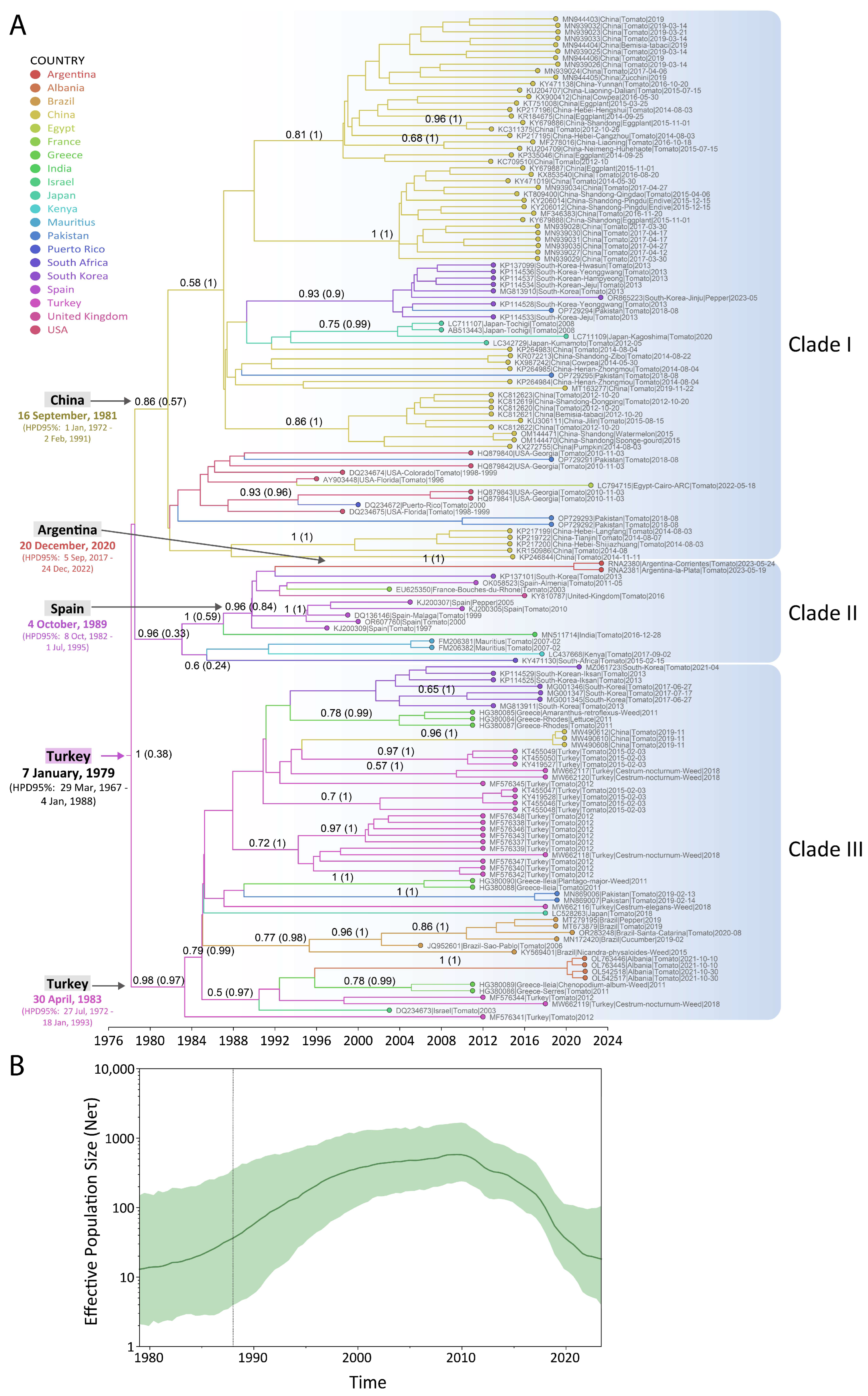
3.3. Spatial and Temporal Dynamics of ToCV
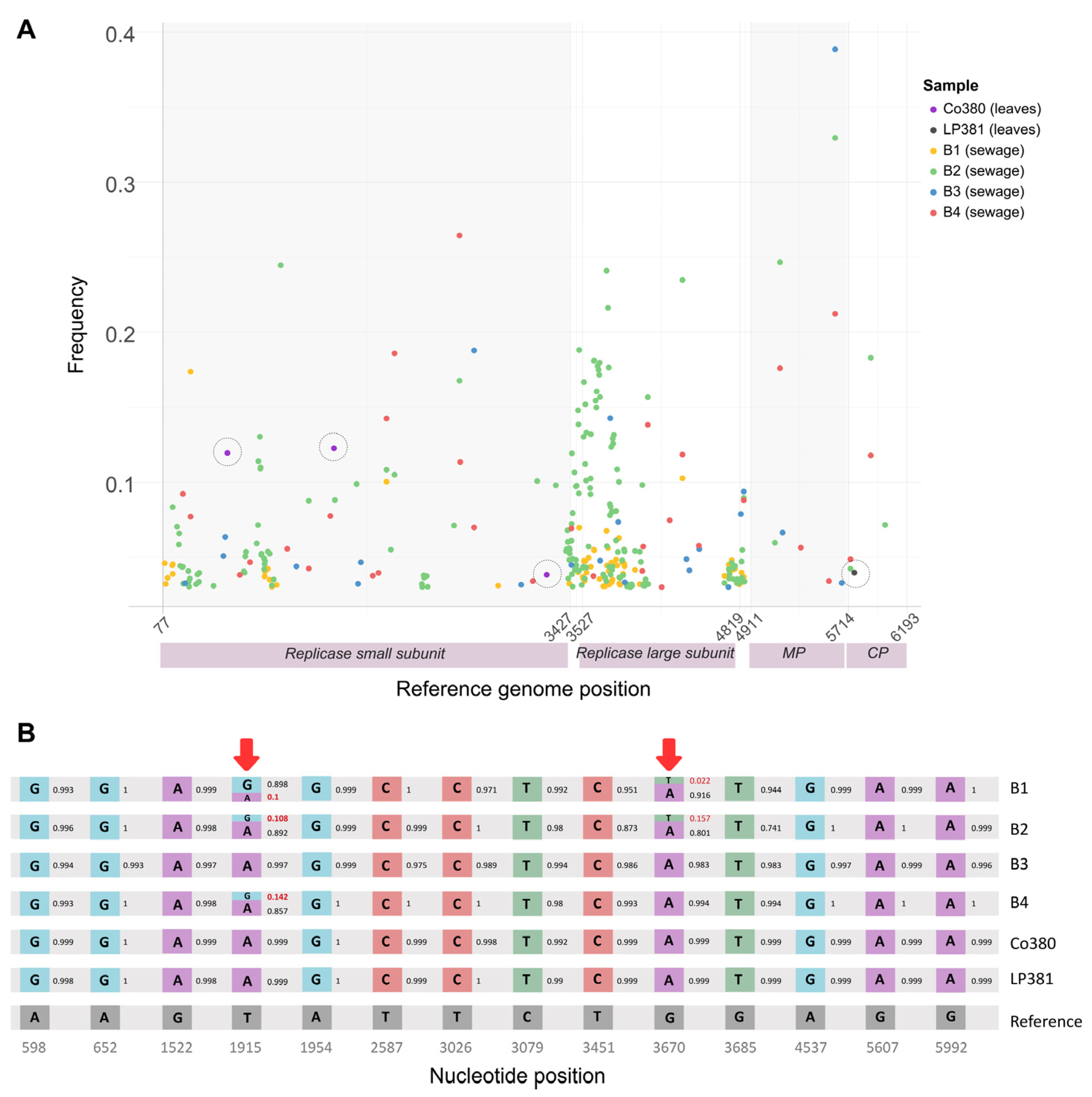
3.4. Intra-Sample Diversity in Argentine ToBRFV Sequences
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luria, N.; Smith, E.; Reingold, V.; Bekelman, I.; Lapidot, M.; Levin, I.; Elad, N.; Tam, Y.; Sela, N.; Abu-Ras, A.; et al. A new israeli tobamovirus isolate infects tomato plants harboring Tm-22 resistance genes. PLoS ONE 2017, 12, e0170429. [Google Scholar] [CrossRef] [PubMed]
- Salem, N.; Mansour, A.; Ciuffo, M.; Falk, B.W.; Turina, M. A new tobamovirus infecting tomato crops in Jordan. Arch. Virol. 2016, 161, 503–506. [Google Scholar] [CrossRef]
- Obregón, V.G.; Ibañez, J.M.; Lattar, T.E.; Juszczak, S.; Groth-Helms, D. First report of tomato brown rugose fruit virus in tomato in Argentina. New Dis. Rep. 2023, 48, 2–3. [Google Scholar] [CrossRef]
- Salem, N.M.; Jewehan, A.; Aranda, M.A.; Fox, A. Tomato brown rugose fruit virus pandemic. Annu. Rev. Phytopathol. 2023, 61, 137–164. [Google Scholar] [CrossRef] [PubMed]
- Molad, O.; Smith, E.; Luria, N.; Bakelman, E.; Lachman, O.; Reches, M.; Dombrovsky, A. Studying tomato brown rugose fruit virus longevity in soil and virion susceptibility to pH treatments helped improve virus control by soil disinfection. Plant Soil. 2024, 505, 543–558. [Google Scholar] [CrossRef]
- Mehle, N.; Bačnik, K.; Bajde, I.; Brodarič, J.; Fox, A.; Gutiérrez-Aguirre, I.; Kitek, M.; Kutnjak, D.; Loh, Y.L.; Maksimović Carvalho Ferreira, O.; et al. Tomato brown rugose fruit virus in aqueous environments–Survival and significance of water-mediated transmission. Front. Plant Sci. 2023, 14, 1187920. [Google Scholar] [CrossRef]
- Skelton, A.; Frew, L.; Ward, R.; Hodgson, R.; Forde, S.; McDonough, S.; Webster, G.; Chisnall, K.; Mynett, M.; Buxton-Kirk, A.; et al. Tomato brown rugose fruit virus: Survival and disinfection efficacy on common glasshouse surfaces. Viruses 2023, 15, 2076. [Google Scholar] [CrossRef]
- Davino, S.; Caruso, A.G.; Bertacca, S.; Barone, S.; Panno, S. Tomato brown rugose fruit virus: Seed transmission rate and efficacy of different seed disinfection treatments. Plants 2020, 9, 1615. [Google Scholar] [CrossRef]
- Salem, N.M.; Sulaiman, A.; Samarah, N.; Turina, M.; Vallino, M. Localization and mechanical transmission of tomato brown rugose fruit virus in tomato seeds. Plant Dis. 2022, 106, 275–281. [Google Scholar] [CrossRef]
- Cultrona, M.; Bonini, N.; Pacifico, D.; Tessitori, M. First Report of convolvulus arvensis and polycarpon tetraphyllum as natural hosts of tomato brown rugose fruit virus. Plant Dis. 2024, 108, 827. [Google Scholar] [CrossRef]
- Salem, N.M.; Abumuslem, M.; Turina, M.; Samarah, N.; Sulaiman, A.; Abu-Irmaileh, B.; Ata, Y. New weed hosts for tomato brown rugose fruit virus in wild mediterranean vegetation. Plants 2022, 11, 2287. [Google Scholar] [CrossRef]
- Chanda, B.; Gilliard, A.; Jaiswal, N.; Ling, K.-S. Comparative analysis of host range, ability to infect tomato cultivars with tm-2 2 gene, and real-time reverse transcription pcr detection of tomato brown rugose fruit virus. Plant Dis. 2021, 105, 3643–3652. [Google Scholar] [CrossRef]
- Zhang, S.; Griffiths, J.S.; Marchand, G.; Bernards, M.A.; Wang, A. Tomato brown rugose fruit virus: An emerging and rapidly spreading plant RNA virus that threatens tomato production worldwide. Mol. Plant Pathol. 2022, 23, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Caruso, A.G.; Tortorici, S.; Davino, S.; Bertacca, S.; Ragona, A.; Lo Verde, G.; Biondi, A.; Noris, E.; Rizzo, R.; Panno, S. The invasive tomato pest tuta absoluta can transmit the emergent tomato brown rugose fruit virus. Entomol. Gen. 2024, 44, 289–296. [Google Scholar] [CrossRef]
- Levitzky, N.; Smith, E.; Lachman, O.; Luria, N.; Mizrahi, Y.; Bakelman, H.; Sela, N.; Laskar, O.; Milrot, E.; Dombrovsky, A. The bumblebee bombus terrestris carries a primary inoculum of tomato brown rugose fruit virus contributing to disease spread in tomatoes. PLoS ONE 2019, 14, e0210871. [Google Scholar] [CrossRef]
- Wisler, G.C.; Li, R.H.; Liu, H.-Y.; Lowry, D.S.; Duffus, J.E. Tomato chlorosis virus: A new whitefly-transmitted, phloem-limited, bipartite closterovirus of tomato. Phytopathology 1998, 88, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Fiallo-Olivé, E.; Navas-Castillo, J. Tomato chlorosis virus, an emergent plant virus still expanding its geographical and host ranges. Mol. Plant Pathol. 2019, 20, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Fiallo-Olivé, E.; Navas-Castillo, J. Tomato chlorosis virus, a promiscuous virus with multiple host plants and whitefly vectors. Ann. Appl. Biol. 2023, 182, 29–36. [Google Scholar] [CrossRef]
- Wintermantel, W.M.; Wisler, G.C.; Anchieta, A.G.; Liu, H.-Y.; Karasev, A.V.; Tzanetakis, I.E. The complete nucleotide sequence and genome organization of Tomato chlorosis virus. Arch. Virol. 2005, 150, 2287–2298. [Google Scholar] [CrossRef]
- Wintermantel, W.M.; Wisler, G.C. Vector Specificity, Host Range, and Genetic Diversity of Tomato Chlorosis Virus. Plant Dis. 2006, 90, 814–819. [Google Scholar] [CrossRef]
- Shi, X.; Tang, X.; Zhang, X.; Zhang, D.; Li, F.; Yan, F.; Zhang, Y.; Zhou, X.; Liu, Y. Transmission efficiency, preference and behavior of bemisia tabaci MEAM1 and MED under the Influence of Tomato Chlorosis Virus. Front. Plant Sci. 2018, 8, 2271. [Google Scholar] [CrossRef]
- Huang, L.; Shi, X.; Shi, J.; Zhang, Z.; Fang, Y.; Zhang, Z.; Pan, Q.; Zheng, L.; Gao, Y.; Zhang, D.; et al. Tomato chlorosis virus infection facilitates bemisia tabaci MED reproduction by elevating vitellogenin expression. Insects 2021, 12, 101. [Google Scholar] [CrossRef]
- Bačnik, K.; Kutnjak, D.; Pecman, A.; Mehle, N.; Žnidarič, M.T.; Gutiérrez Aguirre, I.; Ravnikar, M. Viromics and infectivity analysis reveal the release of infective plant viruses from wastewater into the environment. Water Res. 2020, 177, 115628. [Google Scholar] [CrossRef]
- Duarte, M.F.; de Andrade, I.A.; Silva, J.M.F.; de Melo, F.L.; Machado, A.M.; Inoue-Nagata, A.K.; Nagata, T. Metagenomic analyses of plant virus sequences in sewage water for plant viruses monitoring. Trop. Plant Pathol. 2023, 48, 408–416. [Google Scholar] [CrossRef]
- Nash, D.; Ellmen, I.; Knapp, J.J.; Menon, R.; Overton, A.K.; Cheng, J.; Lynch, M.D.J.; Nissimov, J.I.; Charles, T.C. A novel tiled amplicon sequencing assay targeting the Tomato brown rugose fruit virus (ToBRFV) genome reveals widespread distribution in municipal wastewater treatment systems in the province of Ontario, Canada. Viruses 2024, 16, 460. [Google Scholar] [CrossRef]
- Cuevas-Ferrando, E.; Sánchez, G.; Pérez-Cataluña, A. Exploring plant virus diversity in wastewater and reclaimed water through metagenomic analysis. Water Res. 2025, 270, 122827. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. RnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, J.; Xiao, A.; Gu, X.; Lee, L.; Armas, F.; Kauffman, K. SARS-CoV-2 titers in wastewater are higher than expected. mSystems 2020, 5, 1–9. [Google Scholar]
- Wang, D.; Coscoy, L.; Zylberberg, M.; Avila, P.C.; Boushey, H.A.; Ganem, D.; DeRisi, J.L. Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. USA 2002, 99, 15687–15692. [Google Scholar] [CrossRef]
- Abueg, L.A.L.; Afgan, E.; Allart, O.; Awan, A.H.; Bacon, W.A.; Baker, D.; Bassetti, M.; Batut, B.; Bernt, M.; Blankenberg, D.; et al. The galaxy platform for accessible, reproducible, and collaborative data analyses: 2024 update. Nucleic Acids Res. 2024, 52, W83–W94. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 1–19. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2018, 20, 1160–1166. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Klap, C.; Luria, N.; Smith, E.; Hadad, L.; Bakelman, E.; Sela, N.; Belausov, E.; Lachman, O.; Leibman, D.; Dombrovsky, A. Tomato brown rugose fruit virus contributes to enhanced pepino mosaic virus titers in tomato plants. Viruses 2020, 12, 879. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.; Batuman, O. Co-infection of tomato brown rugose fruit virus and pepino mosaic virus in grocery tomatoes in South Florida: Prevalence and genomic diversity. Viruses 2023, 15, 2305. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Ding, T.; Chu, D. Synergistic effects of a Tomato chlorosis virus and tomato yellow leaf curl virus mixed infection on host tomato plants and the whitefly vector. Front. Plant Sci. 2021, 12, 672400. [Google Scholar] [CrossRef]
- Kumar, M.; Kavalappara, S.R.; McAvoy, T.; Hutton, S.; Simmons, A.M.; Bag, S. Association of Tomato chlorosis virus complicates the management of tomato yellow leaf curl virus in cultivated tomato (Solanum lycopersicum) in the Southern United States. Horticulturae 2023, 9, 948. [Google Scholar] [CrossRef]
- Ontiveros, I.; López-Moya, J.J.; Díaz-Pendón, J.A. Coinfection of tomato plants with tomato yellow leaf curl virus and Tomato chlorosis virus affects the interaction with host and whiteflies. Phytopathology 2022, 112, 944–952. [Google Scholar] [CrossRef]
- Fortes, I.M.; Fernández-Muñoz, R.; Moriones, E. Crinivirus Tomato chlorosis virus compromises the control of tomato yellow leaf curl virus in tomato plants by the Ty-1 gene. Phytopathology 2023, 113, 1347–1359. [Google Scholar] [CrossRef]
- Favara, G.M.; Bampi, D.; Molina, J.P.E.; Rezende, J.A.M. Kinetics of systemic invasion and latent and incubation periods of tomato severe rugose virus and Tomato chlorosis virus in single and co-infections in tomato plants. Phytopathology 2019, 109, 480–487. [Google Scholar] [CrossRef]
- Vaghi Medina, C.G.; Martin, D.P.; López Lambertini, P.M. Tomato mottle wrinkle virus, a recombinant begomovirus infecting tomato in Argentina. Arch. Virol. 2015, 160, 581–585. [Google Scholar] [CrossRef]
- Vaghi Medina, C.G.; López Lambertini, P.M. Tomato dwarf leaf virus, a new world begomovirus infecting tomato in Argentina. Arch. Virol. 2012, 157, 1975–1980. [Google Scholar] [CrossRef]
- Pagán, I.; Firth, C.; Holmes, E.C. Phylogenetic analysis reveals rapid evolutionary dynamics in the plant RNA virus genus tobamovirus. J. Mol. Evol. 2010, 71, 298–307. [Google Scholar] [CrossRef]
- García-Arenal, F.; McDonald, B.A. An analysis of the durability of resistance to plant viruses. Phytopathology 2003, 93, 941–952. [Google Scholar] [CrossRef] [PubMed]
- van de Vossenberg, B.T.L.H.; Visser, M.; Bruinsma, M.; Koenraadt, H.M.S.; Westenberg, M.; Botermans, M. Real-Time tracking of Tomato Brown Rugose Fruit Virus (ToBRFV) outbreaks in the netherlands using nextstrain. PLoS ONE 2020, 15, e0234671. [Google Scholar] [CrossRef] [PubMed]
- Botermans, M.; de Koning, P.P.M.; Oplaat, C.; Fowkes, A.R.; McGreig, S.; Skelton, A.; Adams, I.P.; Fox, A.; De Jonghe, K.; Demers, J.E.; et al. Tomato brown rugose fruit virus nextstrain build version 3: Rise of a novel clade. PhytoFrontiersTM 2023, 3, 442–446. [Google Scholar] [CrossRef]
- van de Vossenberg, B.T.L.H.; Dawood, T.; Woźny, M.; Botermans, M. First expansion of the public Tomato brown rugose fruit virus (ToBRFV) nextstrain build; inclusion of new genomic and epidemiological data. PhytoFrontiersTM 2021, 1, 359–363. [Google Scholar] [CrossRef]
- Abrahamian, P.; Cai, W.; Nunziata, S.O.; Ling, K.-S.; Jaiswal, N.; Mavrodieva, V.A.; Rivera, Y.; Nakhla, M.K. Comparative analysis of Tomato brown rugose fruit virus isolates shows limited genetic diversity. Viruses 2022, 14, 2816. [Google Scholar] [CrossRef] [PubMed]
- Çelik, A.; Coşkan, S.; Morca, A.F.; Santosa, A.I.; Koolivand, D. Insight into population structure and evolutionary analysis of the emerging Tomato brown rugose fruit virus. Plants 2022, 11, 3279. [Google Scholar] [CrossRef]
- Ghorbani, A. Genetic Analysis of tomato brown rugose fruit virus reveals evolutionary adaptation and codon usage bias patterns. Sci. Rep. 2024, 14, 21281. [Google Scholar] [CrossRef]
- EPPO. Updated Version of the Pest Risk Analysis for Tobamovirus Fructirugosum (Tomato Brown Rugose Fruit Virus); EPPO Technical Document No. 1092; EPPO: Paris, France, 2024. [Google Scholar]
- ISF. ISPM 38 on the International Movement of Seed-A Training Manual Prepared by The International Seed Federation (ISF); The International Seed Federation: Nyon, Switzerland, 2017; pp. 1–34. [Google Scholar]
- de Koning, P.P.M.; Fowkes, A.R.; Zisi, Z.; Beris, D.; Oplaat, C.; McGreig, S.; Skelton, A.; Adams, I.P.; van Gemert, J.; de Krom, C.; et al. Tomato brown rugose fruit virus nextstrain build version 4: Pathways of introduction and local spread. PhytoFrontiersTM 2025, 5, 105–111. [Google Scholar] [CrossRef]
- Yeşilyurt, N.; Çevik, B. Genetic diversity and phylogenetic analyses of Tomato chlorosis virus isolates using the coat protein gene sequences. J. Plant Pathol. 2019, 101, 1143–1150. [Google Scholar] [CrossRef]
- Barbosa, J.C.; Teixeira, A.P.M.; Moreira, A.G.; Camargo, L.E.A.; Bergamin Filho, A.; Kitajima, E.W.; Rezende, J.A.M. First report of tomato chlorosis virus infecting tomato crops in Brazil. Plant Dis. 2008, 92, 1709. [Google Scholar] [CrossRef]
- Arruabarrena, A.; Rubio, L.; González-Arcos, M.; Maeso, D.; Fonseca, M.E.N.; Boiteux, L.S. First report of Tomato chlorosis virus infecting tomato crops in uruguay. Plant Dis. 2014, 98, 1445. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.B. Cultural Practices in Disease Control. In Plant Pathology, an Advanced Treatise; Horsfall, J.G., Dimond, A.E., Eds.; Academic Press: New York, NY, USA, 1960; Volume 3, pp. 357–429. [Google Scholar]
- Scholthof, K.-B.G. The disease triangle: Pathogens, the environment and society. Nat. Rev. Microbiol. 2007, 5, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Leveau, J.H.J. Re-envisioning the plant disease triangle: Full integration of the host microbiota and a focal pivot to health outcomes. Annu. Rev. Phytopathol. 2024, 62, 31–47. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibañez, J.M.; Zambrana, R.; Carreras, P.; Obregón, V.; Irazoqui, J.M.; Vera, P.A.; Lattar, T.E.; Blanco Fernández, M.D.; Puebla, A.F.; Amadio, A.F.; et al. Phylodynamic of Tomato Brown Rugose Fruit Virus and Tomato Chlorosis Virus, Two Emergent Viruses in Mixed Infections in Argentina. Viruses 2025, 17, 533. https://doi.org/10.3390/v17040533
Ibañez JM, Zambrana R, Carreras P, Obregón V, Irazoqui JM, Vera PA, Lattar TE, Blanco Fernández MD, Puebla AF, Amadio AF, et al. Phylodynamic of Tomato Brown Rugose Fruit Virus and Tomato Chlorosis Virus, Two Emergent Viruses in Mixed Infections in Argentina. Viruses. 2025; 17(4):533. https://doi.org/10.3390/v17040533
Chicago/Turabian StyleIbañez, Julia M., Romina Zambrana, Pamela Carreras, Verónica Obregón, José M. Irazoqui, Pablo A. Vera, Tatiana E. Lattar, María D. Blanco Fernández, Andrea F. Puebla, Ariel F. Amadio, and et al. 2025. "Phylodynamic of Tomato Brown Rugose Fruit Virus and Tomato Chlorosis Virus, Two Emergent Viruses in Mixed Infections in Argentina" Viruses 17, no. 4: 533. https://doi.org/10.3390/v17040533
APA StyleIbañez, J. M., Zambrana, R., Carreras, P., Obregón, V., Irazoqui, J. M., Vera, P. A., Lattar, T. E., Blanco Fernández, M. D., Puebla, A. F., Amadio, A. F., Torres, C., & López Lambertini, P. M. (2025). Phylodynamic of Tomato Brown Rugose Fruit Virus and Tomato Chlorosis Virus, Two Emergent Viruses in Mixed Infections in Argentina. Viruses, 17(4), 533. https://doi.org/10.3390/v17040533