


Human Papillomavirus Type 16 Stimulates WAVE1- and WAVE2-Dependent Actin Protrusions for Endocytic Entry
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. CRISPR/Cas9 Gene Editing
2.3. Clonal Proliferation Analysis
2.4. Protein Overexpression
2.5. Western Blotting
2.6. Pseudovirion and Virus-Like Particle Production
2.7. Antibodies
2.8. Post-Transcriptional Gene Silencing
2.9. Pseudovirus Infection Assay
2.10. Cell Surface Binding Assay
2.11. Virus Internalization Assays
2.12. Immunofluorescence Microscopy Assays
2.13. Confocal Fluorescence Microscopy and Image Analysis
2.14. Statistics
3. Results
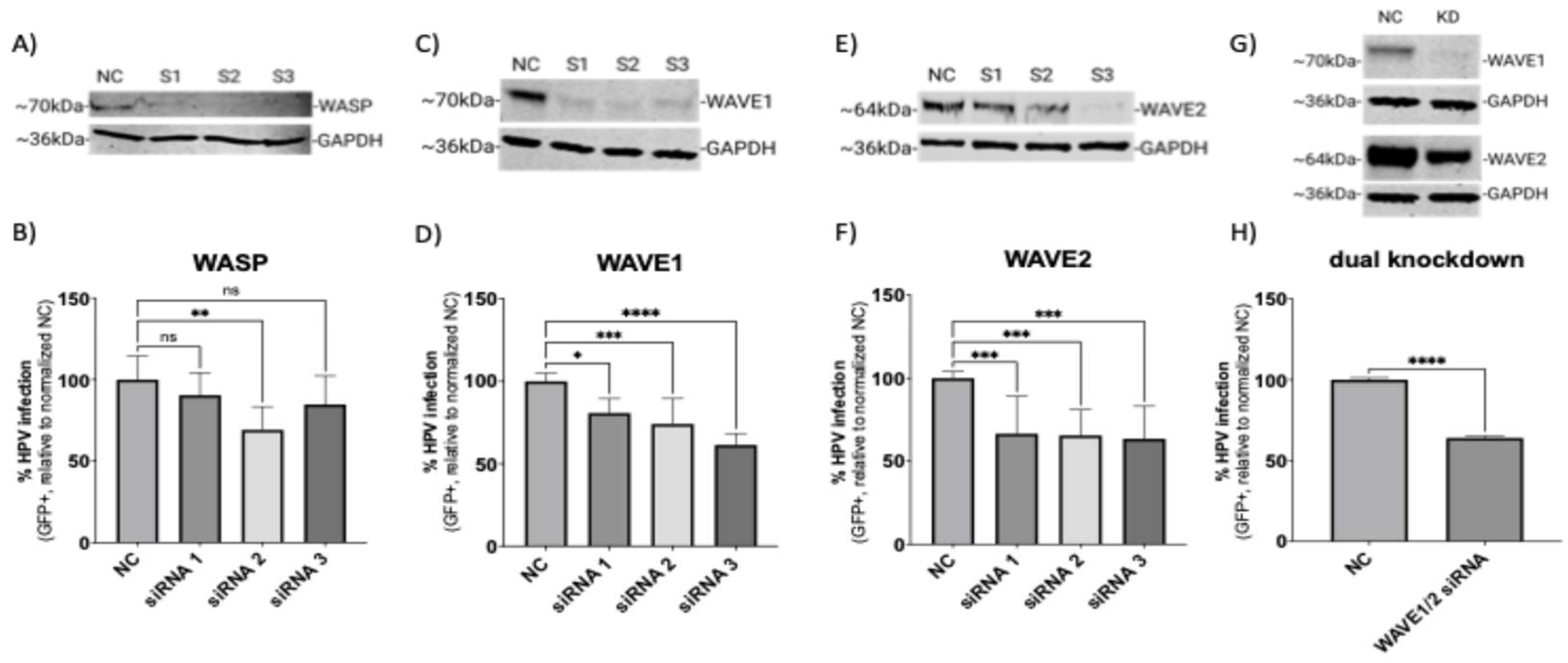
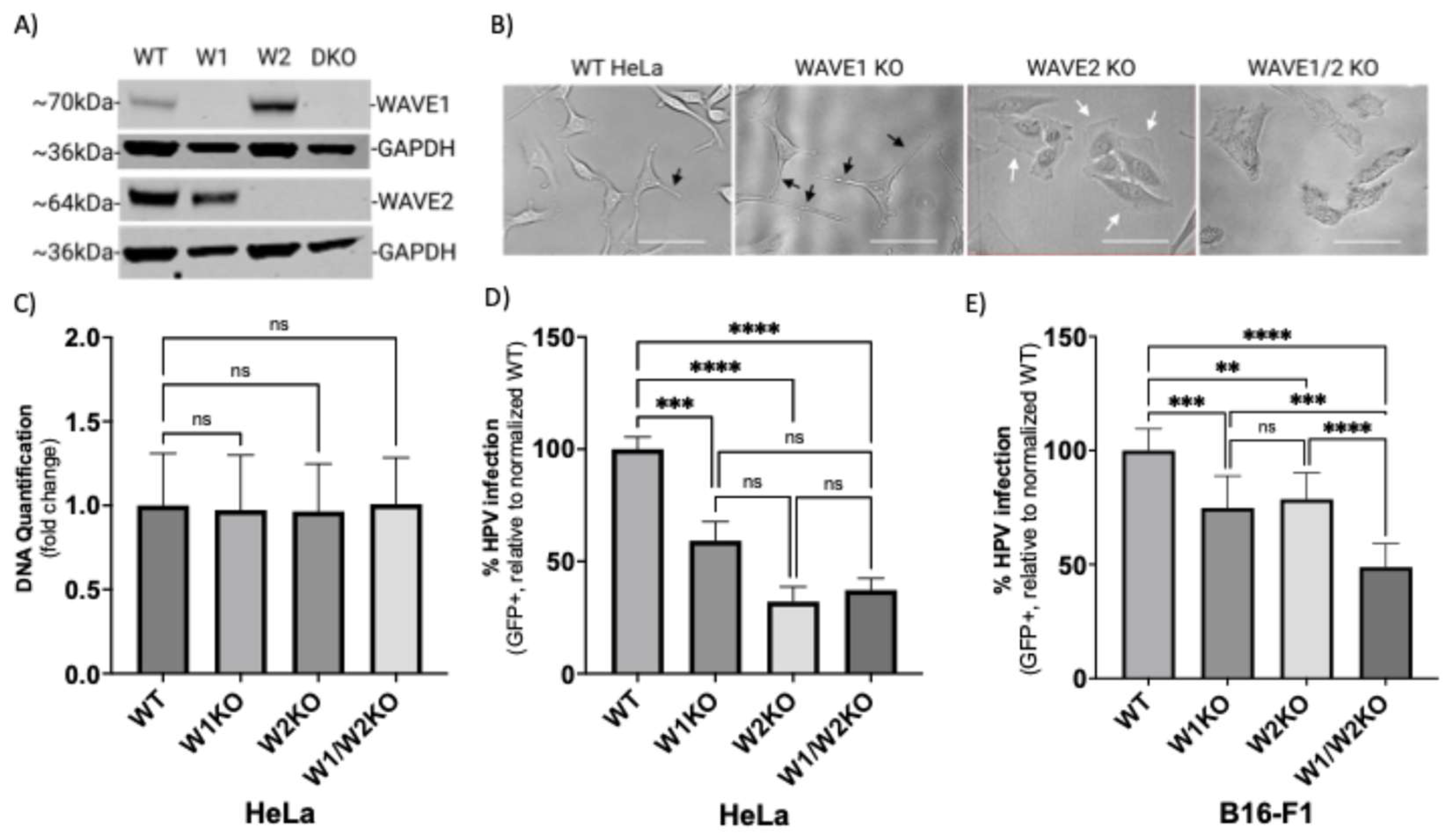
3.1. Wiskott–Aldrich Syndrome Protein Family Members 1 and 2 (WAVE1 and WAVE2) Are Critical for the Infection of HPV16 in HeLa Cells
3.2. WAVE1 and WAVE2 Individually Facilitate HPV16 Entry
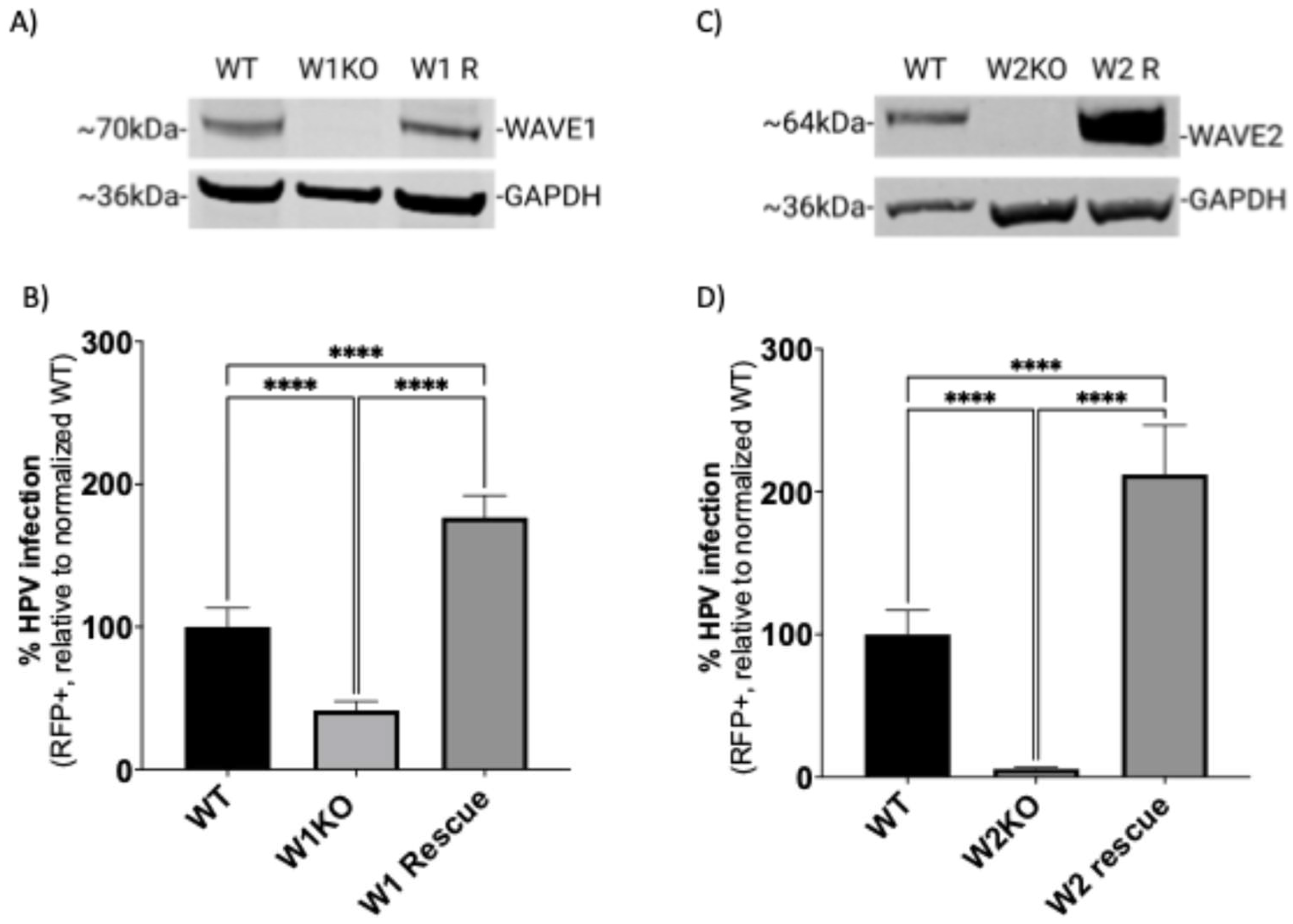
3.3. Restoring WAVE1 or WAVE2 Protein Expression in KO HeLa Cells Rescues HPV16 Infectivity
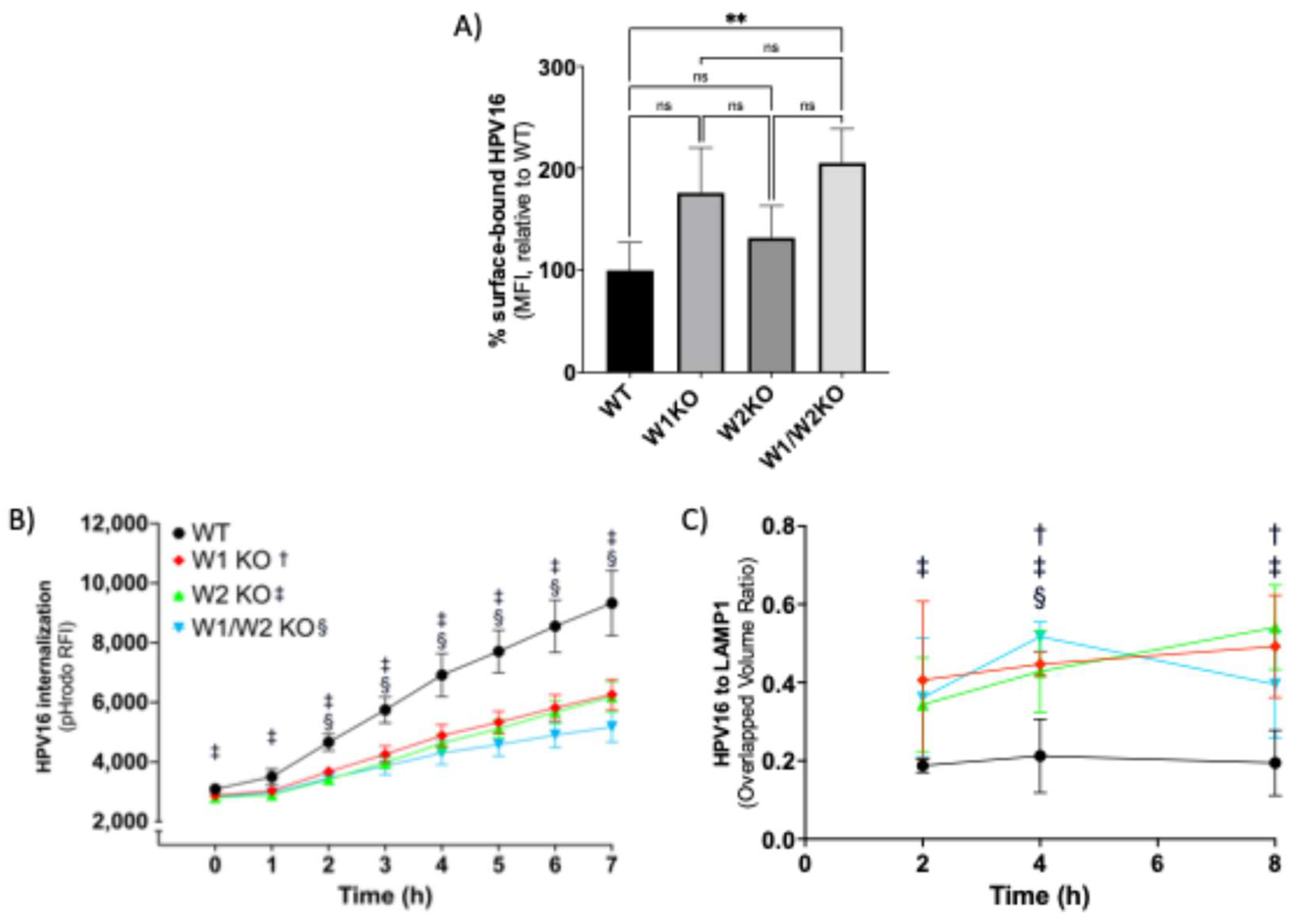
3.4. WAVE1 and WAVE2 Are Required for Proficient HPV16 Internalization and Endocytic Trafficking
3.5. HPV16 Colocalizes with WAVE1 and WAVE2 at the Cellular Dorsal Surface Prior to Entry
3.6. HPV16 Stimulates Dorsal Surface Membrane Protrusions
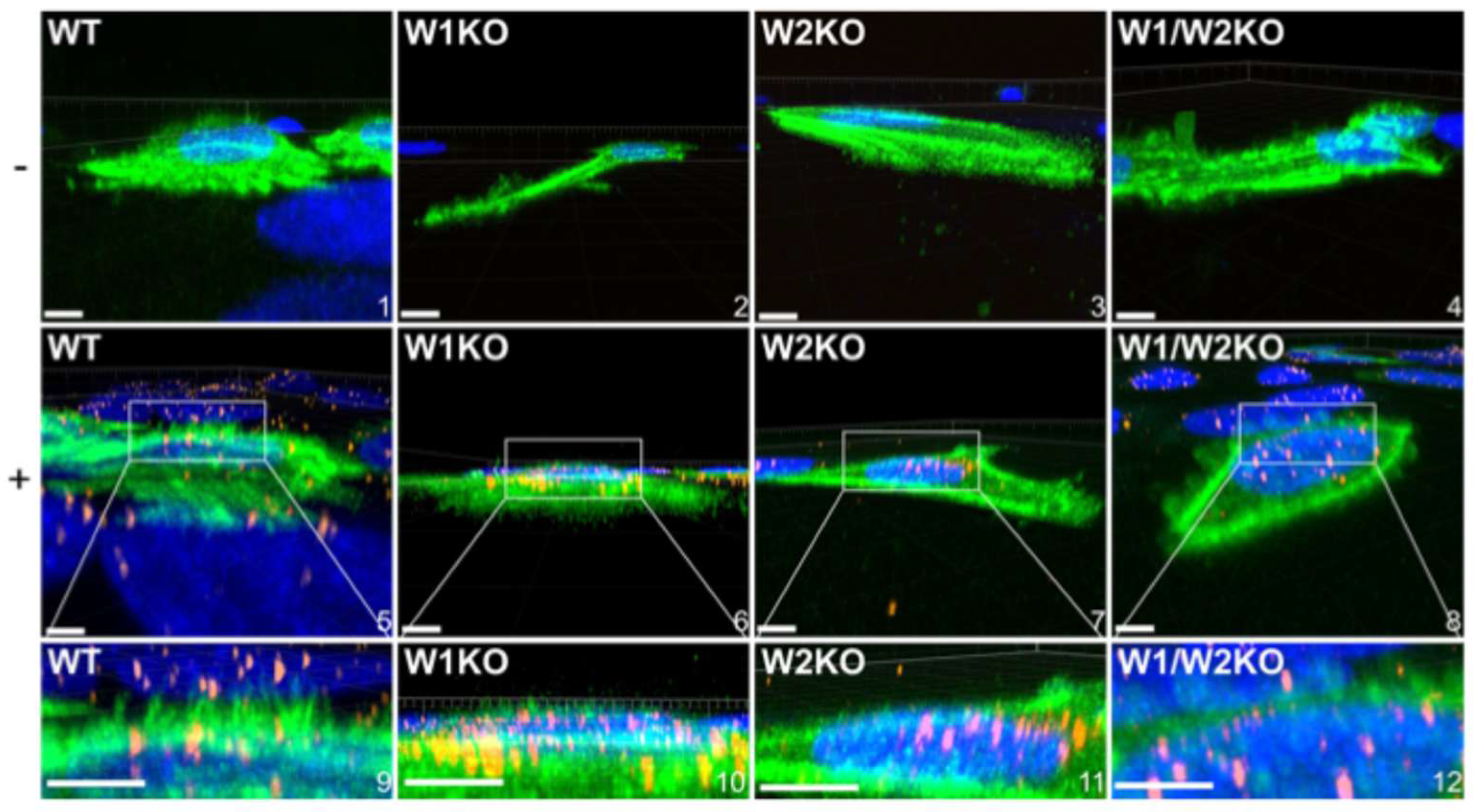
3.7. WAVE1 and WAVE2 Are Necessary for HPV16-Driven Dorsal Surface Actin Protrusions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.-A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front. Public Health 2021, 8, 552028. [Google Scholar] [CrossRef] [PubMed]
- Bruni, L.; Albero, G.; Rowley, J.; Alemany, L.; Arbyn, M.; Giuliano, A.R.; Markowitz, L.E.; Broutet, N.; Taylor, M. Global and regional estimates of genital human papillomavirus prevalence among men: A systematic review and meta-analysis. Lancet Glob. Health 2023, 11, e1345–e1362. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Lang Kuhs, K.A.; Faden, D.L.; Chen, L.; Smith, D.K.; Pinheiro, M.; Wood, C.B.; Davis, S.; Yeager, M.; Boland, J.F.; Cullen, M.; et al. Genetic variation within the human papillomavirus type 16 genome is associated with oropharyngeal cancer prognosis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2022, 33, 638–648. [Google Scholar] [CrossRef]
- Molano, M.; van den Brule, A.; Plummer, M.; Weiderpass, E.; Posso, H.; Arslan, A.; Meijer, C.J.L.M.; Muñoz, N.; Franceschi, S.; the HPV Study Group. Determinants of Clearance of Human Papillomavirus Infections in Colombian Women with Normal Cytology: A Population-based, 5-Year Follow-up Study. Am. J. Epidemiol. 2003, 158, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of Human Papillomaviruses in Differentiating Epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef]
- Mikuličić, S.; Strunk, J.; Florin, L. HPV16 Entry into Epithelial Cells: Running a Gauntlet. Viruses 2021, 13, 2460. [Google Scholar] [CrossRef]
- Fausch, S.C.; Fahey, L.M.; Silva, D.M.D.; Kast, W.M. Human Papillomavirus Can Escape Immune Recognition through Langerhans Cell Phosphoinositide 3-Kinase Activation. J. Immunol. 2005, 174, 7172–7178. [Google Scholar] [CrossRef]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and Caveolin-Independent Entry of Human Papillomavirus Type 16—Involvement of Tetraspanin-Enriched Microdomains (TEMs). PLoS ONE 2008, 3, e3313. [Google Scholar] [CrossRef]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kühling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of Human Papillomavirus Type 16 by Actin-Dependent, Clathrin- and Lipid Raft-Independent Endocytosis. PLOS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef]
- Becker, M.; Greune, L.; Schmidt, M.A.; Schelhaas, M. Extracellular Conformational Changes in the Capsid of Human Papillomaviruses Contribute to Asynchronous Uptake into Host Cells. J. Virol. 2018, 92, e02106. [Google Scholar] [CrossRef] [PubMed]
- Brinkert, P.; Krebs, L.; Ventayol, P.S.; Greune, L.; Bannach, C.; Bucher, D.; Kollasser, J.; Dersch, P.; Boulant, S.; Stradal, T.E.B.; et al. Endocytic vacuole formation by WASH-mediated endocytosis. bioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.06.18.448076v1 (accessed on 30 March 2025).
- Mercer, J.; Helenius, A. Gulping rather than sipping: Macropinocytosis as a way of virus entry. Curr. Opin. Microbiol. 2012, 15, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.H.; Teasdale, R.D.; Liebl, D. Macropinosome quantitation assay. MethodsX 2014, 1, 36–41. [Google Scholar] [CrossRef]
- Lin, X.P.; Mintern, J.D.; Gleeson, P.A. Macropinocytosis in Different Cell Types: Similarities and Differences. Membranes 2020, 10, 177. [Google Scholar] [CrossRef]
- Finke, J.; Hitschler, L.; Boller, K.; Florin, L.; Lang, T. HPV caught in the tetraspanin web? Med. Microbiol. Immunol. 2020, 209, 447–459. [Google Scholar] [CrossRef]
- Finke, J.; Mikuličić, S.; Loster, A.-L.; Gawlitza, A.; Florin, L.; Lang, T. Anatomy of a viral entry platform differentially functionalized by integrins α3 and α6. Sci. Rep. 2020, 10, 5356. [Google Scholar] [CrossRef]
- Biondo, A.; Meneses, P.I. The Process of Filopodia Induction during HPV Infection. Viruses 2022, 14, 1150. [Google Scholar] [CrossRef]
- Schelhaas, M.; Ewers, H.; Rajamäki, M.-L.; Day, P.M.; Schiller, J.T.; Helenius, A. Human Papillomavirus Type 16 Entry: Retrograde Cell Surface Transport along Actin-Rich Protrusions. PLoS Pathog. 2008, 4, e1000148. [Google Scholar] [CrossRef]
- Mooren, O.L.; Galletta, B.J.; Cooper, J.A. Roles for Actin Assembly in Endocytosis. Annu. Rev. Biochem. 2012, 81, 661–686. [Google Scholar] [CrossRef]
- Jin, M.; Shirazinejad, C.; Wang, B.; Yan, A.; Schöneberg, J.; Upadhyayula, S.; Xu, K.; Drubin, D.G. Branched actin networks are organized for asymmetric force production during clathrin-mediated endocytosis in mammalian cells. Nat. Commun. 2022, 13, 3578. [Google Scholar] [CrossRef]
- Lu, R.; Drubin, D.G.; Sun, Y. Clathrin-mediated endocytosis in budding yeast at a glance. J. Cell Sci. 2016, 129, 1531–1536. [Google Scholar] [CrossRef]
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: An actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006, 7, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Galletta, B.J.; Chuang, D.Y.; Cooper, J.A. Distinct Roles for Arp2/3 Regulators in Actin Assembly and Endocytosis. PLOS Biol. 2008, 6, e1. [Google Scholar] [CrossRef]
- Donaldson, J.G. Macropinosome formation, maturation and membrane recycling: Lessons from clathrin-independent endosomal membrane systems. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180148. [Google Scholar] [CrossRef]
- Rottner, K.; Stradal, T.E.B.; Chen, B. WAVE regulatory complex. Curr. Biol. 2021, 31, R512–R517. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.A.; Piper, H.K.; Chen, B. WASP family proteins: Molecular mechanisms and implications in human disease. Eur. J. Cell Biol. 2022, 101, 151244. [Google Scholar] [CrossRef]
- Takenawa, T.; Suetsugu, S. The WASP–WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef]
- Buck, C.B.; Thompson, C.D. Production of papillomavirus-based gene transfer vectors. Curr. Protoc. Cell Biol. 2007, 26, 26.1. [Google Scholar] [CrossRef]
- Cardone, G.; Moyer, A.L.; Cheng, N.; Thompson, C.D.; Dvoretzky, I.; Lowy, D.R.; Schiller, J.T.; Steven, A.C.; Buck, C.B.; Trus, B.L. Maturation of the Human Papillomavirus 16 Capsid. mBio 2014, 5, e01104-14. [Google Scholar] [CrossRef]
- Da Silva, D.M.; Schiller, J.T.; Kast, W.M. Heterologous boosting increases immunogenicity of chimeric papillomavirus virus-like particle vaccines. Vaccine 2003, 21, 3219–3227. [Google Scholar] [CrossRef]
- Kirnbauer, R.; Booy, F.; Cheng, N.; Lowy, D.R.; Schiller, J.T. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. USA 1992, 89, 12180–12184. [Google Scholar]
- Greenstone, H.L.; Nieland, J.D.; de Visser, K.E.; De Bruijn, M.L.; Kirnbauer, R.; Roden, R.B.; Lowy, D.R.; Kast, W.M.; Schiller, J.T. Chimeric papillomavirus virus-like particles elicit antitumor immunity against the E7 oncoprotein in an HPV16 tumor model. Proc. Natl. Acad. Sci. USA 1998, 95, 1800–1805. [Google Scholar] [CrossRef]
- Christensen, N.D.; Reed, C.A.; Cladel, N.M.; Hall, K.; Leiserowitz, G.S. Monoclonal Antibodies to HPV-6 L1 Virus-like Particles Identify Conformational and Linear Neutralizing Epitopes on HPV-11 in Addition to Type-Specific Epitopes on HPV-6. Virology 1996, 224, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Sapp, M.; Kraus, U.; Volpers, C.; Snijders, P.J.F.; Walboomers, J.M.M.; Streeck, R.E. Analysis of type-restricted and cross-reactive epitopes on virus-like particles of human papillomavirus type 33 and in infected tissues using monoclonal antibodies to the major capsid protein. J. Gen. Virol. 1994, 75, 3375–3383. [Google Scholar] [CrossRef]
- Schädlich, L.; Senger, T.; Gerlach, B.; Mücke, N.; Klein, C.; Bravo, I.G.; Müller, M.; Gissmann, L. Analysis of Modified Human Papillomavirus Type 16 L1 Capsomeres: The Ability To Assemble into Larger Particles Correlates with Higher Immunogenicity. J. Virol. 2009, 83, 7690–7705. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.R.; Fernandez, D.J.; Thornton, S.M.; Skeate, J.G.; Lühen, K.P.; Silva, D.M.D.; Langen, R.; Kast, W.M. Heterotetrameric annexin A2/S100A10 (A2t) is essential for oncogenic human papillomavirus trafficking and capsid disassembly, and protects virions from lysosomal degradation. Sci. Rep. 2018, 8, 11642. [Google Scholar] [CrossRef]
- Skeate, J.G.; Segerink, W.H.; Garcia, M.D.; Fernandez, D.J.; Prins, R.; Lühen, K.P.; Voss, F.O.; Da Silva, D.M.; Kast, W.M. Theta-Defensins Inhibit High-Risk Human Papillomavirus Infection Through Charge-Driven Capsid Clustering. Front. Immunol. 2020, 11, 561843. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Tang, Q.; Schaks, M.; Koundinya, N.; Yang, C.; Pollard, L.W.; Svitkina, T.M.; Rottner, K.; Goode, B.L. WAVE1 and WAVE2 have distinct and overlapping roles in controlling actin assembly at the leading edge. Mol. Biol. Cell 2020, 31, 2168–2178. [Google Scholar] [CrossRef]
- Khajah, M.A.; Luqmani, Y.A. Involvement of Membrane Blebbing in Immunological Disorders and Cancer. Med. Princ. Pract. 2016, 25 (Suppl. S2), 18–27. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.; Tobita, C.; Umemoto, E.U.; Starkus, J.; Rysavy, N.M.; Shimoda, L.M.N.; Sung, C.; Stokes, A.J.; Turner, H. Calcium-dependent, non-apoptotic, large plasma membrane bleb formation in physiologically stimulated mast cells and basophils. J. Extracell. Vesicles 2019, 8, 1578589. [Google Scholar] [CrossRef]
- Eisenmann, K.M.; Harris, E.S.; Kitchen, S.M.; Holman, H.A.; Higgs, H.N.; Alberts, A.S. Dia-Interacting Protein Modulates Formin-Mediated Actin Assembly at the Cell Cortex. Curr. Biol. 2007, 17, 579–591. [Google Scholar] [CrossRef]
- Samperio Ventayol, P.; Schelhaas, M. Fluorescently Labeled Human Papillomavirus Pseudovirions for Use in Virus Entry Experiments. Curr. Protoc. Microbiol. 2015, 37, 14B.4.1–14B.4.22. [Google Scholar] [CrossRef] [PubMed]
- Siddiqa, A.; Broniarczyk, J.; Banks, L.; Siddiqa, A.; Broniarczyk, J.; Banks, L. Papillomaviruses and Endocytic Trafficking. Int. J. Mol. Sci. 2018, 19, 2619. [Google Scholar] [CrossRef]
- Smith, J.L.; Lidke, D.S.; Ozbun, M.A. Virus activated filopodia promote human papillomavirus type 31 uptake from the extracellular matrix. Virology 2008, 381, 16–21. [Google Scholar] [CrossRef]
- Valdivia, A.; Goicoechea, S.M.; Awadia, S.; Zinn, A.; Garcia-Mata, R.; Chernoff, J. Regulation of circular dorsal ruffles, macropinocytosis, and cell migration by RhoG and its exchange factor, Trio. Mol. Biol. Cell 2017, 28, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Virus entry by macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef]
- Cerqueira, C.; Samperio Ventayol, P.; Vogeley, C.; Schelhaas, M. Kallikrein-8 Proteolytically Processes Human Papillomaviruses in the Extracellular Space To Facilitate Entry into Host Cells. J. Virol. 2015, 89, 7038–7052. [Google Scholar] [CrossRef]
- Spoden, G.; Kühling, L.; Cordes, N.; Frenzel, B.; Sapp, M.; Boller, K.; Florin, L.; Schelhaas, M. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 2013, 87, 7765–7773. [Google Scholar] [CrossRef]
- Aksoy, P.; Abban, C.Y.; Kiyashka, E.; Qiang, W.; Meneses, P.I. HPV16 infection of HaCaTs is dependent on β4 integrin, and α6 integrin processing. Virology 2014, 449, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef]
- Suetsugu, S.; Yamazaki, D.; Kurisu, S.; Takenawa, T. Differential Roles of WAVE1 and WAVE2 in Dorsal and Peripheral Ruffle Formation for Fibroblast Cell Migration. Dev. Cell 2003, 5, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.D.; Donaldson, J.G. Arf6, JIP3, and dynein shape and mediate macropinocytosis. Mol. Biol. Cell 2019, 30, 1477–1489. [Google Scholar] [CrossRef]
- Li, Y.; Gonzalez, W.G.; Andreev, A.; Tang, W.; Gandhi, S.; Cunha, A.; Prober, D.; Lois, C.; Bronner, M.E. Macropinocytosis-mediated membrane recycling drives neural crest migration by delivering F-actin to the lamellipodium. Proc. Natl. Acad. Sci. USA 2020, 117, 27400–27411. [Google Scholar] [CrossRef]
- Krause, M.; Gautreau, A. Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014, 15, 577–590. [Google Scholar] [CrossRef]
- Valiya Veettil, M.; Sadagopan, S.; Kerur, N.; Chakraborty, S.; Chandran, B. Interaction of c-Cbl with Myosin IIA Regulates Bleb Associated Macropinocytosis of Kaposi’s Sarcoma-Associated Herpesvirus. PLoS Pathog. 2010, 6, e1001238. [Google Scholar] [CrossRef]
- Mercer, J.; Knébel, S.; Schmidt, F.I.; Crouse, J.; Burkard, C.; Helenius, A. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. USA 2010, 107, 9346–9351. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Elde, N.C. A cross-species view on viruses. Curr. Opin. Virol. 2012, 2, 561–568. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Gu, Z.; Noss, E.H.; Hsu, V.W.; Brenner, M.B. Integrins traffic rapidly via circular dorsal ruffles and macropinocytosis during stimulated cell migration. J. Cell Biol. 2011, 193, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef]
- Suzuki, T.; Miki, H.; Takenawa, T.; Sasakawa, C. Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 1998, 17, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Faris, R.; McCullough, A.; Andersen, S.E.; Moninger, T.O.; Weber, M.M. The Chlamydia trachomatis secreted effector TmeA hijacks the N-WASP-ARP2/3 actin remodeling axis to facilitate cellular invasion. PLoS Pathog. 2020, 16, e1008878. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Robbins, D.; Leong, J.M. EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev. Cell 2004, 7, 217–228. [Google Scholar] [CrossRef]
- Bonfim-Melo, A.; Ferreira, É.R.; Mortara, R.A. Rac1/WAVE2 and Cdc42/N-WASP Participation in Actin-Dependent Host Cell Invasion by Extracellular Amastigotes of Trypanosoma cruzi. Front. Microbiol. 2018, 9, 360. [Google Scholar] [CrossRef]
- Guerra, S.; Aracil, M.; Conde, R.; Bernad, A.; Esteban, M. Wiskott-Aldrich Syndrome Protein Is Needed for Vaccinia Virus Pathogenesis. J. Virol. 2005, 79, 2133–2140. [Google Scholar] [CrossRef]
- Swaine, T.; Dittmar, M.T. CDC42 Use in Viral Cell Entry Processes by RNA Viruses. Viruses 2015, 7, 6526–6536. [Google Scholar] [CrossRef]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential Roles for Soluble Virion-Associated Heparan Sulfonated Proteoglycans and Growth Factors in Human Papillomavirus Infections. PLOS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef]
- Zheng, K.; Xiang, Y.; Wang, X.; Wang, Q.; Zhong, M.; Wang, S.; Wang, X.; Fan, J.; Kitazato, K.; Wang, Y. Epidermal Growth Factor Receptor-PI3K Signaling Controls Cofilin Activity To Facilitate Herpes Simplex Virus 1 Entry into Neuronal Cells. mBio 2014, 5, e00958-13. [Google Scholar] [CrossRef]
- Wu, G.-Y.; Deisseroth, K.; Tsien, R.W. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat. Neurosci. 2001, 4, 151–158. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez, D.J.; Cheng, S.; Prins, R.; Hamm-Alvarez, S.F.; Kast, W.M. Human Papillomavirus Type 16 Stimulates WAVE1- and WAVE2-Dependent Actin Protrusions for Endocytic Entry. Viruses 2025, 17, 542. https://doi.org/10.3390/v17040542
Fernandez DJ, Cheng S, Prins R, Hamm-Alvarez SF, Kast WM. Human Papillomavirus Type 16 Stimulates WAVE1- and WAVE2-Dependent Actin Protrusions for Endocytic Entry. Viruses. 2025; 17(4):542. https://doi.org/10.3390/v17040542
Chicago/Turabian StyleFernandez, Daniel J., Stephanie Cheng, Ruben Prins, Sarah F. Hamm-Alvarez, and W. Martin Kast. 2025. "Human Papillomavirus Type 16 Stimulates WAVE1- and WAVE2-Dependent Actin Protrusions for Endocytic Entry" Viruses 17, no. 4: 542. https://doi.org/10.3390/v17040542
APA StyleFernandez, D. J., Cheng, S., Prins, R., Hamm-Alvarez, S. F., & Kast, W. M. (2025). Human Papillomavirus Type 16 Stimulates WAVE1- and WAVE2-Dependent Actin Protrusions for Endocytic Entry. Viruses, 17(4), 542. https://doi.org/10.3390/v17040542