
Neuroinflammation, Blood–Brain Barrier, and HIV Reservoirs in the CNS: An In-Depth Exploration of Latency Mechanisms and Emerging Therapeutic Strategies


Abstract
:
1. Introduction
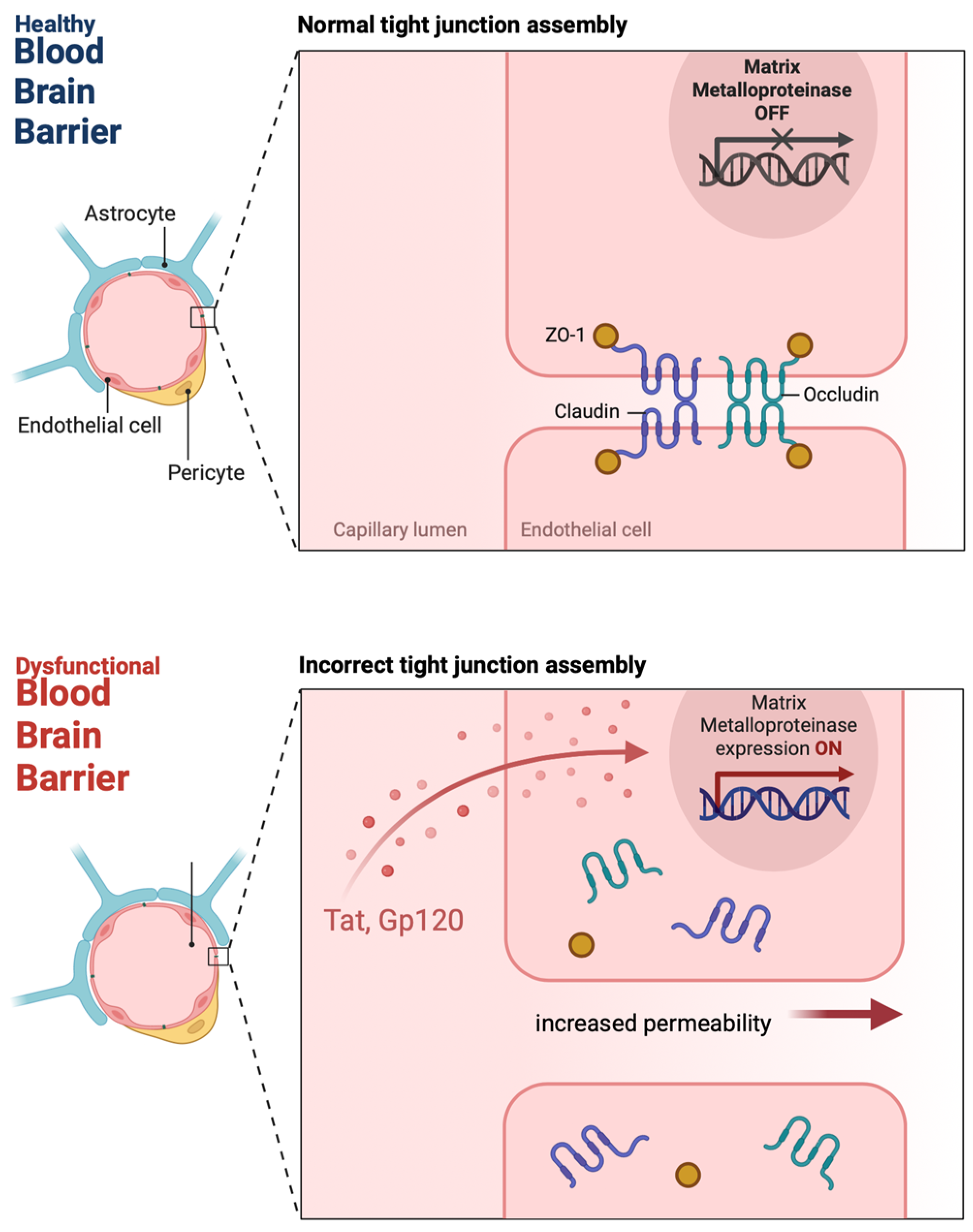
2. HIV and BBB Disruption
3. Monocyte Trafficking and HIV Entry into the CNS
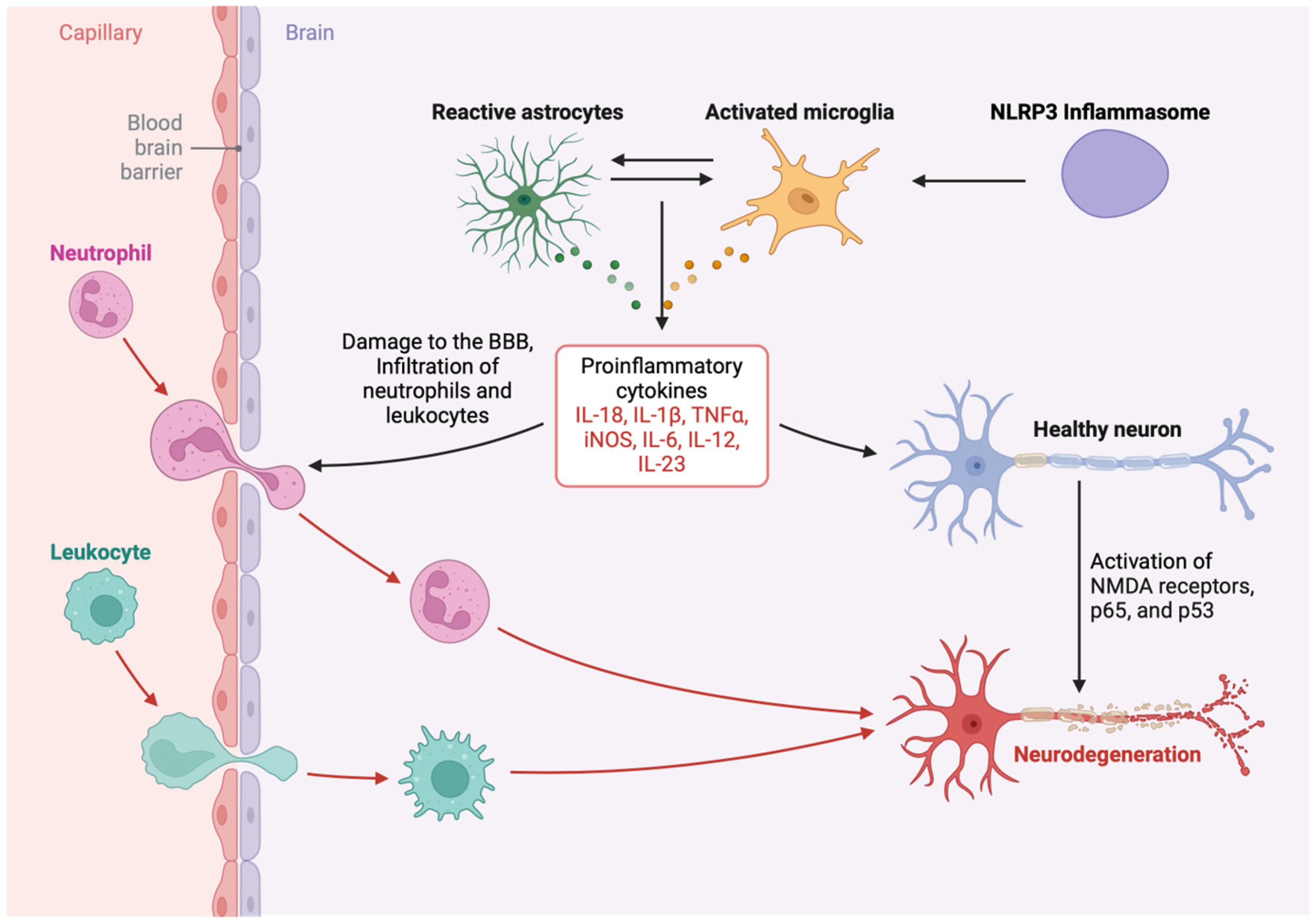
4. Chronic Microglial Activation, Neuroinflammation, and Neuronal Injury
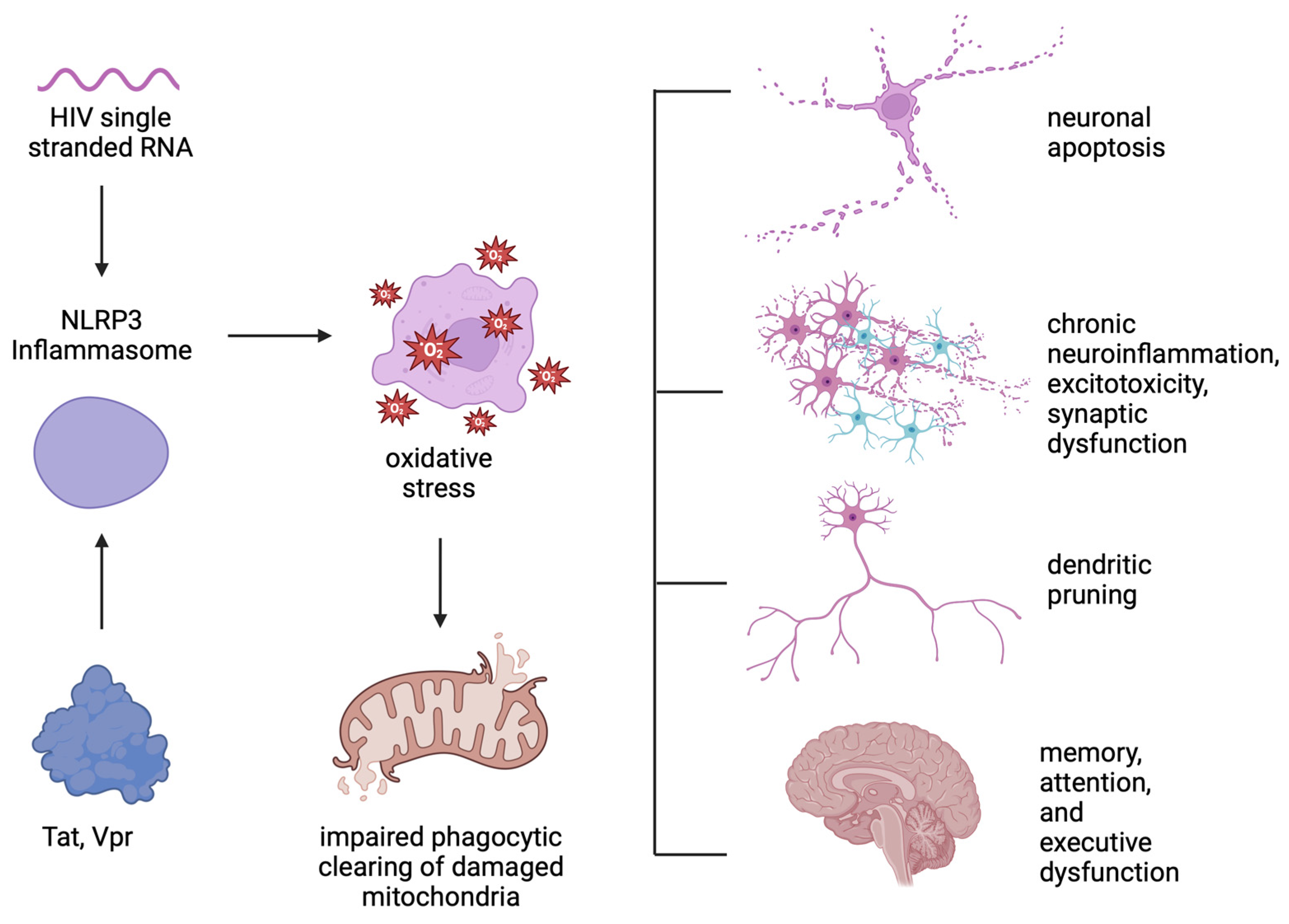
5. NLRP3 Inflammasome Activation
6. HIV Latency Mechanisms in the CNS
7. Latency and the “Kick and Kill” Strategy
7.1. Focused Ultrasound
7.2. Receptor-Mediated Transport
7.3. Exosomes
7.4. Nanoparticles
8. Gene Editing and CRISPR/Cas9
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, S.; Ances, B.; Cinque, P.; Dravid, A.; Dreyer, A.J.; Gisslen, M.; Joska, J.A.; Kwasa, J.; Meyer, A.C.; Mpongo, N.; et al. Cognitive impairment in people living with HIV: Consensus recommendations for a new approach. Nat. Rev. Neurol. 2023, 19, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef]
- Dos Reis, R.S.; Selvam, S.; Wagner, M.C.E.; Ayyavoo, V. Modeling HIV-1 Infection in CNS via Infected Monocytes Using Immunocompetent Brain Organoids. Methods Mol. Biol. 2024, 2807, 261–270. [Google Scholar] [CrossRef]
- Thompson, L.J.; Genovese, J.; Hong, Z.; Singh, M.V.; Singh, V.B. HIV-Associated Neurocognitive Disorder: A Look into Cellular and Molecular Pathology. Int. J. Mol. Sci. 2024, 25, 4697. [Google Scholar] [CrossRef]
- Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The Paradox of HIV Blood-Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci. 2020, 43, 695–708. [Google Scholar] [CrossRef]
- Sonti, S.; Sharma, A.L.; Tyagi, M. HIV-1 persistence in the CNS: Mechanisms of latency, pathogenesis and an update on eradication strategies. Virus Res. 2021, 303, 198523. [Google Scholar] [CrossRef]
- Rahimy, E.; Li, F.Y.; Hagberg, L.; Fuchs, D.; Robertson, K.; Meyerhoff, D.J.; Zetterberg, H.; Price, R.W.; Gisslen, M.; Spudich, S. Blood-Brain Barrier Disruption Is Initiated During Primary HIV Infection and Not Rapidly Altered by Antiretroviral Therapy. J. Infect. Dis. 2017, 215, 1132–1140. [Google Scholar] [CrossRef]
- Pachter, J.S.; de Vries, H.E.; Fabry, Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J. Neuropathol. Exp. Neurol. 2003, 62, 593–604. [Google Scholar] [CrossRef]
- Spudich, S.; Gonzalez-Scarano, F. HIV-1-related central nervous system disease: Current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb. Perspect. Med. 2012, 2, a007120. [Google Scholar] [CrossRef] [PubMed]
- Kanmogne, G.D.; Schall, K.; Leibhart, J.; Knipe, B.; Gendelman, H.E.; Persidsky, Y. HIV-1 gp120 compromises blood-brain barrier integrity and enhances monocyte migration across blood-brain barrier: Implication for viral neuropathogenesis. J. Cereb. Blood Flow. Metab. 2007, 27, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Price, T.O.; Ercal, N.; Nakaoke, R.; Banks, W.A. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005, 1045, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Liao, K.; Hu, G.; Moidunny, S.; Roy, S.; Buch, S. HIV Tat-Mediated Induction of Monocyte Transmigration Across the Blood-Brain Barrier: Role of Chemokine Receptor CXCR3. Front. Cell Dev. Biol. 2021, 9, 724970. [Google Scholar] [CrossRef]
- Xu, R.; Feng, X.; Xie, X.; Zhang, J.; Wu, D.; Xu, L. HIV-1 Tat protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res. 2012, 1436, 13–19. [Google Scholar] [CrossRef]
- Toborek, M.; Lee, Y.W.; Pu, H.; Malecki, A.; Flora, G.; Garrido, R.; Hennig, B.; Bauer, H.C.; Nath, A. HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J. Neurochem. 2003, 84, 169–179. [Google Scholar] [CrossRef]
- Kim, T.A.; Avraham, H.K.; Koh, Y.H.; Jiang, S.; Park, I.W.; Avraham, S. HIV-1 Tat-mediated apoptosis in human brain microvascular endothelial cells. J. Immunol. 2003, 170, 2629–2637. [Google Scholar] [CrossRef]
- Andras, I.E.; Pu, H.; Deli, M.A.; Nath, A.; Hennig, B.; Toborek, M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J. Neurosci. Res. 2003, 74, 255–265. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Clements, J.E.; Zink, M.C.; Berman, J.W. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J. Neurosci. 2011, 31, 9456–9465. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Duan, F.; Morsey, B.; Persidsky, Y.; Kanmogne, G.D. HIV-1 activates proinflammatory and interferon-inducible genes in human brain microvascular endothelial cells: Putative mechanisms of blood-brain barrier dysfunction. J. Cereb. Blood Flow. Metab. 2008, 28, 697–711. [Google Scholar] [CrossRef]
- Wang, H.; Sun, J.; Goldstein, H. Human immunodeficiency virus type 1 infection increases the in vivo capacity of peripheral monocytes to cross the blood-brain barrier into the brain and the in vivo sensitivity of the blood-brain barrier to disruption by lipopolysaccharide. J. Virol. 2008, 82, 7591–7600. [Google Scholar] [CrossRef] [PubMed]
- Cassol, E.; Alfano, M.; Biswas, P.; Poli, G. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J. Leukoc. Biol. 2006, 80, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Catalfamo, M.; Le Saout, C.; Lane, H.C. The role of cytokines in the pathogenesis and treatment of HIV infection. Cytokine Growth Factor. Rev. 2012, 23, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Burdo, T.H.; Miller, A.D. Animal models of HIV peripheral neuropathy. Future Virol. 2014, 9, 465–474. [Google Scholar] [CrossRef]
- Williams, D.W.; Veenstra, M.; Gaskill, P.J.; Morgello, S.; Calderon, T.M.; Berman, J.W. Monocytes mediate HIV neuropathogenesis: Mechanisms that contribute to HIV associated neurocognitive disorders. Curr. HIV Res. 2014, 12, 85–96. [Google Scholar] [CrossRef]
- Campbell, J.H.; Hearps, A.C.; Martin, G.E.; Williams, K.C.; Crowe, S.M. The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure. AIDS 2014, 28, 2175–2187. [Google Scholar] [CrossRef]
- Yadav, A.; Collman, R.G. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J. Neuroimmune Pharmacol. 2009, 4, 430–447. [Google Scholar] [CrossRef]
- Borrajo Lopez, A.; Penedo, M.A.; Rivera-Baltanas, T.; Perez-Rodriguez, D.; Alonso-Crespo, D.; Fernandez-Pereira, C.; Olivares, J.M.; Agis-Balboa, R.C. Microglia: The Real Foe in HIV-1-Associated Neurocognitive Disorders? Biomedicines 2021, 9, 925. [Google Scholar] [CrossRef]
- Crowe, S.; Zhu, T.; Muller, W.A. The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J. Leukoc. Biol. 2003, 74, 635–641. [Google Scholar] [CrossRef]
- Zhu, T. HIV-1 in peripheral blood monocytes: An underrated viral source. J. Antimicrob. Chemother. 2002, 50, 309–311. [Google Scholar] [CrossRef]
- Kedzierska, K.; Crowe, S.M. The role of monocytes and macrophages in the pathogenesis of HIV-1 infection. Curr. Med. Chem. 2002, 9, 1893–1903. [Google Scholar] [CrossRef] [PubMed]
- Conant, K.; Garzino-Demo, A.; Nath, A.; McArthur, J.C.; Halliday, W.; Power, C.; Gallo, R.C.; Major, E.O. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 3117–3121. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Osiecki, K.; Lopez, L.; Goldstein, H.; Calderon, T.M.; Berman, J.W. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: A potential mechanism of HIV-CNS invasion and NeuroAIDS. J. Neurosci. 2006, 26, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Kelder, W.; McArthur, J.C.; Nance-Sproson, T.; McClernon, D.; Griffin, D.E. Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann. Neurol. 1998, 44, 831–835. [Google Scholar] [CrossRef]
- Ansari, A.W.; Bhatnagar, N.; Dittrich-Breiholz, O.; Kracht, M.; Schmidt, R.E.; Heiken, H. Host chemokine (C-C motif) ligand-2 (CCL2) is differentially regulated in HIV type 1 (HIV-1)-infected individuals. Int. Immunol. 2006, 18, 1443–1451. [Google Scholar] [CrossRef]
- Hernandez, C.; Gorska, A.M.; Eugenin, E. Mechanisms of HIV-mediated blood-brain barrier compromise and leukocyte transmigration under the current antiretroviral era. iScience 2024, 27, 109236. [Google Scholar] [CrossRef]
- Williams, D.W.; Anastos, K.; Morgello, S.; Berman, J.W. JAM-A and ALCAM are therapeutic targets to inhibit diapedesis across the BBB of CD14+CD16+ monocytes in HIV-infected individuals. J. Leukoc. Biol. 2015, 97, 401–412. [Google Scholar] [CrossRef]
- D’Antoni, M.L.; Mitchell, B.I.; McCurdy, S.; Byron, M.M.; Ogata-Arakaki, D.; Chow, D.; Mehta, N.N.; Boisvert, W.A.; Lefebvre, E.; Shikuma, C.M.; et al. Cenicriviroc inhibits trans-endothelial passage of monocytes and is associated with impaired E-selectin expression. J. Leukoc. Biol. 2018, 104, 1241–1252. [Google Scholar] [CrossRef]
- Bowler, S.; Siriwardhana, C.; Mitchell, B.I.; D’Antoni, M.L.; Ogata-Arakaki, D.; Souza, S.; Yee, R.; Gangcuangco, L.M.A.; Chow, D.C.; Ndhlovu, L.C.; et al. Cenicriviroc, a dual CCR2 and CCR5 antagonist leads to a reduction in plasma fibrotic biomarkers in persons living with HIV on antiretroviral therapy. HIV Res. Clin. Pract. 2019, 20, 123–129. [Google Scholar] [CrossRef]
- Williams, K.C.; Hickey, W.F. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu. Rev. Neurosci. 2002, 25, 537–562. [Google Scholar] [CrossRef]
- Fantuzzi, L.; Tagliamonte, M.; Gauzzi, M.C.; Lopalco, L. Dual CCR5/CCR2 targeting: Opportunities for the cure of complex disorders. Cell. Mol. Life Sci. 2019, 76, 4869–4886. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Ogle, M.E.; Segar, C.E.; Sridhar, S.; Botchwey, E.A. Monocytes and macrophages in tissue repair: Implications for immunoregenerative biomaterial design. Exp. Biol. Med. 2016, 241, 1084–1097. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Ellis, R.J.; Marquine, M.J.; Kaul, M.; Fields, J.A.; Schlachetzki, J.C. Mechanisms underlying HIV-associated cognitive impairment and emerging therapies for its management. Nat. Rev. Neurol. 2023, 19, 668–687. [Google Scholar] [CrossRef]
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E.; Straube, E.; et al. HIV-1-associated neurocognitive disorder: Epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 2017, 264, 1715–1727. [Google Scholar] [CrossRef]
- Chen, N.C.; Partridge, A.T.; Sell, C.; Torres, C.; Martin-Garcia, J. Fate of microglia during HIV-1 infection: From activation to senescence? Glia 2017, 65, 431–446. [Google Scholar] [CrossRef]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 infection and AIDS: Consequences for the central nervous system. Cell Death Differ. 2005, 12 (Suppl. S1), 878–892. [Google Scholar] [CrossRef]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef]
- Borrajo, A.; Spuch, C.; Penedo, M.A.; Olivares, J.M.; Agis-Balboa, R.C. Important role of microglia in HIV-1 associated neurocognitive disorders and the molecular pathways implicated in its pathogenesis. Ann. Med. 2021, 53, 43–69. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Banks, W.A. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav. Immun. 2015, 45, 1–12. [Google Scholar] [CrossRef]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Katuri, A.; Bryant, J.; Heredia, A.; Makar, T.K. Role of the inflammasomes in HIV-associated neuroinflammation and neurocognitive disorders. Exp. Mol. Pathol. 2019, 108, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Wiley, C.A.; Kofler, J. Imaging microglial activation during neuroinflammation and Alzheimer’s disease. J. Neuroimmune Pharmacol. 2009, 4, 227–243. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Darwish, S.F.; Elbadry, A.M.M.; Elbokhomy, A.S.; Salama, G.A.; Salama, R.M. The dual face of microglia (M1/M2) as a potential target in the protective effect of nutraceuticals against neurodegenerative diseases. Front. Aging 2023, 4, 1231706. [Google Scholar] [CrossRef]
- Argandona Lopez, C.; Brown, A.M. Microglial- neuronal crosstalk in chronic viral infection through mTOR, SPP1/OPN and inflammasome pathway signaling. Front. Immunol. 2024, 15, 1368465. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, L.; Jia, B.; Wu, L.; Li, Y.; Curthoys, N.; Zheng, J.C. Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: Relevant to HIV-1-associated neurocognitive disorders. J. Neurosci. 2011, 31, 15195–15204. [Google Scholar] [CrossRef]
- Ravichandran, K.A.; Heneka, M.T. Inflammasomes in neurological disorders—Mechanisms and therapeutic potential. Nat. Rev. Neurol. 2024, 20, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Mamik, M.K.; Hui, E.; Branton, W.G.; McKenzie, B.A.; Chisholm, J.; Cohen, E.A.; Power, C. HIV-1 Viral Protein R Activates NLRP3 Inflammasome in Microglia: Implications for HIV-1 Associated Neuroinflammation. J. Neuroimmune Pharmacol. 2017, 12, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Chivero, E.T.; Guo, M.L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation. J. Neurosci. 2017, 37, 3599–3609. [Google Scholar] [CrossRef]
- Dutta, D.; Liu, J.; Xu, E.; Xiong, H. Methamphetamine Enhancement of HIV-1 gp120-Mediated NLRP3 Inflammasome Activation and Resultant Proinflammatory Responses in Rat Microglial Cultures. Int. J. Mol. Sci. 2024, 25, 3588. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- He, X.; Yang, W.; Zeng, Z.; Wei, Y.; Gao, J.; Zhang, B.; Li, L.; Liu, L.; Wan, Y.; Zeng, Q.; et al. NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology. Cell. Mol. Immunol. 2020, 17, 283–299. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Rawat, P.; Teodorof-Diedrich, C.; Spector, S.A. Human immunodeficiency virus Type-1 single-stranded RNA activates the NLRP3 inflammasome and impairs autophagic clearance of damaged mitochondria in human microglia. Glia 2019, 67, 802–824. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Kong, W.; Frouard, J.; Xie, G.; Corley, M.J.; Helmy, E.; Zhang, G.; Schwarzer, R.; Montano, M.; Sohn, P.; Roan, N.R.; et al. Neuroinflammation generated by HIV-infected microglia promotes dysfunction and death of neurons in human brain organoids. PNAS Nexus 2024, 3, pgae179. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.H.; Li, T.Q.; Zhang, W.; Zhao, Q.; Wang, Y. Targeting Inflammasome Activation in Viral Infection: A Therapeutic Solution? Viruses 2023, 15, 1451. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, S.E.; de Rivero Vaccari, J.P.; Dale, G.; Brand, F.J., 3rd; Nonner, D.; Bullock, M.R.; Dahl, G.P.; Dietrich, W.D.; Keane, R.W. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J. Cereb. Blood Flow. Metab. 2014, 34, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Coutts, G.; Lenart, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60. [Google Scholar] [CrossRef]
- Tang, Y.; Chaillon, A.; Gianella, S.; Wong, L.M.; Li, D.; Simermeyer, T.L.; Porrachia, M.; Ignacio, C.; Woodworth, B.; Zhong, D.; et al. Brain microglia serve as a persistent HIV reservoir despite durable antiretroviral therapy. J. Clin. Investig. 2023, 133, e167417. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef]
- Rheinberger, M.; Costa, A.L.; Kampmann, M.; Glavas, D.; Shytaj, I.L.; Sreeram, S.; Penzo, C.; Tibroni, N.; Garcia-Mesa, Y.; Leskov, K.; et al. Genomic profiling of HIV-1 integration in microglia cells links viral integration to the topologically associated domains. Cell Rep. 2023, 42, 112110. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Jennings, A.L.; Valada, A.; O’Shea, C.; Iskhakova, M.; Hu, B.; Javidfar, B.; Ben Hutta, G.; Lambert, T.Y.; Murray, J.; Kassim, B.; et al. HIV integration in the human brain is linked to microglial activation and 3D genome remodeling. Mol. Cell 2022, 82, 4647–4663.E8. [Google Scholar] [CrossRef] [PubMed]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Ho, Y.C. Interferon opens up: HIV-induced inflammation reconfigures 3D chromatin conformation and affects where HIV integrates. Mol. Cell 2022, 82, 4585–4587. [Google Scholar] [CrossRef]
- Van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 transcription and latency: An update. Retrovirology 2013, 10, 67. [Google Scholar] [CrossRef]
- Narasipura, S.D.; Kim, S.; Al-Harthi, L. Epigenetic regulation of HIV-1 latency in astrocytes. J. Virol. 2014, 88, 3031–3038. [Google Scholar] [CrossRef]
- Lu, F.; Zankharia, U.; Vladimirova, O.; Yi, Y.; Collman, R.G.; Lieberman, P.M. Epigenetic Landscape of HIV-1 Infection in Primary Human Macrophage. J. Virol. 2022, 96, e0016222. [Google Scholar] [CrossRef]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef]
- Williams, D.W.; Calderon, T.M.; Lopez, L.; Carvallo-Torres, L.; Gaskill, P.J.; Eugenin, E.A.; Morgello, S.; Berman, J.W. Mechanisms of HIV entry into the CNS: Increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS ONE 2013, 8, e69270. [Google Scholar] [CrossRef]
- Farhadian, S.; Patel, P.; Spudich, S. Neurological Complications of HIV Infection. Curr. Infect. Dis. Rep. 2017, 19, 50. [Google Scholar] [CrossRef]
- Sreeram, S.; Ye, F.; Garcia-Mesa, Y.; Nguyen, K.; El Sayed, A.; Leskov, K.; Karn, J. The potential role of HIV-1 latency in promoting neuroinflammation and HIV-1-associated neurocognitive disorder. Trends Immunol. 2022, 43, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Dumaop, W.; Smith, D.; Adame, A.; Everall, I.; Letendre, S.; Ellis, R.; Cherner, M.; Grant, I.; Masliah, E. Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology 2013, 80, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.R.; On, H.; Roberts, E.; Lu, H.K.; Moso, M.A.; Raison, J.A.; Papaioannou, C.; Cheng, W.J.; Ellett, A.M.; Jacobson, J.C.; et al. Toxicity and in vitro activity of HIV-1 latency-reversing agents in primary CNS cells. J. Neurovirol. 2016, 22, 455–463. [Google Scholar] [CrossRef]
- Sadowski, I.; Hashemi, F.B. Strategies to eradicate HIV from infected patients: Elimination of latent provirus reservoirs. Cell. Mol. Life Sci. 2019, 76, 3583–3600. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef]
- Herskovitz, J.; Hasan, M.; Patel, M.; Kevadiya, B.D.; Gendelman, H.E. Pathways Toward a Functional HIV-1 Cure: Balancing Promise and Perils of CRISPR Therapy. Methods Mol. Biol. 2022, 2407, 429–445. [Google Scholar] [CrossRef]
- Terry, R.L.; Getts, D.R.; Deffrasnes, C.; van Vreden, C.; Campbell, I.L.; King, N.J. Inflammatory monocytes and the pathogenesis of viral encephalitis. J. Neuroinflamm. 2012, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Schwab, M.E.; Popovich, P.G. Central nervous system regenerative failure: Role of oligodendrocytes, astrocytes, and microglia. Cold Spring Harb. Perspect. Biol. 2014, 7, a020602. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Clements, J.E. Eradication of HIV from the brain: Reasons for pause. AIDS 2011, 25, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antivir. Res. 2019, 166, 19–34. [Google Scholar] [CrossRef]
- Singh, V.; Dashti, A.; Mavigner, M.; Chahroudi, A. Latency Reversal 2.0: Giving the Immune System a Seat at the Table. Curr. HIV/AIDS Rep. 2021, 18, 117–127. [Google Scholar] [CrossRef]
- Barat, C.; Proust, A.; Deshiere, A.; Leboeuf, M.; Drouin, J.; Tremblay, M.J. Astrocytes sustain long-term productive HIV-1 infection without establishment of reactivable viral latency. Glia 2018, 66, 1363–1381. [Google Scholar] [CrossRef]
- Wong, M.E.; Johnson, C.J.; Hearps, A.C.; Jaworowski, A. Development of a Novel In Vitro Primary Human Monocyte-Derived Macrophage Model To Study Reactivation of HIV-1 Transcription. J. Virol. 2021, 95, e0022721. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef]
- Nuhn, M.M.; Gumbs, S.B.H.; Buchholtz, N.; Jannink, L.M.; Gharu, L.; de Witte, L.D.; Wensing, A.M.J.; Lewin, S.R.; Nijhuis, M.; Symons, J. Shock and kill within the CNS: A promising HIV eradication approach? J. Leukoc. Biol. 2022, 112, 1297–1315. [Google Scholar] [CrossRef]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood-brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9, 2336. [Google Scholar] [CrossRef] [PubMed]
- Aryal, M.; Arvanitis, C.D.; Alexander, P.M.; McDannold, N. Ultrasound-mediated blood-brain barrier disruption for targeted drug delivery in the central nervous system. Adv. Drug Deliv. Rev. 2014, 72, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef]
- Timbie, K.F.; Mead, B.P.; Price, R.J. Drug and gene delivery across the blood-brain barrier with focused ultrasound. J. Control. Release 2015, 219, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.T.; Wei, K.C.; Liu, H.L. Theranostic Strategy of Focused Ultrasound Induced Blood-Brain Barrier Opening for CNS Disease Treatment. Front. Pharmacol. 2019, 10, 86. [Google Scholar] [CrossRef]
- Treat, L.H.; McDannold, N.; Vykhodtseva, N.; Zhang, Y.; Tam, K.; Hynynen, K. Targeted delivery of doxorubicin to the rat brain at therapeutic levels using MRI-guided focused ultrasound. Int. J. Cancer 2007, 121, 901–907. [Google Scholar] [CrossRef]
- Gorick, C.M.; Breza, V.R.; Nowak, K.M.; Cheng, V.W.T.; Fisher, D.G.; Debski, A.C.; Hoch, M.R.; Demir, Z.E.F.; Tran, N.M.; Schwartz, M.R.; et al. Applications of focused ultrasound-mediated blood-brain barrier opening. Adv. Drug Deliv. Rev. 2022, 191, 114583. [Google Scholar] [CrossRef]
- McMahon, D.; O’Reilly, M.A.; Hynynen, K. Therapeutic Agent Delivery Across the Blood-Brain Barrier Using Focused Ultrasound. Annu. Rev. Biomed. Eng. 2021, 23, 89–113. [Google Scholar] [CrossRef]
- Chen, K.T.; Wei, K.C.; Liu, H.L. Focused Ultrasound Combined with Microbubbles in Central Nervous System Applications. Pharmaceutics 2021, 13, 1084. [Google Scholar] [CrossRef]
- Poon, C.; McMahon, D.; Hynynen, K. Noninvasive and targeted delivery of therapeutics to the brain using focused ultrasound. Neuropharmacology 2017, 120, 20–37. [Google Scholar] [CrossRef]
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, D.G.; Razansky, D.; Gotz, J. Ultrasound as a versatile tool for short- and long-term improvement and monitoring of brain function. Neuron 2023, 111, 1174–1190. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Weisholtz, D.S.; Strangman, G.E.; Yoo, S.S. Safety Review and Perspectives of Transcranial Focused Ultrasound Brain Stimulation. Brain Neurorehabilit. 2021, 14, e4. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Hynynen, K.; Lipsman, N. Applications of focused ultrasound in the brain: From thermoablation to drug delivery. Nat. Rev. Neurol. 2021, 17, 7–22. [Google Scholar] [CrossRef]
- Dobrakowski, P.P.; Machowska-Majchrzak, A.K.; Labuz-Roszak, B.; Majchrzak, K.G.; Kluczewska, E.; Pierzchala, K.B. MR-guided focused ultrasound: A new generation treatment of Parkinson’s disease, essential tremor and neuropathic pain. Interv. Neuroradiol. 2014, 20, 275–282. [Google Scholar] [CrossRef]
- Felix, M.S.; Borloz, E.; Metwally, K.; Dauba, A.; Larrat, B.; Matagne, V.; Ehinger, Y.; Villard, L.; Novell, A.; Mensah, S.; et al. Ultrasound-Mediated Blood-Brain Barrier Opening Improves Whole Brain Gene Delivery in Mice. Pharmaceutics 2021, 13, 1245. [Google Scholar] [CrossRef]
- Park, S.H.; Baik, K.; Jeon, S.; Chang, W.S.; Ye, B.S.; Chang, J.W. Extensive frontal focused ultrasound mediated blood-brain barrier opening for the treatment of Alzheimer’s disease: A proof-of-concept study. Transl. Neurodegener. 2021, 10, 44. [Google Scholar] [CrossRef]
- Burgess, A.; Shah, K.; Hough, O.; Hynynen, K. Focused ultrasound-mediated drug delivery through the blood-brain barrier. Expert. Rev. Neurother. 2015, 15, 477–491. [Google Scholar] [CrossRef]
- Simoncicova, E.; Goncalves de Andrade, E.; Vecchiarelli, H.A.; Awogbindin, I.O.; Delage, C.I.; Tremblay, M.E. Present and future of microglial pharmacology. Trends Pharmacol. Sci. 2022, 43, 669–685. [Google Scholar] [CrossRef]
- Savage, J.C.; Picard, K.; Gonzalez-Ibanez, F.; Tremblay, M.E. A Brief History of Microglial Ultrastructure: Distinctive Features, Phenotypes, and Functions Discovered Over the Past 60 Years by Electron Microscopy. Front. Immunol. 2018, 9, 803. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Goncalves de Andrade, E.; Kofoed, R.H.; Matthews, P.M.; Aubert, I.; Tremblay, M.E.; Morse, S.V. Using focused ultrasound to modulate microglial structure and function. Front. Cell. Neurosci. 2023, 17, 1290628. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, W.A.; Brandon, M.R.; Hunt, S.V.; Williams, A.F.; Gatter, K.C.; Mason, D.Y. Transferrin receptor on endothelium of brain capillaries. Nature 1984, 312, 162–163. [Google Scholar] [CrossRef]
- Ulbrich, K.; Hekmatara, T.; Herbert, E.; Kreuter, J. Transferrin- and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood-brain barrier (BBB). Eur. J. Pharm. Biopharm. 2009, 71, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Tashima, T. Smart Strategies for Therapeutic Agent Delivery into Brain across the Blood-Brain Barrier Using Receptor-Mediated Transcytosis. Chem. Pharm. Bull. 2020, 68, 316–325. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Burkhart, A.; Melander, F.; Kempen, P.J.; Vejlebo, J.B.; Siupka, P.; Nielsen, M.S.; Andresen, T.L.; Moos, T. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci. Rep. 2017, 7, 10396. [Google Scholar] [CrossRef]
- Pardridge, W.M. Kinetics of Blood-Brain Barrier Transport of Monoclonal Antibodies Targeting the Insulin Receptor and the Transferrin Receptor. Pharmaceuticals 2021, 15, 3. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Boado, R.J. Reengineering biopharmaceuticals for targeted delivery across the blood-brain barrier. Methods Enzymol. 2012, 503, 269–292. [Google Scholar] [CrossRef]
- Duffy, K.R.; Pardridge, W.M. Blood-brain barrier transcytosis of insulin in developing rabbits. Brain Res. 1987, 420, 32–38. [Google Scholar] [CrossRef]
- Boado, R.J.; Zhang, Y.; Wang, Y.; Pardridge, W.M. Engineering and expression of a chimeric transferrin receptor monoclonal antibody for blood-brain barrier delivery in the mouse. Biotechnol. Bioeng. 2009, 102, 1251–1258. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Jayant, R.D.; Nair, M. Nanomedicine for neuroHIV/AIDS management. Nanomedicine 2018, 13, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug targeting to the brain. Pharm. Res. 2007, 24, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, D.B.; Sandhu, J.K.; Costain, W.J. Emerging Technologies for Delivery of Biotherapeutics and Gene Therapy Across the Blood-Brain Barrier. BioDrugs 2018, 32, 547–559. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Kumar, S.; El-Hage, N.; Batrakova, E. Extracellular Vesicles in HIV, Drug Abuse, and Drug Delivery. J. Neuroimmune Pharmacol. 2020, 15, 387–389. [Google Scholar] [CrossRef]
- Mahajan, S.D.; Ordain, N.S.; Kutscher, H.; Karki, S.; Reynolds, J.L. HIV Neuroinflammation: The Role of Exosomes in Cell Signaling, Prognostic and Diagnostic Biomarkers and Drug Delivery. Front. Cell Dev. Biol. 2021, 9, 637192. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Shahjin, F.; Chand, S.; Yelamanchili, S.V. Extracellular Vesicles as Drug Delivery Vehicles to the Central Nervous System. J. Neuroimmune Pharmacol. 2020, 15, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; Pereira de Almeida, L. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Control. Release 2017, 262, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Doeppner, T.R.; Herz, J.; Gorgens, A.; Schlechter, J.; Ludwig, A.K.; Radtke, S.; de Miroschedji, K.; Horn, P.A.; Giebel, B.; Hermann, D.M. Extracellular Vesicles Improve Post-Stroke Neuroregeneration and Prevent Postischemic Immunosuppression. Stem Cells Transl. Med. 2015, 4, 1131–1143. [Google Scholar] [CrossRef]
- Guo, Y.; Hu, D.; Lian, L.; Zhao, L.; Li, M.; Bao, H.; Xu, S. Stem Cell-derived Extracellular Vesicles: A Promising Nano Delivery Platform to the Brain? Stem Cell Rev. Rep. 2023, 19, 285–308. [Google Scholar] [CrossRef]
- Branscome, H.; Khatkar, P.; Al Sharif, S.; Yin, D.; Jacob, S.; Cowen, M.; Kim, Y.; Erickson, J.; Brantner, C.A.; El-Hage, N.; et al. Retroviral infection of human neurospheres and use of stem Cell EVs to repair cellular damage. Sci. Rep. 2022, 12, 2019. [Google Scholar] [CrossRef]
- Ludwig, A.K.; Giebel, B. Exosomes: Small vesicles participating in intercellular communication. Int. J. Biochem. Cell Biol. 2012, 44, 11–15. [Google Scholar] [CrossRef]
- Fujita, Y.; Kadota, T.; Araya, J.; Ochiya, T.; Kuwano, K. Extracellular Vesicles: New Players in Lung Immunity. Am. J. Respir. Cell Mol. Biol. 2018, 58, 560–565. [Google Scholar] [CrossRef]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef]
- Patters, B.J.; Kumar, S. The role of exosomal transport of viral agents in persistent HIV pathogenesis. Retrovirology 2018, 15, 79. [Google Scholar] [CrossRef]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Columba Cabezas, S.; Federico, M. Sequences within RNA coding for HIV-1 Gag p17 are efficiently targeted to exosomes. Cell Microbiol. 2013, 15, 412–429. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected Cells Stimulate Production of Pro-inflammatory Cytokines through Trans-activating Response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.G.; Booth, A.; Gould, S.J.; Hildreth, J.E. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J. Biol. Chem. 2003, 278, 52347–52354. [Google Scholar] [CrossRef]
- Kadiu, I.; Narayanasamy, P.; Dash, P.K.; Zhang, W.; Gendelman, H.E. Biochemical and biologic characterization of exosomes and microvesicles as facilitators of HIV-1 infection in macrophages. J. Immunol. 2012, 189, 744–754. [Google Scholar] [CrossRef]
- Gould, S.J.; Booth, A.M.; Hildreth, J.E. The Trojan exosome hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 10592–10597. [Google Scholar] [CrossRef]
- Surnar, B.; Shah, A.S.; Park, M.; Kalathil, A.A.; Kamran, M.Z.; Ramirez Jaime, R.; Toborek, M.; Nair, M.; Kolishetti, N.; Dhar, S. Brain-Accumulating Nanoparticles for Assisting Astrocytes to Reduce Human Immunodeficiency Virus and Drug Abuse-Induced Neuroinflammation and Oxidative Stress. ACS Nano 2021, 15, 15741–15753. [Google Scholar] [CrossRef]
- Nowacek, A.; Gendelman, H.E. NanoART, neuroAIDS and CNS drug delivery. Nanomedicine 2009, 4, 557–574. [Google Scholar] [CrossRef]
- Wong, H.L.; Wu, X.Y.; Bendayan, R. Nanotechnological advances for the delivery of CNS therapeutics. Adv. Drug Deliv. Rev. 2012, 64, 686–700. [Google Scholar] [CrossRef]
- Wong, H.L.; Chattopadhyay, N.; Wu, X.Y.; Bendayan, R. Nanotechnology applications for improved delivery of antiretroviral drugs to the brain. Adv. Drug Deliv. Rev. 2010, 62, 503–517. [Google Scholar] [CrossRef]
- Chertok, B.; Moffat, B.A.; David, A.E.; Yu, F.; Bergemann, C.; Ross, B.D.; Yang, V.C. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials 2008, 29, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Saiyed, Z.M.; Gandhi, N.H.; Nair, M.P. Magnetic nanoformulation of azidothymidine 5’-triphosphate for targeted delivery across the blood-brain barrier. Int. J. Nanomed. 2010, 5, 157–166. [Google Scholar] [CrossRef]
- Nair, M.; Guduru, R.; Liang, P.; Hong, J.; Sagar, V.; Khizroev, S. Externally controlled on-demand release of anti-HIV drug using magneto-electric nanoparticles as carriers. Nat. Commun. 2013, 4, 1707. [Google Scholar] [CrossRef]
- Tomitaka, A.; Arami, H.; Huang, Z.; Raymond, A.; Rodriguez, E.; Cai, Y.; Febo, M.; Takemura, Y.; Nair, M. Hybrid magneto-plasmonic liposomes for multimodal image-guided and brain-targeted HIV treatment. Nanoscale 2017, 10, 184–194. [Google Scholar] [CrossRef]
- Jayant, R.D.; Tiwari, S.; Atluri, V.; Kaushik, A.; Tomitaka, A.; Yndart, A.; Colon-Perez, L.; Febo, M.; Nair, M. Multifunctional Nanotherapeutics for the Treatment of neuroAIDS in Drug Abusers. Sci. Rep. 2018, 8, 12991. [Google Scholar] [CrossRef]
- Hamadani, C.M.; Mahdi, F.; Merrell, A.; Flanders, J.; Cao, R.; Vashisth, P.; Dasanayake, G.S.; Darlington, D.S.; Singh, G.; Pride, M.C.; et al. Ionic Liquid Coating-Driven Nanoparticle Delivery to the Brain: Applications for NeuroHIV. Adv. Sci. 2024, 11, e2305484. [Google Scholar] [CrossRef]
- Dou, H.; Grotepas, C.B.; McMillan, J.M.; Destache, C.J.; Chaubal, M.; Werling, J.; Kipp, J.; Rabinow, B.; Gendelman, H.E. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J. Immunol. 2009, 183, 661–669. [Google Scholar] [CrossRef]
- Kanmogne, G.D.; Singh, S.; Roy, U.; Liu, X.; McMillan, J.; Gorantla, S.; Balkundi, S.; Smith, N.; Alnouti, Y.; Gautam, N.; et al. Mononuclear phagocyte intercellular crosstalk facilitates transmission of cell-targeted nanoformulated antiretroviral drugs to human brain endothelial cells. Int. J. Nanomed. 2012, 7, 2373–2388. [Google Scholar] [CrossRef]
- Jayant, R.D.; Atluri, V.S.; Agudelo, M.; Sagar, V.; Kaushik, A.; Nair, M. Sustained-release nanoART formulation for the treatment of neuroAIDS. Int. J. Nanomed. 2015, 10, 1077–1093. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Preat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Pinto, M.; Silva, V.; Barreiro, S.; Silva, R.; Remiao, F.; Borges, F.; Fernandes, C. Brain drug delivery and neurodegenerative diseases: Polymeric PLGA-based nanoparticles as a forefront platform. Ageing Res. Rev. 2022, 79, 101658. [Google Scholar] [CrossRef] [PubMed]
- Guedj, A.S.; Kell, A.J.; Barnes, M.; Stals, S.; Goncalves, D.; Girard, D.; Lavigne, C. Preparation, characterization, and safety evaluation of poly(lactide-co-glycolide) nanoparticles for protein delivery into macrophages. Int. J. Nanomed. 2015, 10, 5965–5979. [Google Scholar] [CrossRef]
- Latronico, T.; Rizzi, F.; Panniello, A.; Laquintana, V.; Arduino, I.; Denora, N.; Fanizza, E.; Milella, S.; Mastroianni, C.M.; Striccoli, M.; et al. Luminescent PLGA Nanoparticles for Delivery of Darunavir to the Brain and Inhibition of Matrix Metalloproteinase-9, a Relevant Therapeutic Target of HIV-Associated Neurological Disorders. ACS Chem. Neurosci. 2021, 12, 4286–4301. [Google Scholar] [CrossRef] [PubMed]
- Shive, M.S.; Anderson, J.M. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Ma, S.; Feng, X.; Liu, F.; Wang, B.; Zhang, H.; Niu, X. The pro-inflammatory response of macrophages regulated by acid degradation products of poly(lactide-co-glycolide) nanoparticles. Eng. Life Sci. 2021, 21, 709–720. [Google Scholar] [CrossRef]
- Puricelli, C.; Gigliotti, C.L.; Stoppa, I.; Sacchetti, S.; Pantham, D.; Scomparin, A.; Rolla, R.; Pizzimenti, S.; Dianzani, U.; Boggio, E.; et al. Use of Poly Lactic-co-glycolic Acid Nano and Micro Particles in the Delivery of Drugs Modulating Different Phases of Inflammation. Pharmaceutics 2023, 15, 1772. [Google Scholar] [CrossRef]
- Bazargani, A.; Hejazi, M.; Fernandez, M.; Cordeiro, A.; Tsala Ebode, J.; Lewinski, N.; da Rocha, S.; Golshahi, L. PEGylated solid lipid nanoparticles for the intranasal delivery of combination antiretroviral therapy composed of Atazanavir and Elvitegravir to treat NeuroAIDS. Int. J. Pharm. 2025, 670, 125166. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Approaches for CNS delivery of drugs—Nose to brain targeting of antiretroviral agents as a potential attempt for complete elimination of major reservoir site of HIV to aid AIDS treatment. Expert Opin. Drug Deliv. 2019, 16, 287–300. [Google Scholar] [CrossRef]
- Abbate, M.T.A.; Ramoller, I.K.; Sabri, A.H.; Paredes, A.J.; Hutton, A.J.; McKenna, P.E.; Peng, K.; Hollett, J.A.; McCarthy, H.O.; Donnelly, R.F. Formulation of antiretroviral nanocrystals and development into a microneedle delivery system for potential treatment of HIV-associated neurocognitive disorder (HAND). Int. J. Pharm. 2023, 640, 123005. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, Y.; Zhang, W.; Xia, X.; Li, H.; Qin, M.; Gao, H. Nanotechnology for enhanced nose-to-brain drug delivery in treating neurological diseases. J. Control. Release 2024, 366, 519–534. [Google Scholar] [CrossRef]
- Goel, H.; Kalra, V.; Verma, S.K.; Dubey, S.K.; Tiwary, A.K. Convolutions in the rendition of nose to brain therapeutics from bench to bedside: Feats & fallacies. J. Control. Release 2022, 341, 782–811. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Chen, C.; Kaminski, R.; Su, H.; Mancuso, P.; Sillman, B.; Zhang, C.; Liao, S.; Sravanam, S.; Liu, H.; et al. CRISPR editing of CCR5 and HIV-1 facilitates viral elimination in antiretroviral drug-suppressed virus-infected humanized mice. Proc. Natl. Acad. Sci. USA 2023, 120, e2217887120. [Google Scholar] [CrossRef]
- Khanal, S.; Cao, D.; Zhang, J.; Zhang, Y.; Schank, M.; Dang, X.; Nguyen, L.N.T.; Wu, X.Y.; Jiang, Y.; Ning, S.; et al. Synthetic gRNA/Cas9 Ribonucleoprotein Inhibits HIV Reactivation and Replication. Viruses 2022, 14, 1902. [Google Scholar] [CrossRef]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef]
- Kimberland, M.L.; Hou, W.; Alfonso-Pecchio, A.; Wilson, S.; Rao, Y.; Zhang, S.; Lu, Q. Strategies for controlling CRISPR/Cas9 off-target effects and biological variations in mammalian genome editing experiments. J. Biotechnol. 2018, 284, 91–101. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Mancuso, P.; Chen, C.; Kaminski, R.; Gordon, J.; Liao, S.; Robinson, J.A.; Smith, M.D.; Liu, H.; Sariyer, I.K.; Sariyer, R.; et al. CRISPR based editing of SIV proviral DNA in ART treated non-human primates. Nat. Commun. 2020, 11, 6065. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, A.K.; Muruve, D.A. Immunity to adeno-associated virus vectors in animals and humans: A continued challenge. Gene Ther. 2008, 15, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Kunze, C.; Borner, K.; Kienle, E.; Orschmann, T.; Rusha, E.; Schneider, M.; Radivojkov-Blagojevic, M.; Drukker, M.; Desbordes, S.; Grimm, D.; et al. Synthetic AAV/CRISPR vectors for blocking HIV-1 expression in persistently infected astrocytes. Glia 2018, 66, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Murlidharan, G.; Sakamoto, K.; Rao, L.; Corriher, T.; Wang, D.; Gao, G.; Sullivan, P.; Asokan, A. CNS-restricted Transduction and CRISPR/Cas9-mediated Gene Deletion with an Engineered AAV Vector. Mol. Ther. Nucleic Acids 2016, 5, e338. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategy | Mechanism of Action | Drug Delivery System | Advantages | Limitations |
|---|---|---|---|---|
| Antiretroviral Therapy (ART) | Suppresses viral replication, reduces systemic viral load | Oral, injectable, nanoparticle formulations | Established efficacy, reduces viral reservoirs | Limited BBB penetration, cannot target latent reservoirs |
| Latency-Reversing Agents (LRAs) | Reactivates latent HIV for immune clearance | Small molecules, histone deacetylase inhibitors (HDACi) | Potential to purge latent HIV reservoirs | Potential off-target effects, inflammation risk |
| Gene Editing (CRISPR/Cas9) | Excises integrated HIV DNA from host genome | AAV-based delivery, Lipid Nanoparticles | Permanent virus removal, potential cure | Delivery challenges, ethical and safety concerns |
| Neuroprotective Agents | Protects neurons from damage and apoptosis | Peptides, growth factors, small molecules | Preserves neuronal integrity, reduces oxidative stress | Limited BBB permeability, needs improved formulations |
| Anti-Inflammatory Drugs | Reduces neuroinflammation and cytokine release | NSAIDs, corticosteroids, IL-1β inhibitors | Targets neuroinflammation, prevents progression of HAND | Potential systemic side effects, incomplete neuroprotection |
| Monocyte/Microglia Modulators | Modulates immune cell trafficking to the CNS | CCR5 inhibitors, CCL2 modulators | Reduces immune cell-mediated neuroinflammation | Incomplete efficacy in reducing viral reservoirs |
| Focused Ultrasound (FUS) | Temporarily disrupts the BBB for targeted drug delivery | Microbubble-assisted ART delivery | Non-invasive, localized CNS drug delivery | Risk of non-specific BBB opening, transient effects |
| Receptor-Mediated Transport (RMT) | Uses transferrin/insulin receptors for BBB penetration | Nanocarriers conjugated to transferrin/insulin | Efficient brain penetration with minimal systemic toxicity | Receptor saturation limits delivery capacity |
| Exosome-Based Delivery | Natural vesicles for targeted drug delivery to CNS | Exosome-loaded ART and anti-inflammatory agents | Biocompatible, targeted drug delivery | Low drug loading capacity, limited scalability |
| Nanoparticle-Based Drug Delivery | Enhances drug penetration across BBB, sustained release | Polymeric, lipid, magnetic, and hybrid nanoparticles | Prolonged drug release, targeted CNS penetration | Potential toxicity, clearance limitations |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Said, N.; Venketaraman, V. Neuroinflammation, Blood–Brain Barrier, and HIV Reservoirs in the CNS: An In-Depth Exploration of Latency Mechanisms and Emerging Therapeutic Strategies. Viruses 2025, 17, 572. https://doi.org/10.3390/v17040572
Said N, Venketaraman V. Neuroinflammation, Blood–Brain Barrier, and HIV Reservoirs in the CNS: An In-Depth Exploration of Latency Mechanisms and Emerging Therapeutic Strategies. Viruses. 2025; 17(4):572. https://doi.org/10.3390/v17040572
Chicago/Turabian StyleSaid, Noor, and Vishwanath Venketaraman. 2025. "Neuroinflammation, Blood–Brain Barrier, and HIV Reservoirs in the CNS: An In-Depth Exploration of Latency Mechanisms and Emerging Therapeutic Strategies" Viruses 17, no. 4: 572. https://doi.org/10.3390/v17040572
APA StyleSaid, N., & Venketaraman, V. (2025). Neuroinflammation, Blood–Brain Barrier, and HIV Reservoirs in the CNS: An In-Depth Exploration of Latency Mechanisms and Emerging Therapeutic Strategies. Viruses, 17(4), 572. https://doi.org/10.3390/v17040572