Role of Lipids on Entry and Exit of Bluetongue Virus, a Complex Non-Enveloped Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. BTV morphology and its entry process into the host cells

3. BTV outer capsid structure


4. BTV replication and assembly

5. Interaction of outer capsid proteins of BTV with lipid rafts

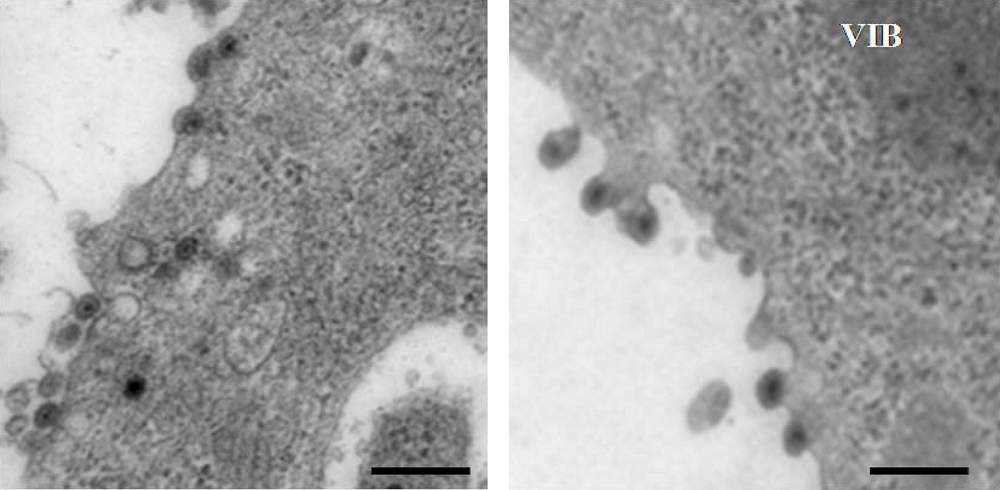
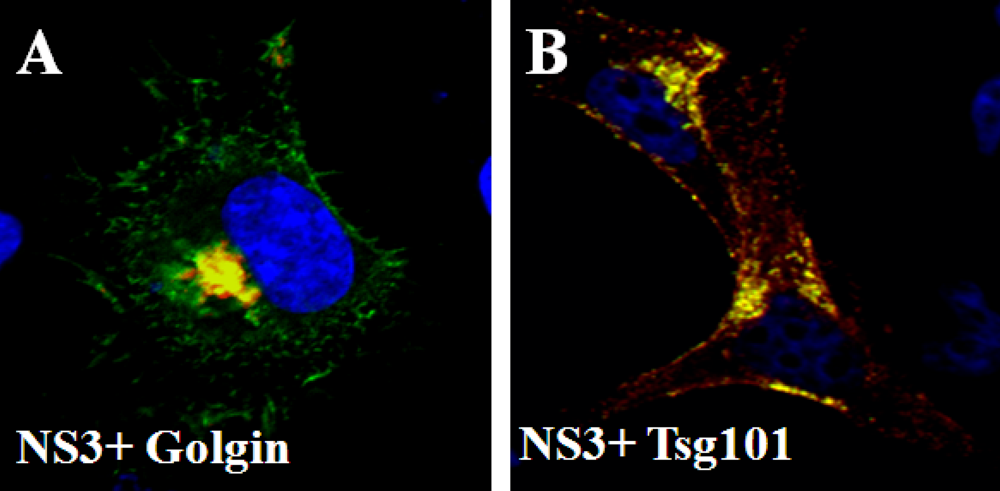
6. Release of BTV from infected cells; role of NS3 protein


7. Conclusions
Acknowledgments
References
- Marsh, M.; Helenius, A. Virus entry: open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Helenius, A. How viruses enter animal cells. Science 2004, 304, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Hirasawa, T.; Roy, P. Complete nucleotide sequence of segment 5 of epizootic haemorrhagic disease virus; the outer capsid protein VP5 is homologous to the VP5 protein of bluetongue virus. Virus Res. 1991, 20, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Hogle, J.M. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu. Rev. Microbiol. 2002, 56, 677–702. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Noad, R. Bluetongue virus assembly and morphogenesis. Curr. Top. Microbiol. Immunol. 2006, 309, 87–116. [Google Scholar] [PubMed]
- Hewat, E.A.; Booth, T.F.; Loudon, P.T.; Roy, P. Three-dimensional reconstruction of baculovirus expressed bluetongue virus core-like particles by cryo-electron microscopy. Virology 1992, 189, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.V.; Yamaguchi, S.; Roy, P. Three-dimensional structure of single-shelled bluetongue virus. J. Virol. 1992, 66, 2135–2142. [Google Scholar] [PubMed]
- Grimes, J.M.; Jakana, J.; Ghosh, M.; Basak, A.K.; Roy, P.; Chiu, W.; Stuart, D.I.; Prasad, B.V. An atomic model of the outer layer of the bluetongue virus core derived from X-ray crystallography and electron cryomicroscopy. Structure 1997, 5, 885–893. [Google Scholar] [CrossRef]
- Grimes, J.M.; Burroughs, J.N.; Gouet, P.; Diprose, J.M.; Malby, R.; Zientara, S.; Mertens, P.P.C.; Stuart, D.I. The atomic structure of the bluetongue virus core. Nature 1998, 395, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Hewat, E.A.; Booth, T.F.; Roy, P. Structure of bluetongue virus particles by cryoelectron microscopy. J. Struct. Biol. 1992, 109, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Nason, E.; Rothnagel, R.; Muknerge, S.K.; Kar, A.K.; Forzan, M.; Prasad, B.V.V.; Roy, P. Interactions between the Inner and Outer Capsids of Bluetongue Virus. J. Virol. 2004, 78, 8059–8067. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.H.; Roy, P. Expression and functional characterization of bluetongue virus VP2 protein: role in cell entry. J. Virol. 1999, 73, 9832–9842. [Google Scholar] [PubMed]
- Eaton, B.T. The replication of bluetongue virus. Curr. Top. Microbiol. Immunol. 1990, 162, 89–118. [Google Scholar] [PubMed]
- Forzan, M.; Wirblich, C.; Roy, P. A capsid protein of nonenveloped Bluetongue virus exhibits membrane fusion activity. Proc. Natl. Acad. Sci. USA 2004, 101, 2100–2105. [Google Scholar] [CrossRef]
- Forzan, M.; Marsh, M.; Roy, P. Bluetongue virus entry into cells. J. Virol. 2007, 81, 4819–4827. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Ramos, A.; Kohout, T.A.; Waldhoer, M.; Marsh, M. Endocytosis of the viral chemokine receptor US28 does not require beta-arrestins but is dependent on the clathrin-mediated pathway. Traffic 2003, 4, 243–253. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Smythe, E. Clathrin-coated vesicle formation: a paradigm for coated-vesicle formation. Biochem. Soc. Trans. 2003, 31, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Meier, O.; Greber, U.F. Adenovirus endocytosis. J. Gene Med. 2004, 6, S152–163. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, A.D.; Eaton, B.T.; Brookes, S.M. The release of bluetongue virus from infected cells and their superinfection by progeny virus. Virology 1989, 173, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.H.; Wirblich, C.; Forzan, M.; Roy, P. Expression and functional characterization of bluetongue virus VP5 protein: role in cellular permeabilization. J. Virol. 2001, 75, 8356–8367. [Google Scholar] [CrossRef] [PubMed]
- Colman, P.M.; Lawrence, M.C. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 2003, 4, 309–319. [Google Scholar] [CrossRef]
- Hewat, E.A.; Booth, T.F.; Roy, P. Structure of correctly self-assembled bluetongue virus-like particles. J. Struct. Biol. 1994, 112, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Bluetongue virus coat protein VP2 contains a sialic acid-binding domain and VP5 has similarities to enveloped virus fusion proteins. Proc. Natl. Academy. Sci. USA 2010, 107, 6292–6297. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Sun, Z.; Wagner, G.; Harrison, S.C. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. Embo J. 2002, 21, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Eaton, B.T.; Crameri, G.S. The site of bluetongue virus attachment to glycophorins from a number of animal erythrocytes. J. Gen. Virol. 1989, 70, 3347–3353. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Liu, J.; Wang, J.; Shen, S.; Lu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 1997, 94, 12303–12308. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997, 387, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Sauter, N.K.; Hanson, J.E.; Glick, G.D.; Brown, J.H.; Crowther, R.L.; Park, S.J.; Skehel, J.J.; Wiley, D.C. Binding of influenza virus hemagglutinin to analogs of its cell-surface receptor, sialic acid: analysis by proton nuclear magnetic resonance spectroscopy and X-ray crystallography. Biochemistry 1992, 31, 9609–9621. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; Rey, F.A.; Gaudin, Y.; Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Dormitzer, P.R.; Nason, E.B.; Prasad, B.V.; Harrison, S.C. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 2004, 430, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.A.; Huismans, H. The in vitro activation and further characterization of the bluetongue virus-associated transcriptase. Virology 1980, 104, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.A.; Huismans, H. In vitro transcription and translation of bluetongue virus mRNA. J. Gen. Virol. 1988, 69, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Stauber, N.; Martinez-Costas, J.; Sutton, G.; Monastyrskaya, K.; Roy, P. Bluetongue virus VP6 protein binds ATP and exhibits an RNA-dependent ATPase function and a helicase activity that catalyze the unwinding of double-stranded RNA substrates. J. Virol. 1997, 71, 7220–7226. [Google Scholar] [PubMed]
- Costas, J.; Sutton, G.; Roy, P. Guanylyltransferase and RNA 5'-triphopatase activities of the purified expressed VP4 protein of bluetongue virus. J. Mol. Biol. 1998, 280, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Ramadevi, N.; Burroughs, J.N.; Mertens, P.P.C.; Jones, I.M.; Roy, P. Capping and methylation of mRNA by purified recombinant VP4 protein of Bluetongue virus. Proc. Natl. Acad. Sci. 1998, 95, 13537–13542. [Google Scholar] [CrossRef]
- Ramadevi, N.; Roy, P. Bluetongue virus core protein VP4 has nucleoside triphosphate phosphohydrolase activity. J. Gen. Virol. 1998, 79, 2475–2480. [Google Scholar] [PubMed]
- Boyce, M.; Wehrfritz, J.; Noad, R.; Roy, P. Purified recombinant bluetongue virus VP1 exhibits RNA replicase activity. J. Virol. 2004, 78, 3994–4002. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.P.; Booth, T.F.; Roy, P. Synthesis of bluetongue virus-encoded phosphoprotein and formation of inclusion bodies by recombinant baculovirus in insect cells: it binds the single-stranded RNA species. J. Gen. Virol. 1990, 71, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Loudon, P.T.; Roy, P. Assembly of five bluetongue virus proteins expressed by recombinant baculoviruses: inclusion of the largest protein VP1 in the core and virus-like proteins. Virology 1991, 180, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, K.; Wirblich, C.; Brierley, I.; Roy, P. Sequence specificity in the interaction of Bluetongue virus non-structural protein 2 (NS2) with viral RNA. J. Biol. Chem. 2003, 278, 31722–31730. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.K.; Ghosh, M.; Roy, P. Mapping the assembly of Bluetongue virus scaffolding protein VP3. Virology 2004, 324, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.K.; Iwatani, N.; Roy, P. Assembly and intracellular localization of the bluetongue virus core protein VP3. J. Virol. 2005, 79, 11487–11495. [Google Scholar] [CrossRef] [PubMed]
- Modrof, J.; Lymperopoulos, K.; Roy, P. Phosphorylation of Bluetongue Virus Nonstructural Protein 2 Is Essential for Formation of Viral Inclusion Bodies. J. Virol. 2005, 79, 10023–10031. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, K.; Noad, R.; Tosi, S.; Nethisinghe, S.; Brierley, I.; Roy, P. Specific binding of Bluetongue virus NS2 to different viral plus-strand RNAs. Virology 2006, 353, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.K.; Bhattacharya, B.; Roy, P. Bluetongue virus RNA binding protein NS2 is a modulator of viral replication and assembly. BMC Mol. Biol. 2007, 8, 4. [Google Scholar] [CrossRef]
- Tanaka, S.; Roy, P. Identification of domains in bluetongue virus VP3 molecules essential for the assembly of virus cores. J. Virol. 1994, 68, 2795–2802. [Google Scholar] [PubMed]
- Limn, C.H.; Staeuber, N.; Monastyrskaya, K.; Gouet, P.; Roy, P. Functional dissection of the major structural protein of bluetongue virus: identification of key residues within VP7 essential for capsid assembly. J. Virol. 2000, 74, 8658–8669. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.S.; Taraporewala, Z.F.; Patton, J.T. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J. Virol. 2004, 78, 7763–7774. [Google Scholar] [CrossRef] [PubMed]
- Broering, T.J.; Kim, J.; Miller, C.L.; Piggott, C.D.; Dinoso, J.B.; Nibert, M. L.; Parker, J. S. Reovirus nonstructural protein mu NS recruits viral core surface proteins and entering core particles to factory-like inclusions. J. Virol. 2004, 78, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Arnold, M.M.; Broering, T.J.; Hastings, C.E.; Nibert, M. L. Localization of mammalian orthoreovirus proteins to cytoplasmic factory-like structures via nonoverlapping regions of microNS. J. Virol. 2010, 84, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Roy, P. Unpublished work . London School of Hygiene and Tripical Medicine: London, UK, 2008. [Google Scholar]
- Bhattacharya, B.; Roy, P. Bluetongue virus outer capsid protein VP5 interacts with membrane lipid rafts via a SNARE domain. J. Virol. 2008, 82, 10600–10612. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.; Simons, K. Caveolae, DIGs and the dynamics of sphingolipid-cholesterol microdomains. Curr. Opin. Cell Biol. 1997, 9, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.; Scheiffele, P.; Verkade, P.; Simons, K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 1998, 141, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.M.; Crowe, S.M.; Mak, J. Lipid rafts and HIV-1: from viral entry to assembly of progeny virions. J. Clin. Virol. 2001, 22, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Wilk, T.; Fuller, S.D. Do lipid rafts mediate virus assembly and pseudotyping? J. Gen. Virol. 2003, 84, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Freed, E.O. Role of lipid rafts in virus replication. Adv Virus Res. 2005, 64, 311–358. [Google Scholar] [CrossRef] [PubMed]
- Kundu, A.; Avalos, R.T.; Sanderson, C.M.; Nayak, D.P. Transmembrane domain of influenza virus neuraminidase, a type II protein, possesses an apical sorting signal in polarized MDCK cells. J Virol. 1996, 70, 6508–6515. [Google Scholar] [PubMed]
- Martin-Belmonte, F.; Lopez-Guerrero, J.A.; Carrasco, L.; Alonso, M.A. The amino-terminal nine amino acid sequence of poliovirus capsid VP4 protein is sufficient to confer N-myristoylation and targeting to detergent-insoluble membranes. Biochemistry 2000, 39, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Moreira, J. E.; Liu, V.; Sugimori, M.; Mikoshiba, K.; Llinas, R. R. Role of the conserved WHXL motif in the C terminus of synaptotagmin in synaptic vesicle docking. Proc. Natl. Acad. Sci. USA 2000, 97, 14715–14719. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Noad, R. J.; Roy, P. Interaction between Bluetongue virus outer capsid protein VP2 and vimentin is necessary for virus egress. J. Virol. 2007, 4, 7. [Google Scholar] [CrossRef]
- Starkey, W.G.; Collins, J.; Wallis, T.S.; Clarke, G.J.; Spencer, A.J.; Haddon, S.J.; Osborne, M. P.; Candy, D. C.; Stephen, J. Kinetics, tissue specificity and pathological changes in murine rotavirus infection of mice. J Gen Virol. 1986, 67, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Pesavento, J.B.; Crawford, S.E.; Estes, M.K.; Prasad, B.V. Rotavirus proteins: structure and assembly. Curr. Top. Microbiol. Immunol. 2006, 309, 189–219. [Google Scholar] [PubMed]
- Storey, S.M.; Gibbons, T.F.; Williams, C.V.; Parr, R.D.; Schroeder, F.; Ball, J.M. Full-length, glycosylated NSP4 is localized to plasma membrane caveolae by a novel raft isolation technique. J. Virol. 2007, 81, 5472–5483. [Google Scholar] [CrossRef] [PubMed]
- Delmas, O.; Breton, M.; Sapin, C.; Le Bivic, A.; Colard, O.; Trugnan, G. Heterogeneity of Raft-type membrane microdomains associated with VP4, the rotavirus spike protein, in Caco-2 and MA 104 cells. J. Virol. 2007, 81, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Mirre, C.; Monlauzeur, L.; Garcia, M.; Delgrossi, M.H.; Le Bivic, A. Detergent-resistant membrane microdomains from Caco-2 cells do not contain caveolin. Am. J. Physiol. 1996, 271, 887–894. [Google Scholar]
- Chwetzoff, S.; Trugnan, G. Rotavirus assembly: an alternative model that utilizes an atypical trafficking pathway. Curr. Top. Microbiol. Immunol. 2006, 309, 245–261. [Google Scholar] [PubMed]
- Sapin, C.; Colard, O.; Delmas, O.; Tessier, C.; Breton, M.; Enouf, V.; Chwetzoff, S.; Ouanich, J.; Cohen, J.; Wolf, C.; Trugnan, G. Rafts promote assembly and atypical targeting of a nonenveloped virus, rotavirus, in Caco-2 cells. J. Virol. 2002, 76, 4591–4602. [Google Scholar] [CrossRef] [PubMed]
- Cuadras, M.A.; Greenberg, H.B. Rotavirus infectious particles use lipid rafts during replication for transport to the cell surface in vitro and in vivo. Virology 2003, 313, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Mohan, K.V.; Muller, J.; Atreya, C.D. Defective rotavirus particle assembly in lovastatin-treated MA104 cells. Arch. Virol. 2008, 153, 2283–2290. [Google Scholar] [CrossRef] [PubMed]
- Eaton, B.T.; Hyatt, A.D.; White, J. R. Association of bluetongue virus with the cytoskeleton. Virology 1987, 157, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, A.D.; Gould, A.R.; Coupar, B.; Eaton, B.T. Localization of the non-structural protein NS3 in bluetongue virus-infected cells. J. Gen. Virol. 1991, 72, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Beaton, A.R.; Rodriguez, J.; Reddy, Y.K.; Roy, P. The membrane trafficking protein calpactin forms a complex with bluetongue virus protein NS3 and mediates virus release. Proc. Natl. Acad. Sci.USA 2002, 99, 13154–13159. [Google Scholar] [CrossRef]
- Wu, X.; Chen, S.Y.; Iwata, H.; Compans, R.W.; Roy, P. Multiple glycoproteins synthesized by the smallest RNA segment (S10) of bluetongue virus. J. Virol. 1992, 66, 7104–7112. [Google Scholar] [PubMed]
- Han, Z.; Harty, R.N. The NS3 protein of bluetongue virus exhibits viroporin-like properties. J. Biol. Chem. 2004, 279, 43092–43097. [Google Scholar] [CrossRef] [PubMed]
- Babst, M.; Katzmann, D.J.; Snyder, W.B.; Wendland, B.; Emr, S.D. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell. 2002, 3, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Gruenberg, J.; Stenmark, H. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 2004, 5, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Wirblich, C.; Bhattacharya, B.; Roy, P. Nonstructural protein 3 of bluetongue virus assists virus release by recruiting ESCRT-I protein Tsg101. J. Virol. 2006, 80, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Celma, C.C.; Roy, P. A viral nonstructural protein regulates bluetongue virus trafficking and release. J. Virol. 2009, 83, 6806–6816. [Google Scholar] [CrossRef] [PubMed]
- Blanchette-Mackie, E.J.; Dwyer, N.K.; Amende, L.M.; Kruth, H.S.; Butler, J.D.; Sokol, J.; Comly, M.E.; Vanier, M.T.; August, J.T.; Brady, R.O. et al. Type-C Niemann-Pick disease: low density lipoprotein uptake is associated with premature cholesterol accumulation in the Golgi complex and excessive cholesterol storage in lysosomes. Proc Natl Acad Sci USA. 1988, 85, 8022–8026. [Google Scholar] [CrossRef]
- Coxey, R.A.; Pentchev, P.G.; Campbell, G.; Blanchette-Mackie, E.J. Differential accumulation of cholesterol in Golgi compartments of normal and Niemann-Pick type C fibroblasts incubated with LDL: a cytochemical freeze-fracture study. J. Lipid Res. 1993, 34, 1165–1176. [Google Scholar] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, G.; Zimmer, K.P.; Trotz, I.; Herrler, G. Vesicular stomatitis virus glycoprotein does not determine the site of virus release in polarized epithelial cells. J. Virol. 2002, 76, 4103–4107. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, J.E.; Fusco, P.J. The M protein of vesicular stomatitis virus associates specifically with the basolateral membranes of polarized epithelial cells independently of the G protein. J. Cell Biol. 1988, 107, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Maisner, A.; Klenk, H.; Herrler, G. Polarized budding of measles virus is not determined by viral surface glycoproteins. J. Virol. 1998, 72, 5276–5278. [Google Scholar] [PubMed]
- Naim, H.Y.; Ehler, E.; Billeter, M.A. Measles virus matrix protein specifies apical virus release and glycoprotein sorting in epithelial cells. EMBO J. 2000, 19, 3576–3585. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, N.; Maurice, M.; Delautier, D.; Quero, A.M.; Servin, A.L.; Trugnan, G. Rotavirus is released from the apical surface of cultured human intestinal cells through nonconventional vesicular transport that bypasses the Golgi apparatus. J. Virol. 1997, 71, 8268–8278. [Google Scholar] [PubMed]
- Sapin, C.; Colard, O.; Delmas, O.; Tessier, C.; Breton, M.; Enouf, V.; Chwetzoff, S.; Ouanich, J.; Cohen, J.; Wolf, C.; Trugnan, G. Rafts promote assembly and atypical targeting of a nonenveloped virus, rotavirus, in Caco-2 cells. J. Virol. 2002, 76, 4591–4602. [Google Scholar] [CrossRef] [PubMed]
- Cuadras, M.A.; Greenberg, H. B. Rotavirus infectious particles use lipid rafts during replication for transport to the cell surface in vitro and in vivo. Virology 2003, 313, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Trask, S.D.; Kim, I.S.; Harrison, S.C.; Dormitzer, P.R. A rotavirus spike protein conformational intermediate binds lipid bilayers. J. Virol. 2010, 84, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Vaney, M.C.; Roussel, A.; Vigouroux, A.; Reilly, B.; Lepault, J.; Kielian, M.; Rey, F.A. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 2004, 427, 320–325. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Bhattacharya, B.; Roy, P. Role of Lipids on Entry and Exit of Bluetongue Virus, a Complex Non-Enveloped Virus. Viruses 2010, 2, 1218-1235. https://doi.org/10.3390/v2051218
Bhattacharya B, Roy P. Role of Lipids on Entry and Exit of Bluetongue Virus, a Complex Non-Enveloped Virus. Viruses. 2010; 2(5):1218-1235. https://doi.org/10.3390/v2051218
Chicago/Turabian StyleBhattacharya, Bishnupriya, and Polly Roy. 2010. "Role of Lipids on Entry and Exit of Bluetongue Virus, a Complex Non-Enveloped Virus" Viruses 2, no. 5: 1218-1235. https://doi.org/10.3390/v2051218
APA StyleBhattacharya, B., & Roy, P. (2010). Role of Lipids on Entry and Exit of Bluetongue Virus, a Complex Non-Enveloped Virus. Viruses, 2(5), 1218-1235. https://doi.org/10.3390/v2051218