The Revolution in Viral Genomics as Exemplified by the Bioinformatic Analysis of Human Adenoviruses


Abstract
:1. Introduction
2. The Labor of Human Adenovirus Genomics
First Example of Comparative Genomics in Human Adenoviruses

3. The Birth of Human Adenovirus Genomics
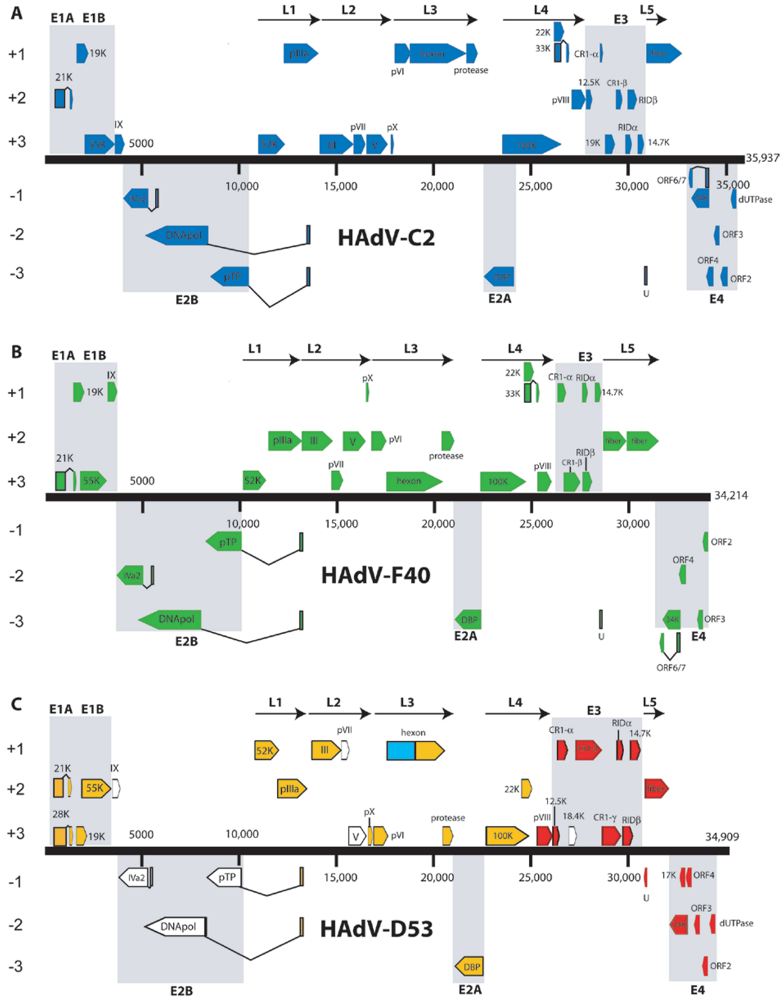
3.1. HAdV-C2: The First HAdV Sequenced

3.2. HAdV-C5
3.3. Illegitimate Recombination Offered as a Possible Mechanism for Molecular Evolution
3.4. HAdV-B11

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HAdV | Year sequenced | Reference |
|---|---|---|
| HAdV-C2 | 1984 | [5] |
| HAdV-C5 | 1992 | [7] |
| HAdV-F40 | 1993 | [9] |
| HAdV-A12 | 1994 | [10] |
| HAdV-D17 | 1999 | NP |
| HAdV-B11 | 2003 | [18] |
| HAdV-B35 | [19] | |
| [28] | ||
| [29] | ||
| HAdV-C1 | 2004 | [27] |
| HAdV-D9 | NP | |
| HAdV-B16 | NP | |
| HAdV-B21 | NP | |
| HAdV-B34 | NP | |
| HAdV-B50 | NP | |
| HAdV-E4 | 2005 | [26] |
| HAdV-B7 | [26] | |
| HAdV-F41 | NP | |
| HAdV-B3 | 2006 | [30] |
| HAdV-D26 | [31] | |
| HAdV-D46 | NP | |
| HAdV-D48 | NP | |
| HAdV-D49 | NP | |
| NP | ||
| HAdV-G52 | 2007 | [32] |
| HAdV-D37 | 2008 | [8] |
| HAdV-D19 | [33] | |
| HAdV-B14 | 2009 | [34] |
| HAdV-D8 | NP | |
| HAdV-D22 | [16] | |
| HAdV-D28 | NP | |
| HAdV-A31 | [35] | |
| HAdV-D53 | [6] | |
| HAdV-D54 | [36] | |
| HAdV-A18 | 2010 | [12] |
| HAdV-D36 | [37] | |
| HAdV-B55 | [17] |
4. Major Advances in the Bioinformatic Revolution of Human Adenoviruses
4.1. HAdV-G52
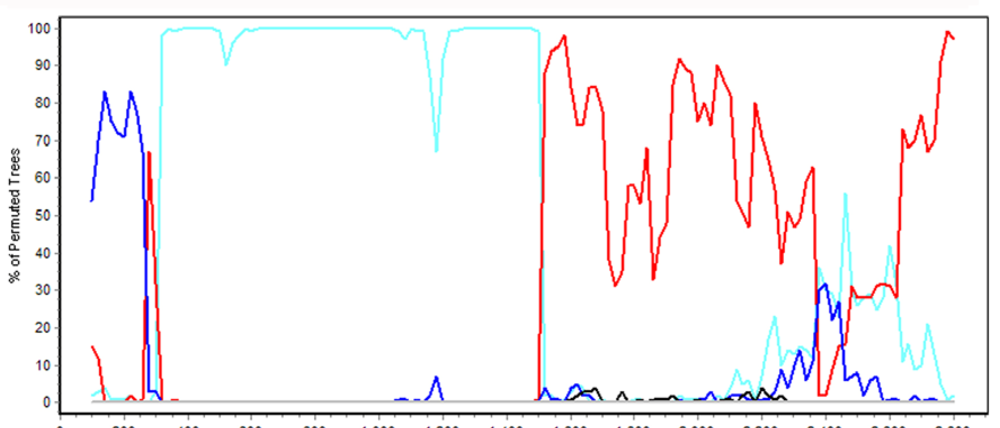
4.2. Simplot Analysis of Novel Adenovirus in Species HAdV-C
4.3. HAdV-D53
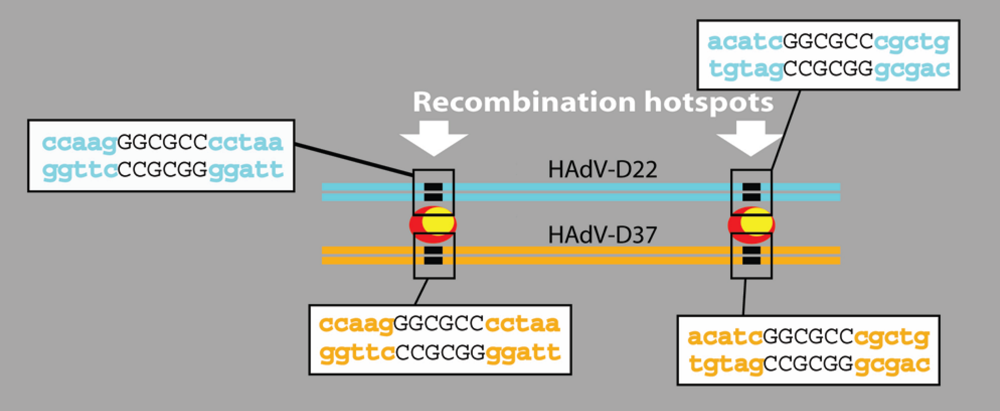
4.4. Recombination is Common in the Penton Gene of Species HAdV-D

4.5. Hypothetical Recognition Sites for Recombination in the Hexon Gene
4.6. HAdV-B55


5. The Future
6. Conclusions
| HAdV | ATCC number |
|---|---|
| HAdV-D10 | VR-1087 |
| HAdV-D13 | VR-14 |
| HAdV-D15 | VR-16 |
| HAdV-D20 | VR-255 |
| HAdV-D23 | VR-258 |
| HAdV-D24 | VR-259 |
| HAdV-D25 | VR-223 |
| HAdV-D27 | VR-1105 |
| HAdV-D28 | VR-226 |
| HAdV-D29 | VR-272 |
| HAdV-D30 | VR-273 |
| HAdV-D32 | VR-625 |
| HAdV-D33 | VR-626 |
| HAdV-D38 | VR-988 |
| HAdV-D39 | VR-932 |
| HAdV-D42 | VR-1304 |
| HAdV-D44 | VR-1306 |
| HAdV-D45 | VR-1307 |
| HAdV-D47 | VR-1309 |
| HAdV-D51 | VR-1603 |
Acknowledgments
References
- Mautner, V.; Williams, J.; Sambrook, J.; Sharp, P.A.; Grodzicker, T. The location of the genes coding for hexon and fiber proteins in adenovirus DNA. Cell 1975, 5, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Akusjarvaiand, G.; Pettersson, U. Nucleotide sequence at the junction between the coding region of the adenovirus 2 hexon messenger RNA and its leader sequence. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 5822–5826. [Google Scholar] [CrossRef] [PubMed]
- Kinloch, R.; Mackay, N.; Mautner, V. Adenovirus hexon sequence comparison of subgroup C serotypes 2 and 5. J. Biol. Chem. 1984, 259, 6431–6436. [Google Scholar] [PubMed]
- Roberts, R.J.; O’Neill, K.E.; Yen, C.T. DNA Sequences from the Adenovirus 2 Genome. J. Biol. Chem. 1984, 259, 13968–13975. [Google Scholar] [PubMed]
- Walsh, M.P.; Chintakuntlawar, A.; Robinson, C.M.; Madisch, I.; Harrach, B.; Hudson, N.R.; Schnurr, D.; Heim, A.; Chodosh, J.; Seto, D.; Jones, M.S. Evidence of molecular evolution driven by recombination events influencing tropism in a novel human adenovirus that causes epidemic keratoconjunctivitis . PLoS One 2009, 4, e5635. [Google Scholar] [CrossRef] [PubMed]
- Chroboczek, J.; Bieber, F.; Jacrot, B. The sequence of the genome of adenovirus type 5 and its comparison with the genome of adenovirus type 2. Virology 1992, 186, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Shariati, F.; Gillaspy, A.F.; Dyer, D.W.; Chodosh, J. Genomic and bioinformatics analysis of human adenovirus type 37: new insights into corneal tropism. BMC Genomics 2008, 9, 213. [Google Scholar] [CrossRef]
- Davison, A.J.; Benkö, M.; Harrach, B. Genetic content and evolution of adenoviruses . J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Sprengel, J.; Schmitz, B.; Heuss-Neitzel, D.; Zock, C.; Doerfler, W. Nucleotide sequence of human adenovirus type 12 DNA: comparative functional analysis. J. Virol. 1994, 68, 379–389. [Google Scholar] [PubMed]
- Hofmayer, S.; Madisch, I.; Darr, S.; Rehren, F.; Heim, A. Unique sequence features of the Human Adenovirus 31 complete genomic sequence are conserved in clinical isolates. BMC Genomics 2009, 10, 557. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.P.; Seto, J; Tirado, D.; Chodosh, J.; Schnurr, D.; Seto, D.; Jones, M.S. Genomic analysis of human adenovirus prototype 18 . Virology In press.
- Crawford-Miksza, L.; Schnurr, D.P. Analysis of 15 adenovirus hexon proteins reveals the location and structure of seven hypervariable regions containing serotype-specific residues. J. Virol. 1996, 70, 1836–1844. [Google Scholar] [PubMed]
- Rux, J.J.; Burnett, R.M. Type-specific epitope locations revealed by X-ray crystallographic study of adenovirus type 5 hexon. Mol. Ther. 2000, 1, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Ivanova, O.E.; Eremeeva, T.P.; Iggo, R.D. Evidence of frequent recombination among human adenoviruses. J. Gen. Virol. 2008, 89, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Rajaiya, J.; Walsh, M.P.; Seto, D.; Dyer, D.W.; Jones, M.S.; Chodosh, J. Computational analysis of human adenovirus type 22 provides evidence for recombination between human adenoviruses species D in the penton base gene. J. Virol. 2009, 83, 8980–8985. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.P.; Seto, J.; Jones, M.S.; Chodosh, J.; Xu, W.; Seto, D. Computational analysis identifies human adenovirus type 55 as a re-emergent acute respiratory disease pathogen. J. Clin. Microbiol. 2010, 48, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Furthmann, A.; Sandig, V.; Lieber, A. The complete nucleotide sequence, genome organization, and origin of human adenovirus type 11. Virology 2003, 309, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.F.; Skog, J.; Lindman, K.; Wadell, G. Comparative analysis of the genome organization of human adenovirus 11, a member of the human adenovirus species B, and the commonly used human adenovirus 5 vector, a member of species C. J. Gen. Virol. 2003, 84, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Hilleman, M.R.; Werner, J.H. Recovery of new agent from patients with acute respiratory illness. Proc. Soc. Exp. Biol. Med. 1954, 851, 183–188. [Google Scholar]
- Hilleman, M.R.; Anderson, S.A.; Levinson, D.J.; Luecking, M.L. Occurrence and significance of adenovirus antibody in sera of recruit and cadre military personnel. Am. J. Hyg. 1958, 67, 179–186. [Google Scholar] [PubMed]
- Rosenbaum, M.J.; Edwards, E.A.; Hoeffler, D.C. Recent experiences with live adenovirus vaccines in Navy recruits. Mil. Med. 1975, 140, 251–257. [Google Scholar] [PubMed]
- Top Jr., F.H.; Grossman, R.A.; Bartelloni, P.J.; Segal, H.; Dudding, B.; Russell, P.; Buescher, E. Immunization with live types 7 and 4 adenovirus vaccines. I. Safety, infectivity, antigenicity, and potency of adenovirus type 7 vaccine in humans . J. Infect. Dis. 1971, 124, 148–154. [Google Scholar] [PubMed]
- Hendrix, R.M.; Lindner, J.L.; Benton, F.R.; Monteith, S.C.; Tuchscherer, M.A.; Gray, G.C.; Gaydos, J.C. Large, Persistent Epidemic of Adenovirus Type 4-Associated Acute Respiratory Disease in U. S. Army Trainees. Emer. Inf. Dis. 1999, 5, 798–801. [Google Scholar] [CrossRef]
- Purkayastha, A.; Su, J.; McGraw, J.; Ditty, S.E.; Hadfield, T.L.; Seto, J.; Russell, K.L.; Tibbetts, C.; Seto, D. Genomic and bioinformatics analyses of HAdV-4vac and HAdV-7vac, two human adenovirus (HAdV) strains that constituted original prophylaxis against HAdV-related acute respiratory disease, a reemerging epidemic disease. J. Clin. Microbiol. 2005, 43, 3083–3094. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, A.; Ditty, S.E.; Su, J.; McGraw, J.; Hadfield, T.L.; Tibbetts, C.; Seto, D. Genomic and bioinformatics analysis of HAdV-4, a human adenovirus causing acute respiratory disease: implications for gene therapy and vaccine vector development. J. Virol. 2005, 79, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Lauer, K.P.; Llorente, I.; Blair, E.; Seto, J.; Krasnov, V.; Purkayastha, A.; Ditty, S.E.; Hadfield, T.L.; Buck, C.; Tibbetts, C.; Seto, D. Natural variation among human adenoviruses: genome sequence and annotation of human adenovirus serotype 1. J. Gen. Virol. 2004, 85, 2615–2625. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Robbins, P.D.; Gambotto, A. Human adenovirus type 35: nucleotide sequence and vector development. Gene Therapy 2003, 10, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Vogels, R.; Zuijdgeest, D.; van Rijnsoever, R.; Hartkoorn, E.; Damen, I.; de Béthune, M.P.; Kostense, S;; Helmus, N.; Koudstaal, W.; et al. Replication-deficient human adenovirus type 35 vectors for gene transfer and vaccination: efficient human cell infection and bypass of preexisting adenovirus immunity . J. Virol. 2002, 77, 8263–8271. [Google Scholar] [CrossRef]
- Zhang, Q.; Su, X.; Gong, S.; Zeng, Q.; Zhu, B.; Wu, Z.; Peng, T.; Zhang, C.; Zhou, R. Comparative genomic analysis of two strains of human adenovirus type 3 isolated from children with acute respiratory infection in southern China. J. Gen. Virol. 2006, 87, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, P.; Seto, J.; Tibbetts, C.; Seto, D. Natural variants of human adenovirus type 3 provide evidence for relative genome stability across time and geographic space . Virology 2010, 397, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.S.; Harrach, B.; Ganac, R.D.; Gozum, M.M.; Dela Cruz, W.P.; Riedel, B.; Pan, C.; Delwart, E.L.; Schnurr, D.P. New adenovirus species found in a patient presenting with gastroenteritis. J. Virol. 2007, 81, 5978–5984. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Shariati, F.; Zaitshik, J.; Gillaspy, A.F.; Dyer, D.W.; Chodosh, J. Human adenovirus type 19: Genomic and bioinformatics analysis of a keratoconjunctivitis isolate. Virus Res. 2009, 139, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Seto, J.; Walsh, M.P.; Mahadevan, P.; Purkayastha, A.; Clark, J.M.; Tibbetts, C.; Seto, D. Genomic and bioinformatics analyses of HAdV-14p, reference strain of are-emerging respiratory pathogen and analysis of B1/B2. Virus Res. 2009, 143, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Darr, S.; Madisch, I.; Hofmayer, S.; Rehren, F.; Heim, A. Phylogeny and primary structure analysis of fiber shafts of all human adenovirus types for rational design of adenoviral gene-therapy vectors. J. Gen.Virol. 2009, 90, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Iida, T.; Ishiko, H.; Ohguchi, T.; Ariga, T.; Tagawa, Y.; Aoki, K.; Ohno, S.; Suzutani, T. Analysis of the complete genome sequence of epidemic keratoconjunctivitis-related human adenovirus type 8, 19, 37 and a novel serotype. J. Gen. Virol. 2009, 90, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.; Jánoska, M.; Kajon, A.E.; Metzgar, D.; Hudson, N.R.; Torres, S.; Harrach, B.; Seto, D.; Chodosh, J.; Jones, M.S. Genomic characterization of human adenovirus 36, a putative obesity agent. Virus Res. 2010, 149, 152–161. [Google Scholar] [CrossRef] [PubMed]
- de Jong, J.C.; Osterhaus, A.D.; Jones, M.S.; Harrach, B. Human adenovirus type 52: a type 41 in disguise? J. Virol. 2008, 82, 3809–3810. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, D.; Dondero, M. Two new candidate adenovirus serotypes. Intervirology 1993, 36, 79–83. [Google Scholar] [PubMed]
- Engelmann, I.; Madisch, I.; Pommer, H.; Heim, A. An outbreak of epidemic keratoconjunctivitis caused by a new intermediate adenovirus 22/H8 identified by molecular typing . Clin. Infect. Dis. 2006, 43, e64–e66. [Google Scholar] [CrossRef] [PubMed]
- Ishiko, H.; Aoki, K. Spread of epidemic keratoconjunctivitis due to a novel serotype of human adenovirus in Japan. J. Clin. Microbiol. 2009, 47, 2678–2679. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhu, Z.; Tang, L.; Wang, L; Tan, X.; Yu, P.; Zhang, Y.; Tian, X.; Wang, J.; Zhang, Y.; Li, D.; Xu, W. Genomic analyses of recombinant adenovirus type 11a in China . J. Clin. Microbiol. 2009, 47, 3082–3090. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Torres, S.; Chodosh, J.; Seto, D.; Jones, M.S. The Revolution in Viral Genomics as Exemplified by the Bioinformatic Analysis of Human Adenoviruses. Viruses 2010, 2, 1367-1381. https://doi.org/10.3390/v2071367
Torres S, Chodosh J, Seto D, Jones MS. The Revolution in Viral Genomics as Exemplified by the Bioinformatic Analysis of Human Adenoviruses. Viruses. 2010; 2(7):1367-1381. https://doi.org/10.3390/v2071367
Chicago/Turabian StyleTorres, Sarah, James Chodosh, Donald Seto, and Morris S. Jones. 2010. "The Revolution in Viral Genomics as Exemplified by the Bioinformatic Analysis of Human Adenoviruses" Viruses 2, no. 7: 1367-1381. https://doi.org/10.3390/v2071367
APA StyleTorres, S., Chodosh, J., Seto, D., & Jones, M. S. (2010). The Revolution in Viral Genomics as Exemplified by the Bioinformatic Analysis of Human Adenoviruses. Viruses, 2(7), 1367-1381. https://doi.org/10.3390/v2071367