Role of Gag in HIV Resistance to Protease Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The HIV-1 protease and its natural substrates

3. HIV resistance to PIs: protease mutations
5. Cleavage site mutations as compensatory changes for resistance-associated loss of viral fitness

6. Cleavage site mutations as resistance mutations
7. Mechanisms of action of Gag cleavage site mutations
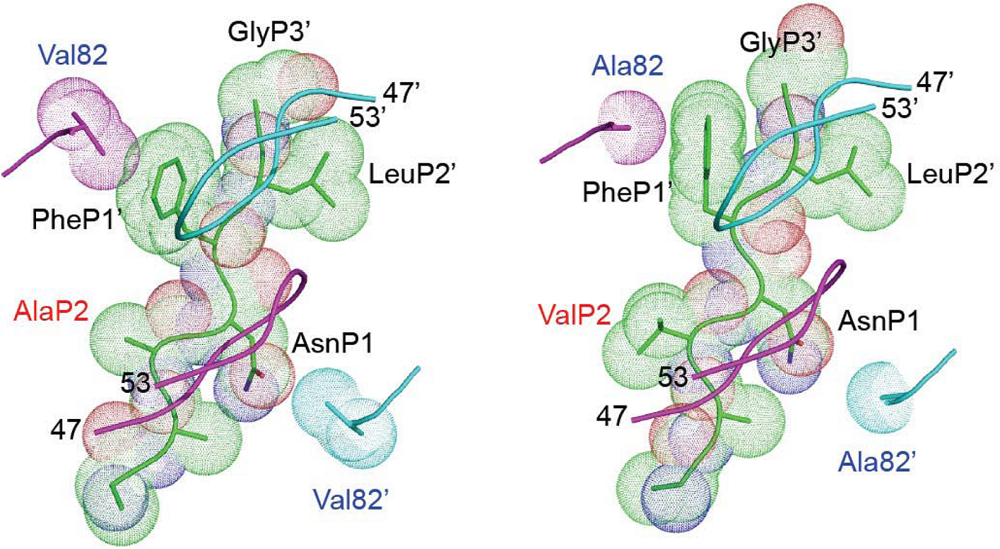
8. Role of other mutations or polymorphisms in Gag in HIV resistance to PIs
9. Gag cleavage site mutations in a clinical context: frequency, kinetics of emergence, and impact on treatment response in vivo
Acknowledgments
References
- Pettit, S.C.; Clemente, J.C.; Jeung, J.A.; Dunn, B.M.; Kaplan, A.H. Ordered processing of the human immunodeficiency virus type 1 GagPol precursor is influenced by the context of the embedded viral protease. J.Virol. 2005, 79, 10601–10607. [Google Scholar] [CrossRef] [PubMed]
- Pettit, S.C.; Lindquist, J.N.; Kaplan, A.H.; Swanstrom, R. Processing sites in the human immunodeficiency virus type 1 (HIV-1) Gag-Pro-Pol precursor are cleaved by the viral protease at different rates. Retrovirology 2005, 2, 66. [Google Scholar] [CrossRef] [PubMed]
- Pettit, S.C.; Moody, M.D.; Wehbie, R.S.; Kaplan, A.H.; Nantermet, P.V.; Klein, C.A.; Swanstrom, R. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 1994, 68, 8017–8027. [Google Scholar] [PubMed]
- Gross, I.; Hohenberg, H.; Wilk, T.; Wiegers, K.; Grattinger, M.; Muller, B.; Fuller, S.; Krausslich, H.G. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 2000, 19, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Krausslich, H.G.; Facke, M.; Heuser, A.M.; Konvalinka, J.; Zentgraf, H. The spacer peptide between human immunodeficiency virus capsid and nucleocapsid proteins is essential for ordered assembly and viral infectivity. J. Virol. 1995, 69, 3407–3419. [Google Scholar] [PubMed]
- Shehu-Xhilaga, M.; Kraeusslich, H.G.; Pettit, S.; Swanstrom, R.; Lee, J.Y.; Marshall, J.A.; Crowe, S.M.; Mak, J. Proteolytic processing of the p2/nucleocapsid cleavage site is critical for human immunodeficiency virus type 1 RNA dimer maturation. J. Virol. 2001, 75, 9156–9164. [Google Scholar] [CrossRef] [PubMed]
- Pettit, S.C.; Henderson, G.J.; Schiffer, C.A.; Swanstrom, R. Replacement of the P1 amino acid of human immunodeficiency virus type 1 Gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J. Virol. 2002, 76, 10226–10233. [Google Scholar] [CrossRef] [PubMed]
- Bally, F.; Martinez, R.; Peters, S.; Sudre, P.; Telenti, A. Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res. Hum. Retroviruses 2000, 16, 1209–1213. [Google Scholar] [PubMed]
- Brumme, Z.L.; Chan, K.J.; Dong, W.W.; Wynhoven, B.; Mo, T.; Hogg, R.S.; Montaner, J.S.; O'Shaughnessy, M.V.; Harrigan, P.R. Prevalence and clinical implications of insertions in the HIV-1 p6Gag N-terminal region in drug-naive individuals initiating antiretroviral therapy. Antivir. Ther. 2003, 8, 91–96. [Google Scholar] [PubMed]
- de Oliveira, T.; Engelbrecht, S.; Janse van Rensburg, E.; Gordon, M.; Bishop, K.; zur Megede, J.; Barnett, S.W.; Cassol, S. Variability at human immunodeficiency virus type 1 subtype C protease cleavage sites: an indication of viral fitness? J. Virol. 2003, 77, 9422–9430. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O.; de Mendoza, C.; Corral, A.; Soriano, V. Changes in the human immunodeficiency virus p7-p1-p6 gag gene in drug-naive and pretreated patients. J. Clin. Microbiol. 2003, 41, 1245–1247. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, J.; Knops, E.; Kupfer, B.; Hamouda, O.; Somogyi, S.; Schuldenzucker, U.; Hoffmann, D.; Kaiser, R.; Pfister, H.; Kucherer, C. Prevalence of C-terminal gag cleavage site mutations in HIV from therapy-naive patients. J. Infect. 2009, 58, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Bandaranayake, R.M.; Prabu-Jeyabalan, M.; Kakizawa, J.; Sugiura, W.; Schiffer, C.A. Structural analysis of human immunodeficiency virus type 1 CRF01_AE protease in complex with the substrate p1-p6. J. Virol. 2008, 82, 6762–6766. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Condra, J.H.; Holder, D.J.; Schleif, W.A.; Blahy, O.M.; Danovich, R.M.; Gabryelski, L.J.; Graham, D.J.; Laird, D.; Quintero, J.C.; Rhodes, A.; Robbins, H.L.; Roth, E.; Shivaprakash, M.; Yang, T.; Chodakewitz, J.A.; Deutsch, P.J.; Leavitt, R.Y.; Massari, F.E.; Mellors, J.W.; Squires, K.E.; Steigbigel, R.T.; Teppler, H.; Emini, E.A. Genetic correlates of in vivo viral resistance to indinavir, a human immunodeficiency virus type 1 protease inhibitor. J. Virol. 1996, 70, 8270–8276. [Google Scholar] [PubMed]
- Molla, A.; Korneyeva, M.; Gao, Q.; Vasavanonda, S.; Schipper, P.J.; Mo, H.M.; Markowitz, M.; Chernyavskiy, T.; Niu, P.; Lyons, N.; Hsu, A.; Granneman, G.R.; Ho, D.D.; Boucher, C.A.; Leonard, J.M.; Norbeck, D.W.; Kempf, D.J. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat. Med. 1996, 2, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; King, N.M.; Schiffer, C.A. Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 2004, 78, 12446–12454. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, B.C.; Vickrey, J.F.; Martin, P.; Proteasa, G.; Koepke, J.I.; Terlecky, S.R.; Wawrzak, Z.; Winters, M.A.; Merigan, T.C.; Kovari, L.C. Crystal structures of a multidrug-resistant human immunodeficiency virus type 1 protease reveal an expanded active-site cavity. J. Virol. 2004, 78, 3123–3132. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; King, N.M.; Nalivaika, E.A.; Heilek-Snyder, G.; Cammack, N.; Schiffer, C.A. Substrate envelope and drug resistance: crystal structure of RO1 in complex with wild-type human immunodeficiency virus type 1 protease. Antimicrob. Agents Chemother. 2006, 50, 1518–1521. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; Romano, K.; Schiffer, C.A. Mechanism of substrate recognition by drug-resistant human immunodeficiency virus type 1 protease variants revealed by a novel structural intermediate. J. Virol. 2006, 80, 3607–3616. [Google Scholar] [CrossRef] [PubMed]
- Mammano, F.; Trouplin, V.; Zennou, V.; Clavel, F. Retracing the evolutionary pathways of human immunodeficiency virus type 1 resistance to protease inhibitors: virus fitness in the absence and in the presence of drug. J. Virol. 2000, 74, 8524–8531. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; Savara, A.V.; Sutton, L.; D'Aquila, R.T. Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1. J. Virol. 1999, 73, 3744–3752. [Google Scholar] [PubMed]
- Zennou, V.; Mammano, F.; Paulous, S.; Mathez, D.; Clavel, F. Loss of viral fitness associated with multiple Gag and Gag-Pol processing defects in human immunodeficiency virus type 1 variants selected for resistance to protease inhibitors in vivo. J. Virol. 1998, 72, 3300–3306. [Google Scholar] [PubMed]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 1999, 13, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Barbour, J.D.; Wrin, T.; Grant, R.M.; Martin, J.N.; Segal, M.R.; Petropoulos, C.J.; Deeks, S.G. Evolution of phenotypic drug susceptibility and viral replication capacity during long-term virologic failure of protease inhibitor therapy in human immunodeficiency virus-infected adults. J. Virol. 2002, 76, 11104–11112. [Google Scholar] [CrossRef] [PubMed]
- Bleiber, G.; Munoz, M.; Ciuffi, A.; Meylan, P.; Telenti, A. Individual contributions of mutant protease and reverse transcriptase to viral infectivity, replication, and protein maturation of antiretroviral drug-resistant human immunodeficiency virus type 1. J. Virol. 2001, 75, 3291–3300. [Google Scholar] [CrossRef] [PubMed]
- Carron de la Carriere, L.; Paulous, S.; Clavel, F.; Mammano, F. Effects of human immunodeficiency virus type 1 resistance to protease inhibitors on reverse transcriptase processing, activity, and drug sensitivity. J. Virol. 1999, 73, 3455–3459. [Google Scholar] [PubMed]
- Mammano, F.; Petit, C.; Clavel, F. Resistance-associated loss of viral fitness in human immunodeficiency virus type 1: phenotypic analysis of protease and gag coevolution in protease inhibitor-treated patients. J. Virol. 1998, 72, 7632–7637. [Google Scholar] [PubMed]
- Doyon, L.; Croteau, G.; Thibeault, D.; Poulin, F.; Pilote, L.; Lamarre, D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 1996, 70, 3763–3769. [Google Scholar] [PubMed]
- Zhang, Y.M.; Imamichi, H.; Imamichi, T.; Lane, H.C.; Falloon, J.; Vasudevachari, M.B.; Salzman, N.P. Drug resistance during indinavir therapy is caused by mutations in the protease gene and in its Gag substrate cleavage sites. J. Virol. 1997, 71, 6662–6670. [Google Scholar] [PubMed]
- Banke, S.; Lillemark, M.R.; Gerstoft, J.; Obel, N.; Jorgensen, L.B. Positive selection pressure introduces secondary mutations at Gag cleavage sites in human immunodeficiency virus type 1 harboring major protease resistance mutations. J. Virol. 2009, 83, 8916–8924. [Google Scholar] [CrossRef] [PubMed]
- Brann, T.W.; Dewar, R.L.; Jiang, M.K.; Shah, A.; Nagashima, K.; Metcalf, J.A.; Falloon, J.; Lane, H.C.; Imamichi, T. Functional correlation between a novel amino acid insertion at codon 19 in the protease of human immunodeficiency virus type 1 and polymorphism in the p1/p6 Gag cleavage site in drug resistance and replication fitness. J. Virol. 2006, 80, 6136–6145. [Google Scholar] [CrossRef] [PubMed]
- Cote, H.C.; Brumme, Z.L.; Harrigan, P.R. Human immunodeficiency virus type 1 protease cleavage site mutations associated with protease inhibitor cross-resistance selected by indinavir, ritonavir, and/or saquinavir. J. Virol. 2001, 75, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.F.; Guinea, R.; Griffin, P.; Macmanus, S.; Elston, R.C.; Wolfram, J.; Richards, N.; Hanlon, M.H.; Porter, D.J.; Wrin, T.; Parkin, N.; Tisdale, M.; Furfine, E.; Petropoulos, C.; Snowden, B.W.; Kleim, J.P. Changes in human immunodeficiency virus type 1 Gag at positions L449 and P453 are linked to I50V protease mutants in vivo and cause reduction of sensitivity to amprenavir and improved viral fitness in vitro. J. Virol. 2002, 76, 7398–7406. [Google Scholar] [CrossRef] [PubMed]
- Malet, I.; Roquebert, B.; Dalban, C.; Wirden, M.; Amellal, B.; Agher, R.; Simon, A.; Katlama, C.; Costagliola, D.; Calvez, V.; Marcelin, A.G. Association of Gag cleavage sites to protease mutations and to virological response in HIV-1 treated patients. J. Infect. 2007, 54, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.H.; Myers, R.E.; Snowden, B.W.; Tisdale, M.; Blair, E.D. HIV type 1 protease cleavage site mutations and viral fitness: implications for drug susceptibility phenotyping assays. AIDS Res. Hum. Retroviruses 2000, 16, 1149–1156. [Google Scholar] [PubMed]
- Verheyen, J.; Litau, E.; Sing, T.; Daumer, M.; Balduin, M.; Oette, M.; Fatkenheuer, G.; Rockstroh, J.K.; Schuldenzucker, U.; Hoffmann, D.; Pfister, H.; Kaiser, R. Compensatory mutations at the HIV cleavage sites p7/p1 and p1/p6-gag in therapy-naive and therapy-experienced patients. Antiv. Ther. 2006, 11, 879–887. [Google Scholar]
- Launay, O.; Duval, X.; Dalban, C.; Descamps, D.; Peytavin, G.; Certain, A.; Mouajjah, S.; Ralaimazava, P.; Verdon, R.; Costagliola, D.; Clavel, F. Lamivudine and indinavir/ritonavir maintenance therapy in highly pretreated HIV-infected patients (Vista ANRS 109). Antivir. Ther. 2006, 11, 889–899. [Google Scholar] [PubMed]
- Prado, J.G.; Wrin, T.; Beauchaine, J.; Ruiz, L.; Petropoulos, C.J.; Frost, S.D.W.; Clotet, B.; D'Aquila, R.T.; Martinez-Picado, J. Amprenavir-resistant HIV-1 exhibits lopinavir cross-resistance and reduced replication capacity. AIDS 2002, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Dam, E.; Quercia, R.; Glass, B.; Descamps, D.; Launay, O.; Duval, X.; Krausslich, H.G.; Hance, A.J.; Clavel, F. Gag mutations strongly contribute to HIV-1 resistance to protease inhibitors in highly drug-experienced patients besides compensating for fitness loss . PLoS Path. 2009, 5, e1000345. [Google Scholar] [CrossRef]
- Nijhuis, M.; van Maarseveen, N.M.; Lastere, S.; Schipper, P.; Coakley, E.; Glass, B.; Rovenska, M.; de Jong, D.; Chappey, C.; Goedegebuure, I.W.; Heilek-Snyder, G.; Dulude, D.; Cammack, N.; Brakier-Gingras, L.; Konvalinka, J.; Parkin, N.; Krausslich, H.G.; Brun-Vezinet, F.; Boucher, C.A. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism . PLoS Med. 2007, 4, e36. [Google Scholar] [CrossRef] [PubMed]
- Coren, L.V.; Thomas, J.A.; Chertova, E.; Sowder, R.C.; Gagliardi, T.D.; Gorelick, R.J.; Ott, D.E. Mutational analysis of the C-terminal gag cleavage sites in human immunodeficiency virus type 1. J. Virol. 2007, 81, 10047–10054. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.L.; Bryan, W.M.; Fakhoury, S.A.; Magaard, V.W.; Huffman, W.F.; Dayton, B.D.; Meek, T.D.; Hyland, L.; Dreyer, G.B.; Metcalf, B.W. Peptide substrates and inhibitors of the HIV-1 protease. Biochem. Biophys. Res. Commun. 1989, 159, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Doyon, L.; Payant, C.; Brakier-Gingras, L.; Lamarre, D. Novel Gag-Pol frameshift site in human immunodeficiency virus type 1 variants resistant to protease inhibitors. J. Virol. 1998, 72, 6146–6150. [Google Scholar] [PubMed]
- Girnary, R.; King, L.; Robinson, L.; Elston, R.; Brierley, I. Structure-function analysis of the ribosomal frameshifting signal of two human immunodeficiency virus type 1 isolates with increased resistance to viral protease inhibitors. J. Gen. Virol. 2007, 88, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Parry, C.M.; Kohli, A.; Boinett, C.J.; Towers, G.J.; McCormick, A.L.; Pillay, D. Gag determinants of fitness and drug susceptibility in protease inhibitor-resistant human immunodeficiency virus type 1. J. Virol. 2009, 83, 9094–9101. [Google Scholar] [CrossRef] [PubMed]
- Gatanaga, H.; Suzuki, Y.; Tsang, H.; Yoshimura, K.; Kavlick, M.F.; Nagashima, K.; Gorelick, R.J.; Mardy, S.; Tang, C.; Summers, M.F.; Mitsuya, H. Amino acid substitutions in Gag protein at non-cleavage sites are indispensable for the development of a high multitude of HIV-1 resistance against protease inhibitors. J. Biol. Chem. 2002, 277, 5952–5961. [Google Scholar] [CrossRef] [PubMed]
- Myint, L.; Matsuda, M.; Matsuda, Z.; Yokomaku, Y.; Chiba, T.; Okano, A.; Yamada, K.; Sugiura, W. Gag non-cleavage site mutations contribute to full recovery of viral fitness in protease inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2004, 48, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Knops, E.; Daumer, M.; Awerkiew, S.; Kartashev, V.; Schulter, E.; Kutsev, S.; Brakier-Gingras, L.; Kaiser, R.; Pfister, H.; Verheyen, J. Evolution of protease inhibitor resistance in the gag and pol genes of HIV subtype G isolates. J. Antimicrob. Chemother. 2010, 65, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Lambert-Niclot, S.; Flandre, P.; Malet, I.; Canestri, A.; Soulie, C.; Tubiana, R.; Brunet, C.; Wirden, M.; Katlama, C.; Calvez, V.; Marcelin, A.G. Impact of gag mutations on selection of darunavir resistance mutations in HIV-1 protease. J. Antimicrob. Chemother. 2008, 62, 905–908. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Clavel, F.; Mammano, F. Role of Gag in HIV Resistance to Protease Inhibitors. Viruses 2010, 2, 1411-1426. https://doi.org/10.3390/v2071411
Clavel F, Mammano F. Role of Gag in HIV Resistance to Protease Inhibitors. Viruses. 2010; 2(7):1411-1426. https://doi.org/10.3390/v2071411
Chicago/Turabian StyleClavel, François, and Fabrizio Mammano. 2010. "Role of Gag in HIV Resistance to Protease Inhibitors" Viruses 2, no. 7: 1411-1426. https://doi.org/10.3390/v2071411
APA StyleClavel, F., & Mammano, F. (2010). Role of Gag in HIV Resistance to Protease Inhibitors. Viruses, 2(7), 1411-1426. https://doi.org/10.3390/v2071411