Intracellular Events and Cell Fate in Filovirus Infection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Central Players: Virus and Target Cells
2.1. Filovirus Structure
2.2. Target Cells in Filovirus Infection
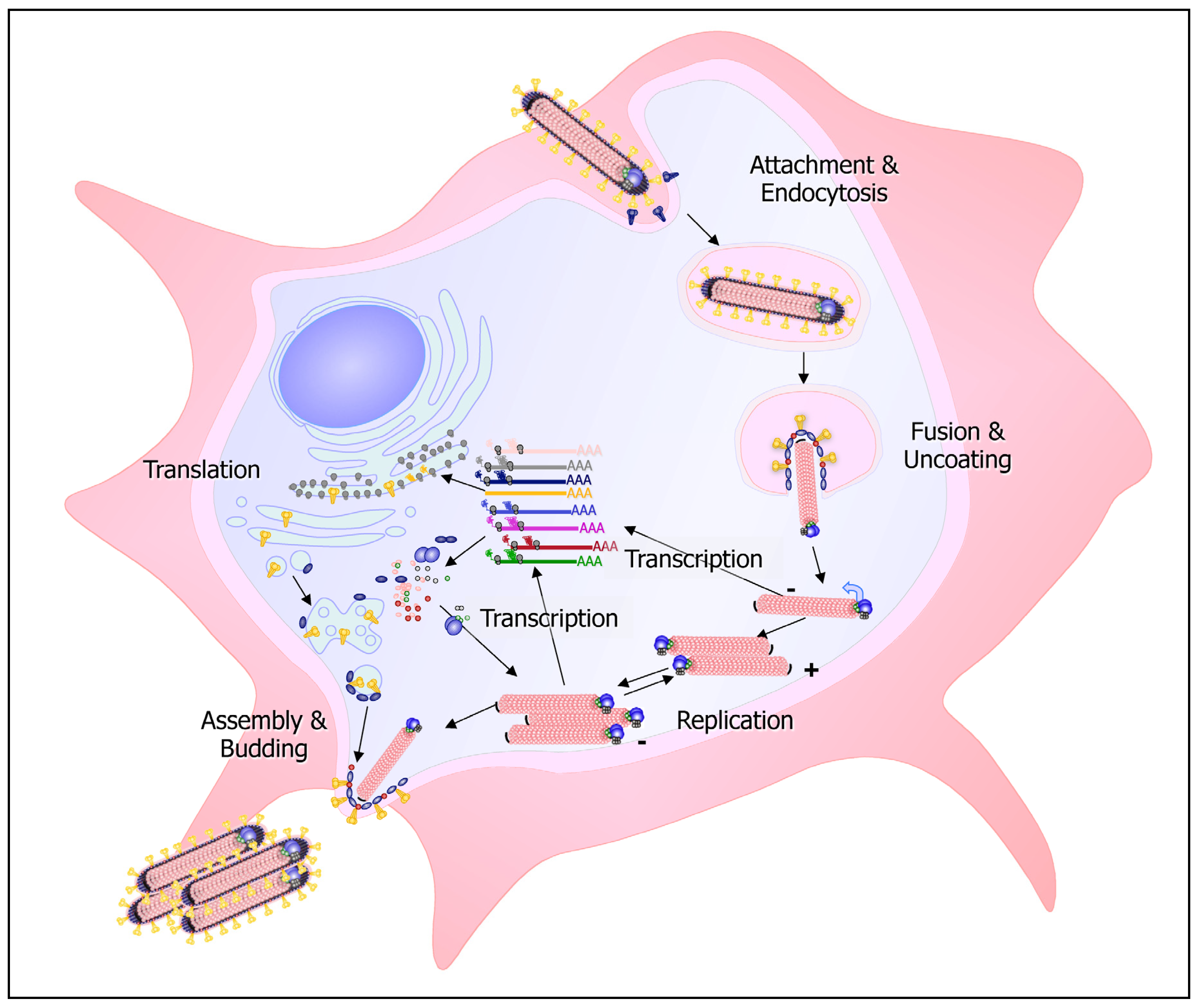
3. What Happens in the Infected Cell?
3.1. Entry: Speak Friend and Enter
3.2. Replication: The Intruders Take Over
3.3. Exit: Rats Abandon a Sinking Ship
4. To Live or Die: The Fate of Infected and Non-Infected Cells in Filovirus Infection
4.1. Mechanisms of Cell Death
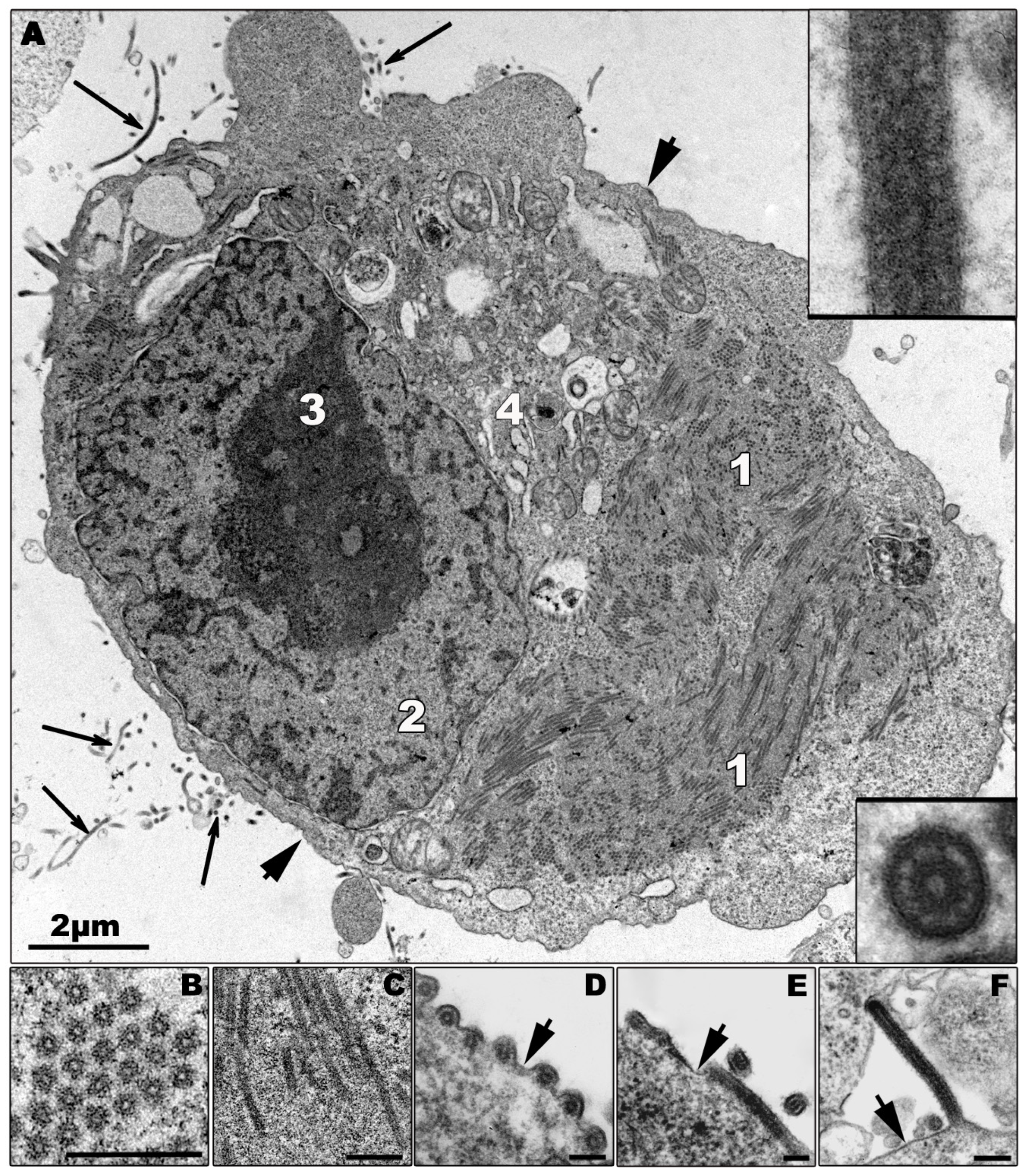
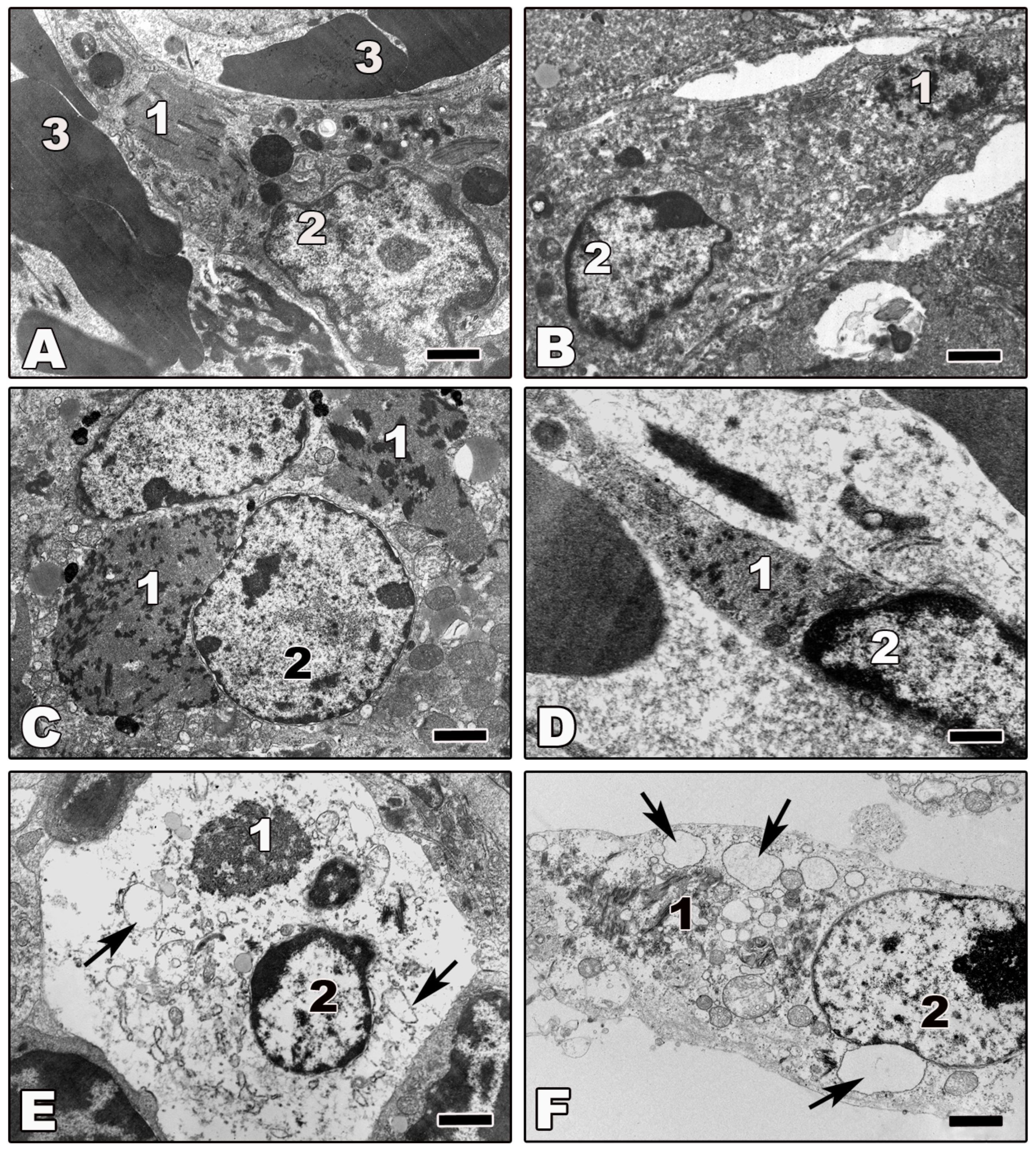
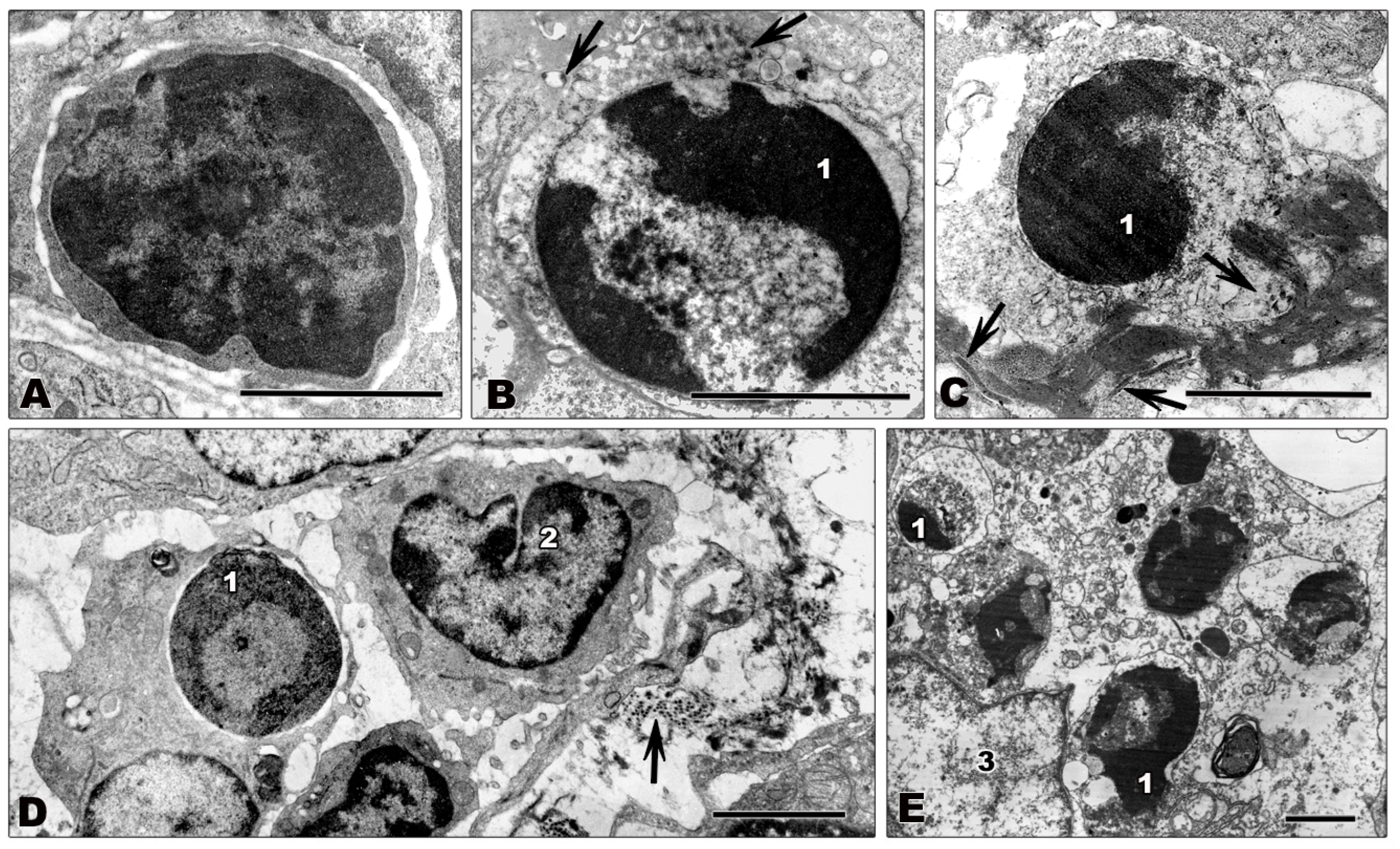
4.2. Infected Cells
4.2.1. Interaction of Filoviruses with Cellular Pathways
4.2.2. The Cytopathic Factor GP
4.3. Non-Infected Cells
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Kuhn, J.H. Filoviruses. A compendium of 40 years of epidemiological, clinical, and laboratory studies. Arch. Virol. Suppl. 2008, 20, 13–360. [Google Scholar] [PubMed]
- Mohamadzadeh, M.; Chen, L.; Schmaljohn, A.L. How Ebola and Marburg viruses battle the immune system. Nat. Rev. Immunol. 2007, 7, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Bradfute, S.B.; Swanson, P.E.; Smith, M.A.; Watanabe, E.; McDunn, J.E.; Hotchkiss, R.S.; Bavari, S. Mechanisms and consequences of ebolavirus-induced lymphocyte apoptosis. J. Immunol. 2010, 184, 327–335. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; West, C.P.; Rollin, P.E.; Jahrling, P.B.; Peters, C.J. ELISA for the detection of antibodies to Ebola viruses. J. Infect. Dis. 1999, 179, S192–S198. [Google Scholar] [CrossRef]
- Onyango, C.O.; Opoka, M.L.; Ksiazek, T.G.; Formenty, P.; Ahmed, A.; Tukei, P.M.; Sang, R.C.; Ofula, V.O.; Konongoi, S.L.; Coldren, R.L.; et al. Laboratory diagnosis of Ebola hemorrhagic fever during an outbreak in Yambio, Sudan, 2004. J. Infect. Dis. 2007, 196, S193–S198. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Rollin, P.E.; Bausch, D.G.; Sanchez, A.; Crary, S.M.; Vincent, M.; Lee, W.F.; Spiropoulou, C.F.; Ksiazek, T.G.; Lukwiya, M.; et al. Rapid diagnosis of Ebola hemorrhagic fever by reverse transcription-PCR in an outbreak setting and assessment of patient viral load as a predictor of outcome. J. Virol. 2004, 78, 4330–4341. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges-Courbot, M.C.; Capron, M.; Lansoud-Soukate, J.; Debre, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999, 5, 423–426. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges, A.J.; Georges-Courbot, M.C.; Capron, M.; Bedjabaga, I.; Lansoud-Soukate, J.; Mavoungou, E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002, 128, 163–168. [Google Scholar] [CrossRef]
- Mahanty, S.; Gupta, M.; Paragas, J.; Bray, M.; Ahmed, R.; Rollin, P.E. Protection from lethal infection is determined by innate immune responses in a mouse model of Ebola virus infection. Virology 2003, 312, 415–424. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Johnson, K.M.; Kawaoka, Y.; Lipkin, W.I.; Negredo, A.I.; Netesov, S.V.; Nichol, S.T.; et al. Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010, 155, 2083–2103. [Google Scholar] [CrossRef]
- Hutchinson, K.L.; Rollin, P.E. Cytokine and chemokine expression in humans infected with Sudan Ebola virus. J. Infect. Dis. 2007, 196, S357–S363. [Google Scholar] [CrossRef]
- Towner, J.S.; Sealy, T.K.; Khristova, M.L.; Albarino, C.G.; Conlan, S.; Reeder, S.A.; Quan, P.L.; Lipkin, W.I.; Downing, R.; Tappero, J.W.; et al. Newly discovered ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008, 4, e1000212. [Google Scholar] [CrossRef] [PubMed]
- Formenty, P.; Hatz, C.; Le Guenno, B.; Stoll, A.; Rogenmoser, P.; Widmer, A. Human infection due to Ebola virus, subtype Cote d'Ivoire: Clinical and biologic presentation. J. Infect. Dis. 1999, 179, S48–S53. [Google Scholar] [CrossRef] [PubMed]
- Le Guenno, B.; Formenty, P.; Boesch, C. Ebola virus outbreaks in the Ivory Coast and Liberia, 1994–1995. Curr. Top. Microbiol. Immunol. 1999, 235, 77–84. [Google Scholar] [PubMed]
- Barrette, R.W.; Metwally, S.A.; Rowland, J.M.; Xu, L.; Zaki, S.R.; Nichol, S.T.; Rollin, P.E.; Towner, J.S.; Shieh, W.J.; Batten, B.; et al. Discovery of swine as a host for the Reston ebolavirus. Science 2009, 325, 204–206. [Google Scholar] [CrossRef]
- Miranda, M.E.; Ksiazek, T.G.; Retuya, T.J.; Khan, A.S.; Sanchez, A.; Fulhorst, C.F.; Rollin, P.E.; Calaor, A.B.; Manalo, D.L.; Roces, M.C.; et al. Epidemiology of Ebola (subtype Reston) virus in the Philippines, 1996. J. Infect. Dis. 1999, 179, S115–S119. [Google Scholar] [CrossRef]
- Rollin, P.E.; Williams, R.J.; Bressler, D.S.; Pearson, S.; Cottingham, M.; Pucak, G.; Sanchez, A.; Trappier, S.G.; Peters, R.L.; Greer, P.W.; et al. Ebola (subtype Reston) virus among quarantined nonhuman primates recently imported from the Philippines to the United States. J. Infect. Dis. 1999, 179, S108–S114. [Google Scholar] [CrossRef]
- Slenczka, W.; Klenk, H.D. Forty years of marburg virus. J. Infect. Dis. 2007, 196, S131–S135. [Google Scholar] [CrossRef]
- Slenczka, W.G. The Marburg virus outbreak of 1967 and subsequent episodes. Curr. Top. Microbiol. Immunol. 1999, 235, 49–75. [Google Scholar]
- Feldmann, H. Marburg hemorrhagic fever—The forgotten cousin strikes. N. Engl. J. Med. 2006, 355, 866–869. [Google Scholar] [CrossRef]
- Towner, J.S.; Khristova, M.L.; Sealy, T.K.; Vincent, M.J.; Erickson, B.R.; Bawiec, D.A.; Hartman, A.L.; Comer, J.A.; Zaki, S.R.; Ströher, U.; et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar] [CrossRef] [PubMed]
- Bausch, D.G.; Nichol, S.T.; Muyembe-Tamfum, J.J.; Borchert, M.; Rollin, P.E.; Sleurs, H.; Campbell, P.; Tshioko, F.K.; Roth, C.; Colebunders, R.; et al. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N. Engl. J. Med. 2006, 355, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Imported case of Marburg hemorrhagic fever—Colorado, 2008. MMWR Morb. Mortal Wkly. Rep. 2009, 58, 1377–1381.
- Timen, A.; Koopmans, M.P.; Vossen, A.C.; van Doornum, G.J.; Gunther, S.; van den Berkmortel, F.; Verduin, K.M.; Dittrich, S.; Emmerich, P.; Osterhaus, A.D.; et al. Response to imported case of Marburg hemorrhagic fever, the Netherland. Emerg. Infect. Dis. 2009, 15, 1171–1175. [Google Scholar] [CrossRef]
- Mühlberger, E. Filovirus replication and transcription. Future Virology 2007, 2, 205–215. [Google Scholar] [CrossRef]
- Hartlieb, B.; Weissenhorn, W. Filovirus assembly and budding. Virology 2006, 344, 64–70. [Google Scholar] [CrossRef]
- Bamberg, S.; Kolesnikova, L.; Möller, P.; Klenk, H.D.; Becker, S. VP24 of Marburg virus influences formation of infectious particles. J. Virol. 2005, 79, 13421–13433. [Google Scholar] [CrossRef]
- Han, Z.; Boshra, H.; Sunyer, J.O.; Zwiers, S.H.; Paragas, J.; Harty, R.N. Biochemical and functional characterization of the Ebola virus VP24 protein: Implications for a role in virus assembly and budding. J. Virol. 2003, 77, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Halfmann, P.; Sagara, H.; Kawaoka, Y. Regions in Ebola virus VP24 that are important for nucleocapsid formation. J. Infect. Dis. 2007, 196, S247–S250. [Google Scholar] [CrossRef]
- Watanabe, S.; Noda, T.; Halfmann, P.; Jasenosky, L.; Kawaoka, Y. Ebola virus (EBOV) VP24 inhibits transcription and replication of the EBOV genome. J. Infect. Dis. 2007, 196, S284–S290. [Google Scholar] [CrossRef]
- Hoenen, T.; Jung, S.; Herwig, A.; Groseth, A.; Becker, S. Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology 2010, 403, 56–66. [Google Scholar] [CrossRef]
- Ebihara, H.; Takada, A.; Kobasa, D.; Jones, S.; Neumann, G.; Theriault, S.; Bray, M.; Feldmann, H.; Kawaoka, Y. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2006, 2, e73. [Google Scholar] [CrossRef]
- Reid, S.P.; Leung, L.W.; Hartman, A.L.; Martinez, O.; Shaw, M.L.; Carbonnelle, C.; Volchkov, V.E.; Nichol, S.T.; Basler, C.F. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 2006, 80, 5156–5167. [Google Scholar] [CrossRef] [PubMed]
- Valmas, C.; Grosch, M.N.; Schümann, M.; Olejnik, J.; Martinez, O.; Best, S.M.; Krähling, V.; Basler, C.F.; Mühlberger, E. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. 2010, 6, e1000721. [Google Scholar] [CrossRef] [PubMed]
- Valmas, C.; Basler, C.F. Marburg virus VP40 antagonizes interferon signaling in a species-specific manner. J. Virol. 2011, 85, 4309–4317. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, H.; Volchkov, V.E.; Volchkova, V.A.; Klenk, H.D. The glycoproteins of Marburg and Ebola virus and their potential roles in pathogenesis. Arch. Virol. Suppl. 1999, 15, 159–169. [Google Scholar]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 5762–5767. [Google Scholar] [CrossRef]
- Volchkov, V.E.; Becker, S.; Volchkova, V.A.; Ternovoj, V.A.; Kotov, A.N.; Netesov, S.V.; Klenk, H.D. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 1995, 214, 421–430. [Google Scholar] [CrossRef]
- Sanchez, A.; Trappier, S.G.; Mahy, B.W.; Peters, C.J.; Nichol, S.T. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 3602–3607. [Google Scholar] [CrossRef]
- Wahl-Jensen, V.; Kurz, S.K.; Hazelton, P.R.; Schnittler, H.J.; Stroher, U.; Burton, D.R.; Feldmann, H. Role of Ebola virus secreted glycoproteins and virus-like particles in activation of human macrophages. J. Virol. 2005, 79, 2413–2419. [Google Scholar] [CrossRef]
- Volchkova, V.A.; Feldmann, H.; Klenk, H.D.; Volchkov, V.E. The nonstructural small glycoprotein sGP of Ebola virus is secreted as an antiparallel-orientated homodimer. Virology 1998, 250, 408–414. [Google Scholar] [CrossRef]
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.J.; Feldmann, H. A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J. Virol. 2011, 85, 5406–5414. [Google Scholar] [CrossRef] [PubMed]
- Bowen, E.T.; Platt, G.S.; Simpson, D.I.; McArdell, L.B.; Raymond, R.T. Ebola haemorrhagic fever: Experimental infection of monkeys. Trans. R. Soc. Trop. Med. Hyg. 1978, 72, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.S.; Simpson, I.H.; Francis, D.P.; Knobloch, J.; Bowen, E.T.; Lolik, P.; Deng, I.M. Ultrastructure of Ebola virus particles in human liver. J. Clin. Pathol. 1978, 31, 201–208. [Google Scholar] [CrossRef]
- Baskerville, A.; Fisher-Hoch, S.P.; Neild, G.H.; Dowsett, A.B. Ultrastructural pathology of experimental Ebola haemorrhagic fever virus infection. J. Pathol. 1985, 147, 199–209. [Google Scholar] [CrossRef]
- Johnson, E.; Jaax, N.; White, J.; Jahrling, P. Lethal experimental infections of rhesus monkeys by aerosolized Ebola virus. Int. J. Exp. Pathol. 1995, 76, 227–236. [Google Scholar] [PubMed]
- Ryabchikova, E.; Strelets, L.; Kolesnikova, L.; Pyankov, O.; Sergeev, A. Respiratory Marburg virus infection in guinea pigs. Arch. Virol. 1996, 141, 2177–2190. [Google Scholar] [CrossRef]
- Ryabchikova, E.; Kolesnikova, L.; Smolina, M.; Tkachev, V.; Pereboeva, L.; Baranova, S.; Grazhdantseva, A.; Rassadkin, Y. Ebola virus infection in guinea pigs: Presumable role of granulomatous inflammation in pathogenesis. Arch. Virol. 1996, 141, 909–921. [Google Scholar] [CrossRef]
- Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 1999, 179, S199–S202. [Google Scholar] [CrossRef]
- Ryabchikova, E.; Price, B.B.S. Ebola and Marburg Viruses: A View of Infection Using Electron Microscopy; Battelle Press: Columbus, Ohio, USA, 2004. [Google Scholar]
- Jaax, N.K.; Davis, K.J.; Geisbert, T.J.; Vogel, P.; Jaax, G.P.; Topper, M.; Jahrling, P.B. Lethal experimental infection of rhesus monkeys with Ebola-Zaire (Mayinga) virus by the oral and conjunctival route of exposure. Arch. Pathol. Lab. Med. 1996, 120, 140–155. [Google Scholar]
- Davis, K.J.; Anderson, A.O.; Geisbert, T.W.; Steele, K.E.; Geisbert, J.B.; Vogel, P.; Connolly, B.M.; Huggins, J.W.; Jahrling, P.B.; Jaax, N.K. Pathology of experimental Ebola virus infection in African green monkeys. Involvement of fibroblastic reticular cells. Arch. Pathol. Lab. Med. 1997, 121, 805–819. [Google Scholar] [PubMed]
- Wyers, M.; Formenty, P.; Cherel, Y.; Guigand, L.; Fernandez, B.; Boesch, C.; Le Guenno, B. Histopathological and immunohistochemical studies of lesions associated with Ebola virus in a naturally infected chimpanzee. J. Infect. Dis. 1999, 179, S54–S59. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Young, H.A.; Jahrling, P.B.; Davis, K.J.; Larsen, T.; Kagan, E.; Hensley, L.E. Pathogenesis of Ebola hemorrhagic fever in primate models: Evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am. J. Pathol. 2003, 163, 2371–2382. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar] [CrossRef] [PubMed]
- Zaki, S.R.; Goldsmith, C.S. Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 1999, 235, 97–116. [Google Scholar] [PubMed]
- Steele, K.E.; Anderson, A.O.; Mohamadzadeh, M. Fibroblastic reticular cell infection by hemorrhagic fever viruses. Immunotherapy 2009, 1, 187–197. [Google Scholar] [CrossRef]
- Reis e Sousa, C. Toll-like receptors and dendritic cells: for whom the bug tolls. Semin. Immunol. 2004, 16, 27–34. [Google Scholar] [CrossRef]
- Reis e Sousa, C. Activation of dendritic cells: translating innate into adaptive immunity. Curr. Opin. Immunol. 2004, 16, 21–25. [Google Scholar] [CrossRef]
- Murphy, F.A.; Simpson, D.I.; Whitfield, S.G.; Zlotnik, I.; Carter, G.B. Marburg virus infection in monkeys. Ultrastructural studies. Lab. Invest. 1971, 24, 279–291. [Google Scholar]
- Bray, M.; Davis, K.; Geisbert, T.; Schmaljohn, C.; Huggins, J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 1999, 179, S248–S258. [Google Scholar] [CrossRef]
- Connolly, B.M.; Steele, K.E.; Davis, K.J.; Geisbert, T.W.; Kell, W.M.; Jaax, N.K.; Jahrling, P.B. Pathogenesis of experimental Ebola virus infection in guinea pigs. J. Infect. Dis. 1999, 179, S203–S217. [Google Scholar] [CrossRef] [PubMed]
- Gibb, T.R.; Bray, M.; Geisbert, T.W.; Steele, K.E.; Kell, W.M.; Davis, K.J.; Jaax, N.K. Pathogenesis of experimental Ebola Zaire virus infection in BALB/c mice. J. Comp. Pathol. 2001, 125, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Jahrling, P.B.; Hanes, M.A.; Zack, P.M. Association of Ebola-related Reston virus particles and antigen with tissue lesions of monkeys imported to the United States. J. Comp. Pathol. 1992, 106, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Riabchikova, E.I.; Baranova, S.G.; Tkachev, V.K.; Grazhdantseva, A.A. The morphological changes in Ebola infection in guinea pigs. Vopr. Virusol. 1993, 38, 176–179. [Google Scholar]
- Geisbert, T.W.; Jaax, N.K. Marburg hemorrhagic fever: Report of a case studied by immunohistochemistry and electron microscopy. Ultrastruct. Pathol. 1998, 22, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.; Geisbert, T.W. Ebola virus: The role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 2005, 37, 1560–1566. [Google Scholar] [CrossRef]
- Schnittler, H.J.; Feldmann, H. Marburg and Ebola hemorrhagic fevers: Does the primary course of infection depend on the accessibility of organ-specific macrophages? Clin. Infect. Dis. 1998, 27, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Baskerville, A.; Bowen, E.T.; Platt, G.S.; McArdell, L.B.; Simpson, D.I. The pathology of experimental Ebola virus infection in monkeys. J. Pathol. 1978, 125, 131–138. [Google Scholar] [CrossRef]
- Martini, G.A.; Siegert, R. Marburg Virus Disease; Springer: New York, NY, USA, 1971. [Google Scholar]
- Zaki, S.R.; Kilmarx, P.H. Ebola virus hemorrhagic fever. In Pathology of Emerging Infections; Horsburgh, C.R., Nelson, A.M., Eds.; ASM (USA): Washington, DC, USA, 1998. [Google Scholar]
- Bavari, S.; Bosio, C.M.; Wiegand, E.; Ruthel, G.; Will, A.B.; Geisbert, T.W.; Hevey, M.; Schmaljohn, C.; Schmaljohn, A.; Aman, M.J. Lipid raft microdomains: A gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 2002, 195, 593–602. [Google Scholar] [CrossRef]
- Empig, C.J.; Goldsmith, M.A. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 2002, 76, 5266–5270. [Google Scholar] [CrossRef]
- Sanchez, A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J. Infect. Dis. 2007, 196, S251–S258. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.L.; Kolokoltsov, A.A.; Davey, R.A.; Maury, W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 2011, 85, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010, 6, pii: e1001110. [Google Scholar] [CrossRef]
- Chan, S.Y.; Empig, C.J.; Welte, F.J.; Speck, R.F.; Schmaljohn, A.; Kreisberg, J.F.; Goldsmith, M.A. Folate receptor-alpha is a cofactor for cellular entry by Marburg and Ebola viruses. Cell 2001, 106, 117–126. [Google Scholar] [CrossRef]
- Simmons, G.; Rennekamp, A.J.; Chai, N.; Vandenberghe, L.H.; Riley, J.L.; Bates, P. Folate receptor alpha and caveolae are not required for Ebola virus glycoprotein-mediated viral infection. J. Virol. 2003, 77, 13433–13438. [Google Scholar] [CrossRef]
- Sinn, P.L.; Hickey, M.A.; Staber, P.D.; Dylla, D.E.; Jeffers, S.A.; Davidson, B.L.; Sanders, D.A.; McCray, P.B., Jr. Lentivirus vectors pseudotyped with filoviral envelope glycoproteins transduce airway epithelia from the apical surface independently of folate receptor alpha. J. Virol. 2003, 77, 5902–5910. [Google Scholar] [CrossRef]
- Sanchez, A.; Yang, Z.Y.; Xu, L.; Nabel, G.J.; Crews, T.; Peters, C.J. Biochemical analysis of the secreted and virion glycoproteins of Ebola virus. J. Virol. 1998, 72, 6442–6447. [Google Scholar] [CrossRef]
- Feldmann, H.; Will, C.; Schikore, M.; Slenczka, W.; Klenk, H.D. Glycosylation and oligomerization of the spike protein of Marburg virus. Virology 1991, 182, 353–356. [Google Scholar] [CrossRef]
- Feldmann, H.; Nichol, S.T.; Klenk, H.D.; Peters, C.J.; Sanchez, A. Characterization of filoviruses based on differences in structure and antigenicity of the virion glycoprotein. Virology 1994, 199, 469–473. [Google Scholar] [CrossRef]
- Becker, S.; Spiess, M.; Klenk, H.D. The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol. 1995, 76, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.P.; Lasala, F.; Carrillo, J.; Muniz, O.; Corbi, A.L.; Delgado, R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002, 76, 6841–6844. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Reeves, J.D.; Grogan, C.C.; Vandenberghe, L.H.; Baribaud, F.; Whitbeck, J.C.; Burke, E.; Buchmeier, M.J.; Soilleux, E.J.; Riley, J.L.; et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 2003, 305, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Baribaud, F.; Doms, R.W.; Pohlmann, S. The role of DC-SIGN and DC-SIGNR in HIV and Ebola virus infection: can potential therapeutics block virus transmission and dissemination? Expert. Opin. Ther. Targets 2002, 6, 423–431. [Google Scholar] [CrossRef]
- Marzi, A.; Gramberg, T.; Simmons, G.; Moller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef]
- Takada, A.; Fujioka, K.; Tsuiji, M.; Morikawa, A.; Higashi, N.; Ebihara, H.; Kobasa, D.; Feldmann, H.; Irimura, T.; Kawaoka, Y. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Virol. 2004, 78, 2943–2947. [Google Scholar] [CrossRef]
- Gramberg, T.; Hofmann, H.; Moller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef]
- Matsuno, K.; Kishida, N.; Usami, K.; Igarashi, M.; Yoshida, R.; Nakayama, E.; Shimojima, M.; Feldmann, H.; Irimura, T.; Kawaoka, Y.; et al. Different potential of C-type lectin-mediated entry between Marburg virus strains. J. Virol. 2010, 84, 5140–5147. [Google Scholar] [CrossRef]
- Takada, A.; Watanabe, S.; Ito, H.; Okazaki, K.; Kida, H.; Kawaoka, Y. Downregulation of beta1 integrins by Ebola virus glycoprotein: Implication for virus entry. Virology 2000, 278, 20–26. [Google Scholar] [CrossRef]
- Schornberg, K.L.; Shoemaker, C.J.; Dube, D.; Abshire, M.Y.; Delos, S.E.; Bouton, A.H.; White, J.M. Alpha5beta1-integrin controls ebolavirus entry by regulating endosomal cathepsins. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8003–8008. [Google Scholar] [CrossRef]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. U. S. A. 2011. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Takada, A.; Ebihara, H.; Neumann, G.; Fujioka, K.; Irimura, T.; Jones, S.; Feldmann, H.; Kawaoka, Y. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 2006, 80, 10109–10116. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Ikeda, Y.; Kawaoka, Y. The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 2007, 196, S259–S263. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Calder, L.J.; Wharton, S.A.; Skehel, J.J.; Wiley, D.C. The central structural feature of the membrane fusion protein subunit from the Ebola virus glycoprotein is a long triple-stranded coiled coil. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 6032–6036. [Google Scholar] [CrossRef]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar] [CrossRef]
- Hood, C.L.; Abraham, J.; Boyington, J.C.; Leung, K.; Kwong, P.D.; Nabel, G.J. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: Implications for viral entry and immunogenicity. J. Virol. 2010, 84, 2972–2982. [Google Scholar] [CrossRef]
- Martinez, O.; Johnson, J.; Manicassamy, B.; Rong, L.; Olinger, G.G.; Hensley, L.E.; Basler, C.F. Zaire Ebola virus entry into human dendritic cells is insensitive to cathepsin L inhibition. Cell Microbiol. 2010, 12, 148–157. [Google Scholar] [CrossRef]
- Kolesnikova, L.; Mühlberger, E.; Ryabchikova, E.; Becker, S. Ultrastructural organization of recombinant Marburg virus nucleoprotein: Comparison with Marburg virus inclusions. J. Virol. 2000, 74, 3899–3904. [Google Scholar] [CrossRef]
- Mavrakis, M.; Kolesnikova, L.; Schoehn, G.; Becker, S.; Ruigrok, R.W. Morphology of Marburg virus NP-RNA. Virology 2002, 296, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Ebihara, H.; Muramoto, Y.; Fujii, K.; Takada, A.; Sagara, H.; Kim, J.H.; Kida, H.; Feldmann, H.; Kawaoka, Y. Assembly and budding of Ebolavirus. PLoS Pathog. 2006, 2, e99. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, L.; Sun, Y.; Nabel, G.J. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol. Cell 2002, 10, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Aoyama, K.; Sagara, H.; Kida, H.; Kawaoka, Y. Nucleocapsid-like structures of Ebola virus reconstructed using electron tomography. J. Vet. Med. Sci. 2005, 67, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Rinne, C.; Hofsäss, U.; Klenk, H.-D.; Mühlberger, E. Interactions of Marburg virus nucleocapsid proteins. Virology 1998, 249, 406–417. [Google Scholar] [CrossRef]
- Schmidt, K.M.; Schümann, M.; Olejnik, J.; Krähling, V.; Mühlberger, E. Recombinant Marburg virus expressing EGFP allows rapid screening of virus growth and real time visualization of virus spread. J. Infect. Dis. 2011, in press. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jahrling, P.B. Differentiation of filoviruses by electron microscopy. Virus Res. 1995, 39, 129–150. [Google Scholar] [CrossRef]
- Liu, Y.; Harty, R.N. Viral and host proteins that modulate filovirus budding. Future Virol. 2010, 5, 481–491. [Google Scholar] [CrossRef]
- Dolnik, O.; Kolesnikova, L.; Becker, S. Filoviruses: Interactions with the host cell. Cell Mol. Life Sci. 2008, 65, 756–776. [Google Scholar] [CrossRef]
- Dolnik, O.; Kolesnikova, L.; Stevermann, L.; Becker, S. Tsg101 is recruited by a late domain of the nucleocapsid protein to support budding of Marburg virus-like particles. J. Virol. 2010, 84, 7847–7856. [Google Scholar] [CrossRef]
- Roxrud, I.; Stenmark, H.; Malerod, L. ESCRT & Co. Biol. Cell 2010, 102, 293–318. [Google Scholar] [PubMed]
- Hurley, J.H.; Boura, E.; Carlson, L.A.; Rozycki, B. Membrane budding. Cell 2010, 143, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, L.; Berghofer, B.; Bamberg, S.; Becker, S. Multivesicular bodies as a platform for formation of the Marburg virus envelope. J. Virol. 2004, 78, 12277–12287. [Google Scholar] [CrossRef]
- Kolesnikova, L.; Bohil, A.B.; Cheney, R.E.; Becker, S. Budding of Marburgvirus is associated with filopodia. Cell Microbiol. 2007, 9, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Kolesnikova, L.; Krahling, V.; Riches, J.D.; Becker, S.; Briggs, J.A. Electron tomography reveals the steps in filovirus budding. PLoS Pathog. 2010, 6, e1000875. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, V.; Zhivotovsky, B. To kill or be killed: How viruses interact with the cell death machinery. J. Intern. Med. 2010, 267, 473–482. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Walker, N.I.; Harmon, B.V.; Gobe, G.C.; Kerr, J.F. Patterns of cell death. Methods Achiev. Exp. Pathol. 1988, 13, 18–54. [Google Scholar] [PubMed]
- Gonzalvez, F.; Ashkenazi, A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 2010, 29, 4752–4765. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Boyce, M.; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef] [PubMed]
- Burlacu, A. Regulation of apoptosis by Bcl-2 family proteins. J. Cell Mol. Med. 2003, 7, 249–257. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef]
- Moquin, D.; Chan, F.K. The molecular regulation of programmed necrotic cell injury. Trends Biochem. Sci. 2010, 35, 434–441. [Google Scholar] [CrossRef]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Alazard-Dany, N.; Volchkova, V.; Reynard, O.; Carbonnelle, C.; Dolnik, O.; Ottmann, M.; Khromykh, A.; Volchkov, V.E. Ebola virus glycoprotein GP is not cytotoxic when expressed constitutively at a moderate level. J. Gen. Virol. 2006, 87, 1247–1257. [Google Scholar] [CrossRef]
- Barrientos, L.G.; Rollin, P.E. Release of cellular proteases into the acidic extracellular milieu exacerbates Ebola virus-induced cell damage. Virology 2007, 358, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Boehmann, Y.; Enterlein, S.; Randolf, A.; Mühlberger, E. A reconstituted replication and transcription system for Ebola virus Reston and comparison with Ebola virus Zaire. Virology 2005, 332, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Goldsmith, C.S.; Metcalfe, M.G.; Spiropoulou, C.F.; Rollin, P.E. Reduced virus replication, proinflammatory cytokine production, and delayed macrophage cell death in human PBMCs infected with the newly discovered Bundibugyo ebolavirus relative to Zaire ebolavirus. Virology 2010, 402, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Spiropoulou, C.; Rollin, P.E. Ebola virus infection of human PBMCs causes massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Virology 2007, 364, 45–54. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Gibb, T.R.; Steele, K.E.; Jaax, N.K.; Jahrling, P.B. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 2000, 80, 171–186. [Google Scholar] [CrossRef]
- Bente, D.; Gren, J.; Strong, J.E.; Feldmann, H. Disease modeling for Ebola and Marburg viruses. Dis. Model Mech. 2009, 2, 12–17. [Google Scholar] [CrossRef]
- Rosenblum, W.I. Cytotoxic edema: Monitoring its magnitude and contribution to brain swelling. J. Neuropathol. Exp. Neurol. 2007, 66, 771–778. [Google Scholar] [CrossRef]
- Saeed, M.F.; Kolokoltsov, A.A.; Freiberg, A.N.; Holbrook, M.R.; Davey, R.A. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 2008, 4, e1000141. [Google Scholar] [CrossRef]
- Cooray, S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 2004, 85, 1065–1076. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Ludwig, S. A new player in a deadly game: Influenza viruses and the PI3K/Akt signalling pathway. Cell Microbiol. 2009, 11, 863–871. [Google Scholar] [CrossRef]
- Brennan, K.; Bowie, A.G. Activation of host pattern recognition receptors by viruses. Curr. Opin. Microbiol. 2010, 13, 503–507. [Google Scholar] [CrossRef]
- Baum, A.; Garcia-Sastre, A. Induction of type I interferon by RNA viruses: Cellular receptors and their substrates. Amino Acids 2010, 38, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schümann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Mühlberger, E.; et al. Processing of genome 5' termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Amarasinghe, G.K. Evasion of interferon responses by Ebola and Marburg viruses. J. Interferon. Cytokine Res. 2009, 29, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Prins, K.C.; Basler, C.F.; Amarasinghe, G.K. Ebolavirus VP35 is a multifunctional virulence factor. Virulence 2010, 1, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Yamashita, M.; Zhang, Y.; Sen, G.C. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J. Virol. 2011, 85, 3708–3716. [Google Scholar] [CrossRef]
- Sharif-Askari, E.; Nakhaei, P.; Oliere, S.; Tumilasci, V.; Hernandez, E.; Wilkinson, P.; Lin, R.; Bell, J.; Hiscott, J. Bax-dependent mitochondrial membrane permeabilization enhances IRF3-mediated innate immune response during VSV infection. Virology 2007, 365, 20–33. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010, 29, 1762–1773. [Google Scholar] [CrossRef]
- Lei, Y.; Moore, C.B.; Liesman, R.M.; O'Connor, B.P.; Bergstralh, D.T.; Chen, Z.J.; Pickles, R.J.; Ting, J.P. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS ONE 2009, 4, e5466. [Google Scholar] [CrossRef]
- Scott, I.; Norris, K.L. The mitochondrial antiviral signaling protein, MAVS, is cleaved during apoptosis. Biochem. Biophys. Res. Commun. 2008, 375, 101–106. [Google Scholar] [CrossRef]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol. Cell Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sugiyama, M.; Yamamoto, M.; Watanabe, Y.; Kawai, T.; Takeda, K.; Akira, S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 2003, 171, 4304–4310. [Google Scholar] [PubMed]
- Kumar, A.; Haque, J.; Lacoste, J.; Hiscott, J.; Williams, B.R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 6288–6292. [Google Scholar] [CrossRef]
- Unterholzner, L.; Bowie, A.G. The interplay between viruses and innate immune signaling: Recent insights and therapeutic opportunities. Biochem. Pharmacol. 2008, 75, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Katze, M.G. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther. 1998, 78, 29–46. [Google Scholar] [CrossRef]
- Feng, Z.; Cerveny, M.; Yan, Z.; He, B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J. Virol. 2007, 81, 182–192. [Google Scholar] [CrossRef]
- Schümann, M.; Gantke, T.; Mühlberger, E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J. Virol. 2009, 83, 8993–8997. [Google Scholar] [CrossRef]
- Zhang, P.; Jacobs, B.L.; Samuel, C.E. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J. Virol. 2008, 82, 840–848. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Kwieciszewski, B.; Dossett, M.; Nakao, H.; Katze, M.G. Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J. Virol. 1999, 73, 6506–6516. [Google Scholar] [CrossRef]
- Garcia, M.A.; Guerra, S.; Gil, J.; Jimenez, V.; Esteban, M. Anti-apoptotic and oncogenic properties of the dsRNA-binding protein of vaccinia virus, E3L. Oncogene 2002, 21, 8379–8387. [Google Scholar] [CrossRef] [PubMed]
- Francois, S.; El Benna, J.; Dang, P.M.; Pedruzzi, E.; Gougerot-Pocidalo, M.A.; Elbim, C. Inhibition of neutrophil apoptosis by TLR agonists in whole blood: Involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J. Immunol. 2005, 174, 3633–3642. [Google Scholar] [CrossRef]
- Aliprantis, A.O.; Yang, R.B.; Mark, M.R.; Suggett, S.; Devaux, B.; Radolf, J.D.; Klimpel, G.R.; Godowski, P.; Zychlinsky, A. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 1999, 285, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Yan, Z.; Prabhakar, B.S.; Feng, Z.; Ma, Y.; Verpooten, D.; Ganesh, B.; He, B. The VP35 protein of Ebola virus impairs dendritic cell maturation induced by virus and lipopolysaccharide. J. Gen. Virol. 2010, 91, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.H.; Kubota, T.; Matsuoka, M.; Jones, S.; Bradfute, S.B.; Bray, M.; Ozato, K. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009, 5, e1000493. [Google Scholar] [CrossRef]
- Leung, L.W.; Park, M.S.; Martinez, O.; Valmas, C.; Lopez, C.B.; Basler, C.F. Ebolavirus VP35 suppresses IFN production from conventional but not plasmacytoid dendritic cells. Immunol. Cell Biol. 2011. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Duckers, H.J.; Sullivan, N.J.; Sanchez, A.; Nabel, E.G.; Nabel, G.J. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000, 6, 886–889. [Google Scholar] [CrossRef]
- Chan, S.Y.; Ma, M.C.; Goldsmith, M.A. Differential induction of cellular detachment by envelope glycoproteins of Marburg and Ebola (Zaire) viruses. J. Gen. Virol. 2000, 81, 2155–2159. [Google Scholar] [CrossRef]
- Volchkov, V.E.; Volchkova, V.A.; Mühlberger, E.; Kolesnikova, L.V.; Weik, M.; Dolnik, O.; Klenk, H.D. Recovery of infectious Ebola virus from complementary DNA: RNA editing of the GP gene and viral cytotoxicity. Science 2001, 291, 1965–1969. [Google Scholar] [CrossRef]
- Simmons, G.; Wool-Lewis, R.J.; Baribaud, F.; Netter, R.C.; Bates, P. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J. Virol. 2002, 76, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Basu, A.; Steele, R.; Beyene, A.; McHowat, J.; Meyer, K.; Ghosh, A.K.; Ray, R. Ebola virus glycoprotein-mediated anoikis of primary human cardiac microvascular endothelial cells. Virology 2004, 321, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Peterson, M.; Yang, Z.Y.; Kong, W.P.; Duckers, H.; Nabel, E.; Nabel, G.J. Ebola virus glycoprotein toxicity is mediated by a dynamin-dependent protein-trafficking pathway. J. Virol. 2005, 79, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Licata, J.M.; Paragas, J.; Harty, R.N. Permeabilization of the plasma membrane by Ebola virus GP2. Virus Genes 2007, 34, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Francica, J.R.; Matukonis, M.K.; Bates, P. Requirements for cell rounding and surface protein down-regulation by Ebola virus glycoprotein. Virology 2009, 383, 237–247. [Google Scholar] [CrossRef]
- Zampieri, C.A.; Fortin, J.F.; Nolan, G.P.; Nabel, G.J. The ERK Mitogen-Activated Protein Kinase Pathway Contributes to Ebola Glycoprotein-Induced Cytotoxicity. J. Virol. 2006. [Google Scholar] [CrossRef]
- Martinez, O.; Valmas, C.; Basler, C.F. Ebola virus-like particle-induced activation of NF-kappaB and Erk signaling in human dendritic cells requires the glycoprotein mucin domain. Virology 2007, 364, 342–354. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Hope, T.J. Full-length Ebola glycoprotein accumulates in the endoplasmic reticulum. Virol. J. 2011, 8, 11. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Young, H.A.; Jahrling, P.B.; Davis, K.J.; Kagan, E.; Hensley, L.E. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: Overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 2003, 188, 1618–1629. [Google Scholar] [CrossRef]
- Aleksandrowicz, P.; Wolf, K.; Falzarano, D.; Feldmann, H.; Seebach, J.; Schnittler, H. Viral haemorrhagic fever and vascular alterations. Hamostaseologie 2008, 28, 77–84. [Google Scholar] [CrossRef]
- Wool-Lewis, R.J.; Bates, P. Characterization of Ebola virus entry by using pseudotyped viruses: Identification of receptor-deficient cell lines. J. Virol. 1998, 72, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Delgado, R.; Xu, L.; Todd, R.F.; Nabel, E.G.; Sanchez, A.; Nabel, G.J. Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science 1998, 279, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.S.; Hensley, L.E.; Geisbert, J.B.; Jahrling, P.B.; Geisbert, T.W. Depletion of peripheral blood T lymphocytes and NK cells during the course of ebola hemorrhagic Fever in cynomolgus macaques. Viral. Immunol. 2004, 17, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Bradfute, S.B.; Braun, D.R.; Shamblin, J.D.; Geisbert, J.B.; Paragas, J.; Garrison, A.; Hensley, L.E.; Geisbert, T.W. Lymphocyte death in a mouse model of Ebola virus infection. J. Infect. Dis. 2007, 196, S296–S304. [Google Scholar] [CrossRef] [PubMed]
- Wauquier, N.; Becquart, P.; Padilla, C.; Baize, S.; Leroy, E.M. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl. Trop. Dis. 2010, 4, e837. [Google Scholar] [CrossRef]
- Zarozinski, C.C.; McNally, J.M.; Lohman, B.L.; Daniels, K.A.; Welsh, R.M. Bystander sensitization to activation-induced cell death as a mechanism of virus-induced immune suppression. J. Virol. 2000, 74, 3650–3658. [Google Scholar] [CrossRef]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef]
- Inoue, Y.; Yasukawa, M.; Fujita, S. Induction of T-cell apoptosis by human herpesvirus 6. J. Virol. 1997, 71, 3751–3759. [Google Scholar] [CrossRef]
- Yates, N.L.; Yammani, R.D.; Alexander-Miller, M.A. Dose-dependent lymphocyte apoptosis following respiratory infection with Vaccinia virus. Virus Res. 2008, 137, 198–205. [Google Scholar] [CrossRef]
- Hilleman, M.R. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14560–14566. [Google Scholar] [CrossRef]
- Gougeon, M.L. Apoptosis as an HIV strategy to escape immune attack. Nat. Rev. Immunol. 2003, 3, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Jekle, A.; Keppler, O.T.; De Clercq, E.; Schols, D.; Weinstein, M.; Goldsmith, M.A. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J. Virol. 2003, 77, 5846–5854. [Google Scholar] [CrossRef]
- Schindler, M.; Munch, J.; Kutsch, O.; Li, H.; Santiago, M.L.; Bibollet-Ruche, F.; Muller-Trutwin, M.C.; Novembre, F.J.; Peeters, M.; Courgnaud, V.; Bailes, E.; Roques, P.; Sodora, D.L.; Silvestri, G.; Sharp, P.M.; Hahn, B.H.; Kirchhoff, F. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 2006, 125, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Westendorp, M.O.; Frank, R.; Ochsenbauer, C.; Stricker, K.; Dhein, J.; Walczak, H.; Debatin, K.M.; Krammer, P.H. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 1995, 375, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Chen, B.K.; Straus, S.E.; Dale, J.K.; Lenardo, M.J.; Baltimore, D. HIV-1 directly kills CD4+ T cells by a Fas-independent mechanism. J. Exp. Med. 1998, 187, 1113–1122. [Google Scholar] [CrossRef]
- Herbeuval, J.P.; Grivel, J.C.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef]
- Hevey, M.; Negley, D.; Pushko, P.; Smith, J.; Schmaljohn, A. Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology 1998, 251, 28–37. [Google Scholar] [CrossRef]
- Ignatyev, G.M. Immune response to filovirus infections. Curr. Top. Microbiol. Immunol. 1999, 235, 205–217. [Google Scholar] [PubMed]
- Leroy, E.M.; Baize, S.; Volchkov, V.E.; Fisher-Hoch, S.P.; Georges-Courbot, M.C.; Lansoud-Soukate, J.; Capron, M.; Debre, P.; McCormick, J.B.; Georges, A.J. Human asymptomatic Ebola infection and strong inflammatory response. Lancet 2000, 355, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Hensley, L.E.; Young, H.A.; Jahrling, P.B.; Geisbert, T.W. Proinflammatory response during Ebola virus infection of primate models: Possible involvement of the tumor necrosis factor receptor superfamily. Immunol. Lett. 2002, 80, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Park, C.G.; Lee, S.Y.; Kandala, G.; Choi, Y. A novel gene product that couples TCR signaling to Fas(CD95) expression in activation-induced cell death. Immunity 1996, 4, 583–591. [Google Scholar] [CrossRef]
- Bosio, C.M.; Aman, M.J.; Grogan, C.; Hogan, R.; Ruthel, G.; Negley, D.; Mohamadzadeh, M.; Bavari, S.; Schmaljohn, A. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J. Infect. Dis. 2003, 188, 1630–1638. [Google Scholar] [CrossRef]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; McRae, M.; Rollin, P.E.; Pulendran, B. Cutting edge: Impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar] [CrossRef]
- Mohamadzadeh, M. Potential factors induced by filoviruses that lead to immune supression. Curr. Mol. Med. 2009, 9, 174–185. [Google Scholar] [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; Lennon, V.A.; Celis, E.; Chen, L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Badovinac, V.P.; Harty, J.T. Programming, demarcating, and manipulating CD8+ T-cell memory. Immunol. Rev. 2006, 211, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Blinov, V.M.; Netesov, S.V. The envelope glycoprotein of Ebola virus contains an immunosuppressive- like domain similar to oncogenic retroviruses. FEBS Lett. 1992, 305, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Bukreyev, A.; Volchkov, V.E.; Blinov, V.M.; Netesov, S.V. The GP-protein of Marburg virus contains the region similar to the 'immunosuppressive domain' of oncogenic retrovirus P15E proteins. FEBS Lett. 1993, 323, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Yaddanapudi, K.; Palacios, G.; Towner, J.S.; Chen, I.; Sariol, C.A.; Nichol, S.T.; Lipkin, W.I. Implication of a retrovirus-like glycoprotein peptide in the immunopathogenesis of Ebola and Marburg viruses. Faseb J. 2006, 20, 2519–2530. [Google Scholar] [CrossRef]




© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Olejnik, J.; Ryabchikova, E.; Corley, R.B.; Mühlberger, E. Intracellular Events and Cell Fate in Filovirus Infection. Viruses 2011, 3, 1501-1531. https://doi.org/10.3390/v3081501
Olejnik J, Ryabchikova E, Corley RB, Mühlberger E. Intracellular Events and Cell Fate in Filovirus Infection. Viruses. 2011; 3(8):1501-1531. https://doi.org/10.3390/v3081501
Chicago/Turabian StyleOlejnik, Judith, Elena Ryabchikova, Ronald B. Corley, and Elke Mühlberger. 2011. "Intracellular Events and Cell Fate in Filovirus Infection" Viruses 3, no. 8: 1501-1531. https://doi.org/10.3390/v3081501