Restricted Access to Myeloid Cells Explained

{kind=link}
{kind=link}
Abstract
:1. Introduction
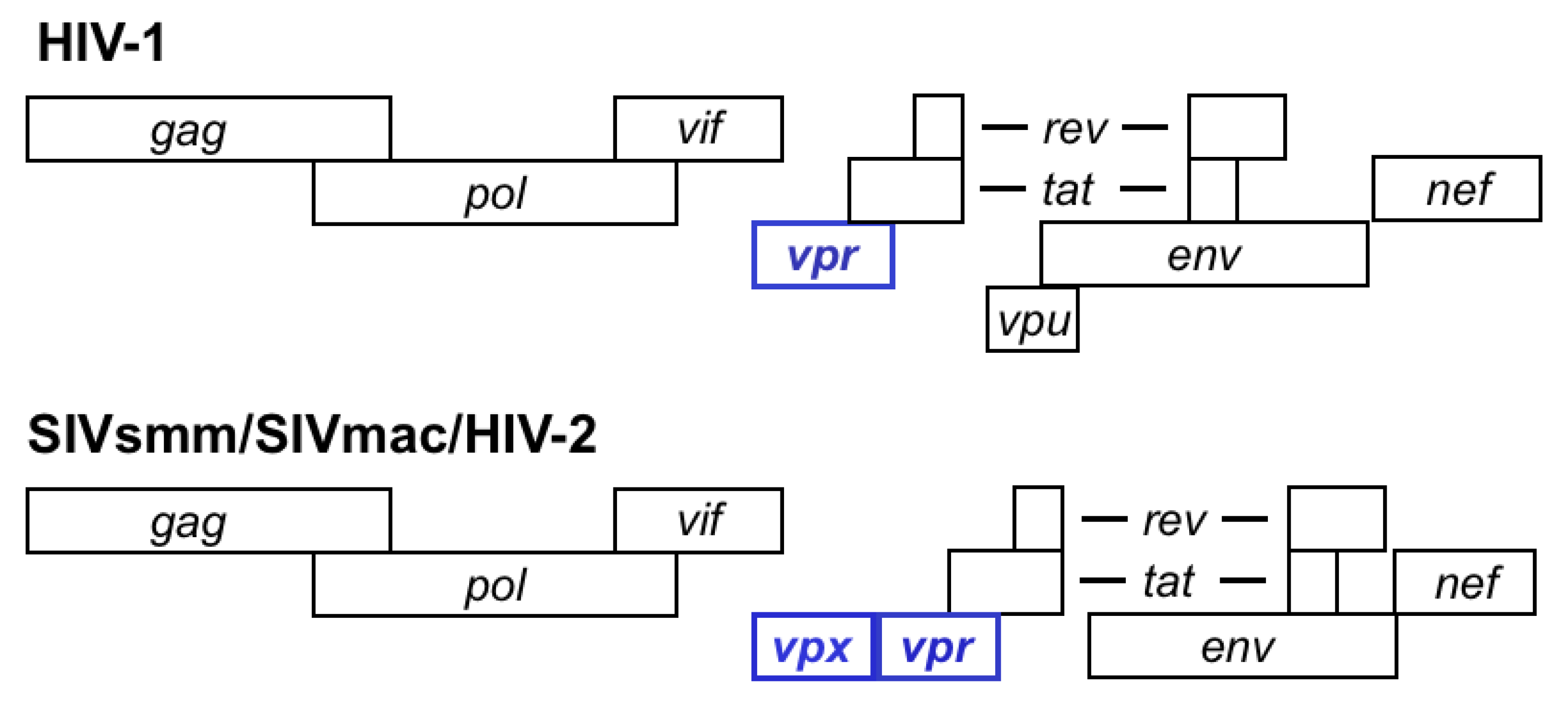
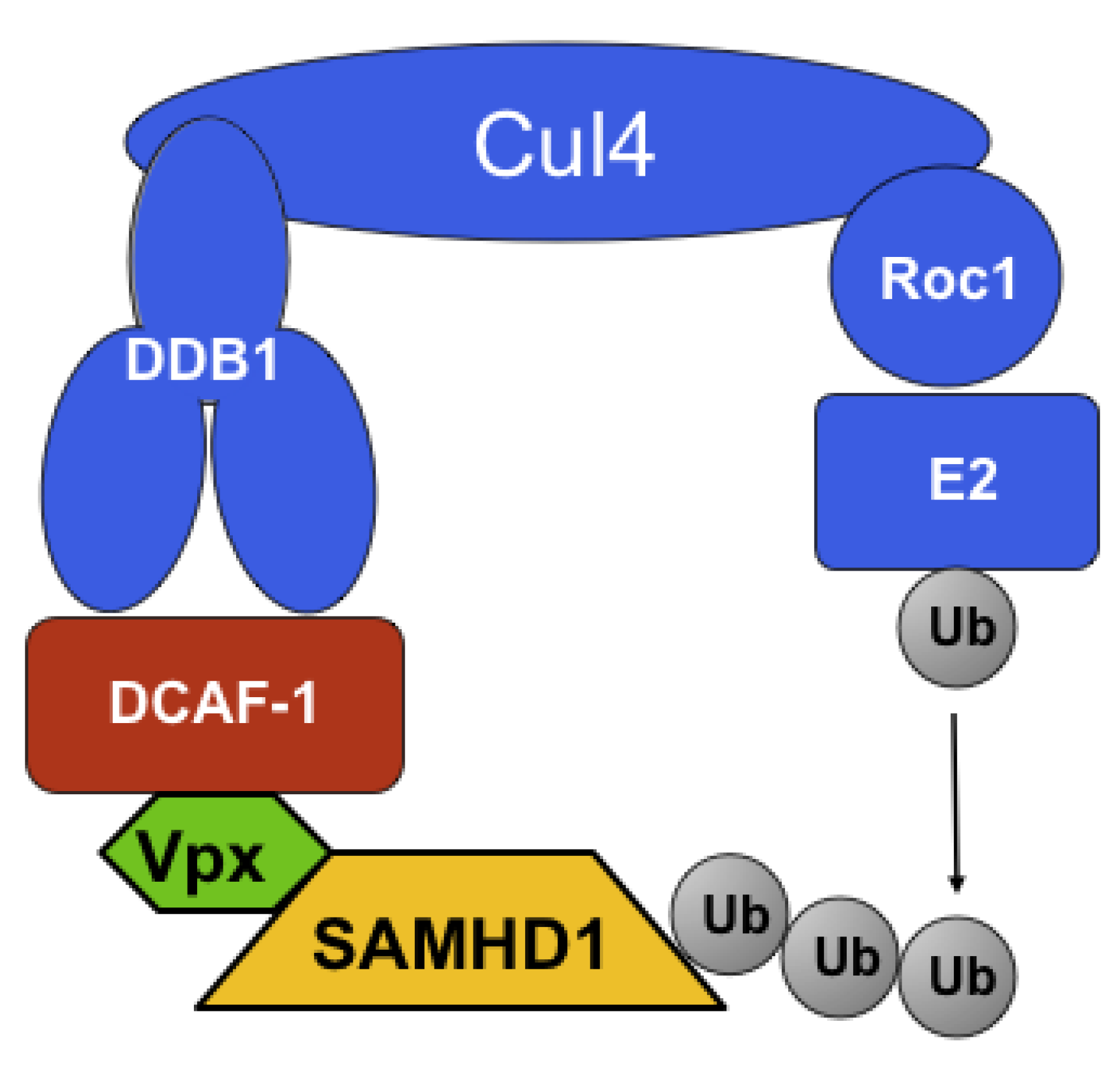
2. Identification of SAMHD1 as the Vpx-Sensitive Restriction Factor
3. SAMHD1 and the Aicardi-Goutières Syndrome
4. Yet Another Restriction
5. Conclusions and Future Directions
Conflict of Interest
References and Notes
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.T. Targeting early infection to prevent HIV-1 mucosal transmission. Nature 2010, 464, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Aghokeng, A.F.; Ayouba, A.; Mpoudi-Ngole, E.; Loul, S.; Liegeois, F.; Delaporte, E.; Peeters, M. Extensive survey on the prevalence and genetic diversity of SIVs in primate bushmeat provides insights into risks for potential new cross-species transmissions. Infect. Genet. Evol. 2010, 10, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Tristem, M.; Marshall, C.; Karpas, A.; Hill, F. Evolution of the primate lentiviruses: Evidence from vpx and vpr. EMBO J. 1992, 11, 3405–3412. [Google Scholar] [CrossRef]
- Sharp, P.M.; Bailes, E.; Stevenson, M.; Emerman, M.; Hahn, B.H. Gene acquisition in HIV and SIV. Nature 1996, 383, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Rogel, M.E.; Wu, L.I.; Emerman, M. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 1995, 69, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Planelles, V.; Jowett, J.B.; Li, Q.X.; Xie, Y.; Hahn, B.; Chen, I.S. Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J. Virol. 1996, 70, 2516–2524. [Google Scholar] [CrossRef]
- Fletcher, T.M., 3rd; Brichacek, B.; Sharova, N.; Newman, M.A.; Stivahtis, G.; Sharp, P.M.; Emerman, M.; Hahn, B.H.; Stevenson, M. Nuclear import and cell cycle arrest functions of the HIV-1 vpr protein are encoded by two separate genes in HIV-2/SIV(sm). EMBO J. 1996, 15, 6155–6165. [Google Scholar] [CrossRef]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [CrossRef]
- Re, F.; Braaten, D.; Franke, E.K.; Luban, J. Human immunodeficiency virus type 1 vpr arrests the cell cycle in g2 by inhibiting the activation of p34cdc2-cyclin b. J. Virol. 1995, 69, 6859–6864. [Google Scholar] [CrossRef]
- Jowett, J.B.; Planelles, V.; Poon, B.; Shah, N.P.; Chen, M.L.; Chen, I.S. The human immunodeficiency virus type 1 vpr gene arrests infected t cells in the G2 + M phase of the cell cycle. J. Virol. 1995, 69, 6304–6313. [Google Scholar] [CrossRef]
- Stewart, S.A.; Poon, B.; Jowett, J.B.; Chen, I.S. Human immunodeficiency virus type 1 vpr induces apoptosis following cell cycle arrest. J. Virol. 1997, 71, 5579–5592. [Google Scholar] [CrossRef] [PubMed]
- Shostak, L.D.; Ludlow, J.; Fisk, J.; Pursell, S.; Rimel, B.J.; Nguyen, D.; Rosenblatt, J.D.; Planelles, V. Roles of p53 and caspases in the induction of cell cycle arrest and apoptosis by HIV-1 vpr. Exp. Cell Res. 1999, 251, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Pancio, H.A.; Vander Heyden, N.; Ratner, L. The c-terminal proline-rich tail of human immunodeficiency virus type 2 vpx is necessary for nuclear localization of the viral preintegration complex in nondividing cells. J. Virol. 2000, 74, 6162–6167. [Google Scholar] [CrossRef] [PubMed]
- Guyader, M.; Emerman, M.; Montagnier, L.; Peden, K. Vpx mutants of HIV-2 are infectious in established cell lines but display a severe defect in peripheral blood lymphocytes. EMBO J. 1989, 8, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.F.; Yu, Q.C.; Essex, M.; Lee, T.H. The vpx gene of simian immunodeficiency virus facilitates efficient viral replication in fresh lymphocytes and macrophage. J. Virol. 1991, 65, 5088–5091. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Riviere, L.; Jarrosson-Wuilleme, L.; Bernaud, J.; Rigal, D.; Darlix, J.L.; Cimarelli, A. SIVsm/HIV-2 vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology 2007, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Otsuka, M.; Miyoshi, M.; Khamsri, B.; Nomaguchi, M.; Adachi, A. Vpx is critical for reverse transcription of the human immunodeficiency virus type 2 genome in macrophages. J. Virol. 2008, 82, 7752–7756. [Google Scholar] [CrossRef]
- Sharova, N.; Wu, Y.; Zhu, X.; Stranska, R.; Kaushik, R.; Sharkey, M.; Stevenson, M. Primate lentiviral vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 2008, 4, e1000057. [Google Scholar] [CrossRef]
- Srivastava, S.; Swanson, S.K.; Manel, N.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral vpx accessory factor targets VPRBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008, 4, e1000059. [Google Scholar] [CrossRef]
- Manel, N.; Hogstad, B.; Wang, Y.; Levy, D.E.; Unutmaz, D.; Littman, D.R. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 2010, 467, 214–217. [Google Scholar] [CrossRef]
- Goujon, C.; Arfi, V.; Pertel, T.; Luban, J.; Lienard, J.; Rigal, D.; Darlix, J.L.; Cimarelli, A. Characterization of simian immunodeficiency virus SIVsm/human immunodeficiency virus type 2 vpx function in human myeloid cells. J. Virol. 2008, 82, 12335–12345. [Google Scholar] [CrossRef]
- Goujon, C.; Jarrosson-Wuilleme, L.; Bernaud, J.; Rigal, D.; Darlix, J.L.; Cimarelli, A. With a little help from a friend: Increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(mac). Gene Ther. 2006, 13, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Cimarelli, A. University of Lyon, Lyon, France. Personal communication. 2011. [Google Scholar]
- DeHart, J.L.; Planelles, V. Human immunodeficiency virus type 1 vpr links proteasomal degradation and checkpoint activation. J. Virol. 2008, 82, 1066–1072. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Belaidouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C.; Margottin-Goguet, F. HIV1 vpr arrests the cell cycle by recruiting DCAF1/VPRBP, a receptor of the cul4-DDB1 ubiquitin ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Ayinde, D.; Maudet, C.; Transy, C.; Margottin-Goguet, F. Limelight on two HIV/siv accessory proteins in macrophage infection: Is vpx overshadowing vpr? Retrovirology 2010, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Sunseri, N.; O'Brien, M.; Bhardwaj, N.; Landau, N.R. Human immunodeficiency virus type 1 modified to package simian immunodeficiency virus vpx efficiently infects macrophages and dendritic cells. J. Virol. 2011, 85, 6263–6274. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. Samhd1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, W.; Cao, X. Identification of human homologue of mouse ifn-gamma induced protein from human dendritic cells. Immunol. Lett. 2000, 74, 221–224. [Google Scholar] [CrossRef]
- Lafuse, W.P.; Brown, D.; Castle, L.; Zwilling, B.S. Cloning and characterization of a novel cDNA that is IFN-gamma-induced in mouse peritoneal macrophages and encodes a putative GTP-binding protein. J. Leukoc. Biol. 1995, 57, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.D.; Proudfoot, M.; Yakunin, A.; Minor, W. Structural insight into the mechanism of substrate specificity and catalytic activity of an hd-domain phosphohydrolase: The 5'-deoxyribonucleotidase yfbr from Escherichia coli. J. Mol. Biol. 2008, 378, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef]
- Oussenko, I.A.; Sanchez, R.; Bechhofer, D.H. Bacillus subtilis YHAM, a member of a new family of 3'-to-5' exonucleases in gram-positive bacteria. J. Bacteriol. 2002, 184, 6250–6259. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Bowie, J.U. The many faces of sam. Sci. STKE 2005, 2005, re7. [Google Scholar] [CrossRef]
- Oberstrass, F.C.; Lee, A.; Stefl, R.; Janis, M.; Chanfreau, G.; Allain, F.H. Shape-specific recognition in the structure of the Vts1p SAM domain with RNA. Nat. Struct. Mol. Biol. 2006, 13, 160–167. [Google Scholar] [CrossRef]
- Rice, G.I.; Bond, J.; Asipu, A.; Brunette, R.L.; Manfield, I.W.; Carr, I.M.; Fuller, J.C.; Jackson, R.M.; Lamb, T.; Briggs, T.A.; et al. Mutations involved in aicardi-goutieres syndrome implicate samhd1 as regulator of the innate immune response. Nat. Genet. 2009, 41, 829–832. [Google Scholar] [CrossRef]
- Crow, Y.J.; Livingston, J.H. Aicardi-goutieres syndrome: An important mendelian mimic of congenital infection. Dev. Med. Child Neurol. 2008, 50, 410–416. [Google Scholar] [CrossRef]
- Yang, Y.G.; Lindahl, T.; Barnes, D.E. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef]
- Yan, N.; Regalado-Magdos, A.D.; Stiggelbout, B.; Lee-Kirsch, M.A.; Lieberman, J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010, 11, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Reinhard, C.; Luban, J. Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. Retrovirology 2011, 8, 49. [Google Scholar] [CrossRef]
- Wiegand, H.L.; Cullen, B.R. Inhibition of alpharetrovirus replication by a range of human APOBEC3 proteins. J. Virol. 2007, 81, 13694–13699. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Durand, S.; Fargier, G.; Nguyen, X.N.; Cordeil, S.; Bouaziz, S.; Muriaux, D.; Darlix, J.L.; Cimarelli, A. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PlosPathog 2011, in press. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Munk, C.; Schweizer, M.; Cichutek, K.; Schule, S.; Flory, E. Interaction of vpx and apolipoprotein B mRNA-editing catalytic polypeptide 3 family member A (APOBEC3A) correlates with efficient lentivirus infection of monocytes. J. Biol. Chem. 2010, 285, 12248–12254. [Google Scholar] [CrossRef]


© 2011 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Planelles, V. Restricted Access to Myeloid Cells Explained. Viruses 2011, 3, 1624-1633. https://doi.org/10.3390/v3091624
Planelles V. Restricted Access to Myeloid Cells Explained. Viruses. 2011; 3(9):1624-1633. https://doi.org/10.3390/v3091624
Chicago/Turabian StylePlanelles, Vicente. 2011. "Restricted Access to Myeloid Cells Explained" Viruses 3, no. 9: 1624-1633. https://doi.org/10.3390/v3091624
APA StylePlanelles, V. (2011). Restricted Access to Myeloid Cells Explained. Viruses, 3(9), 1624-1633. https://doi.org/10.3390/v3091624