Multifunctional Nature of the Arenavirus RING Finger Protein Z

Abstract
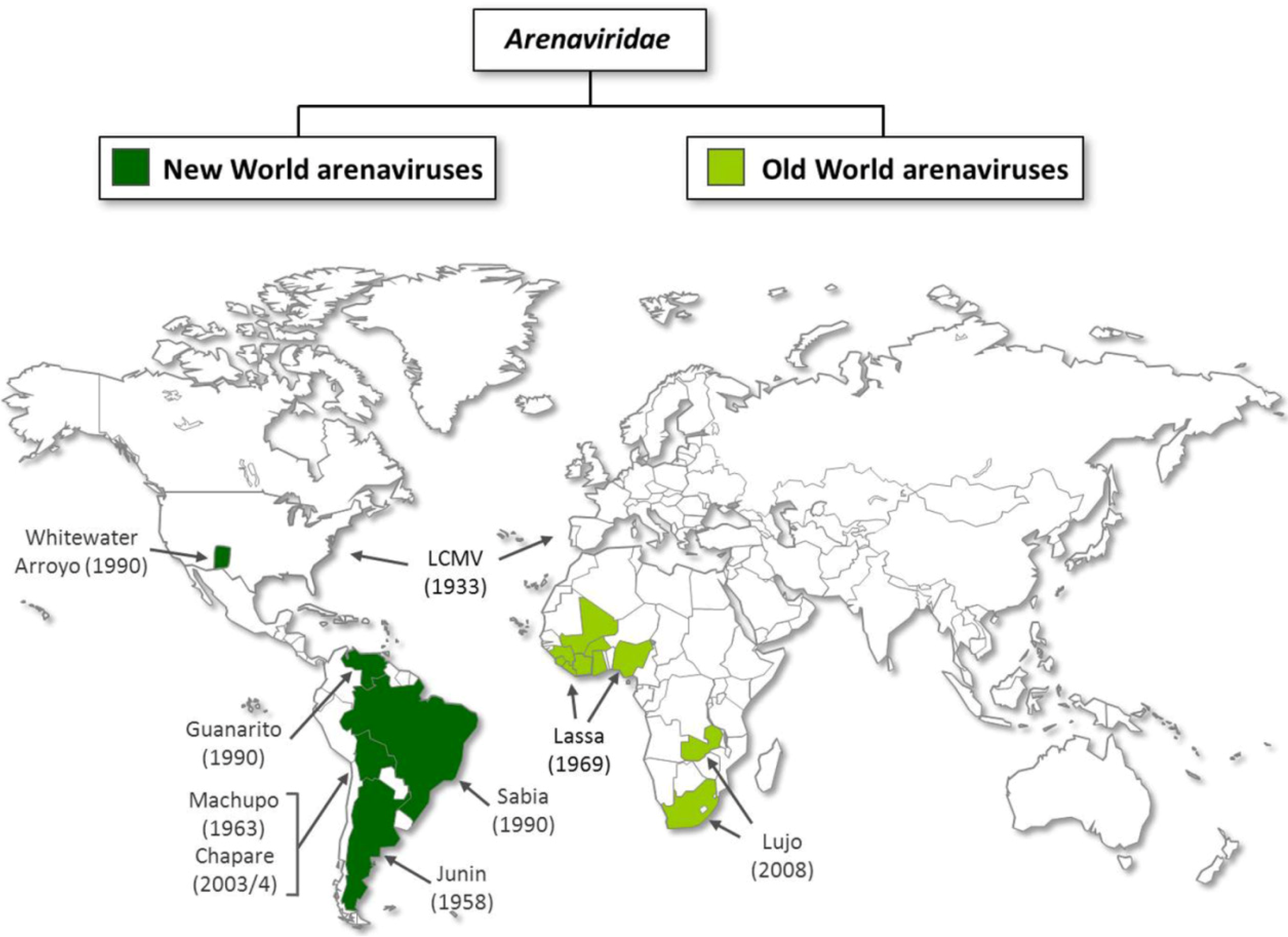
:1. Introduction
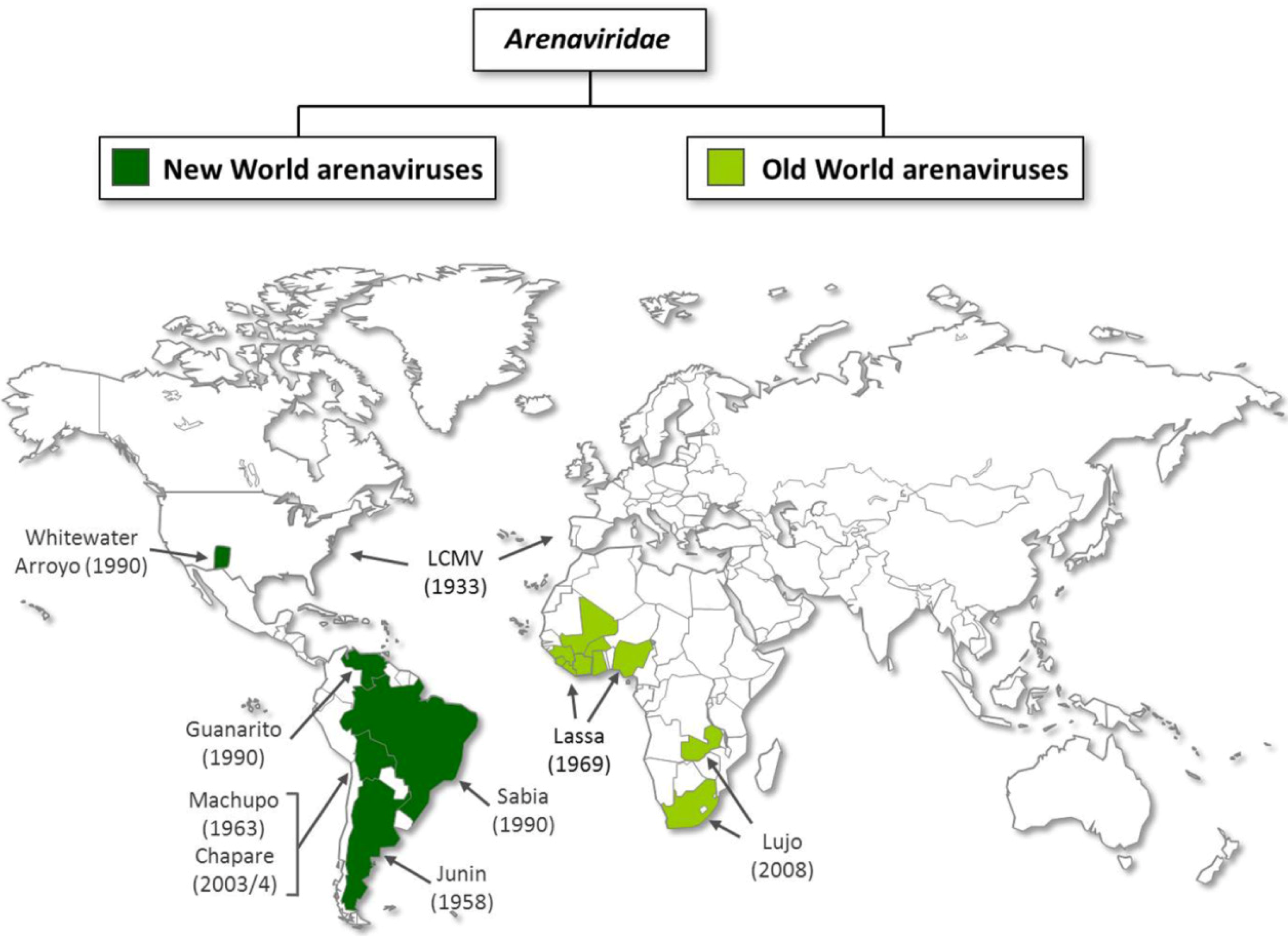
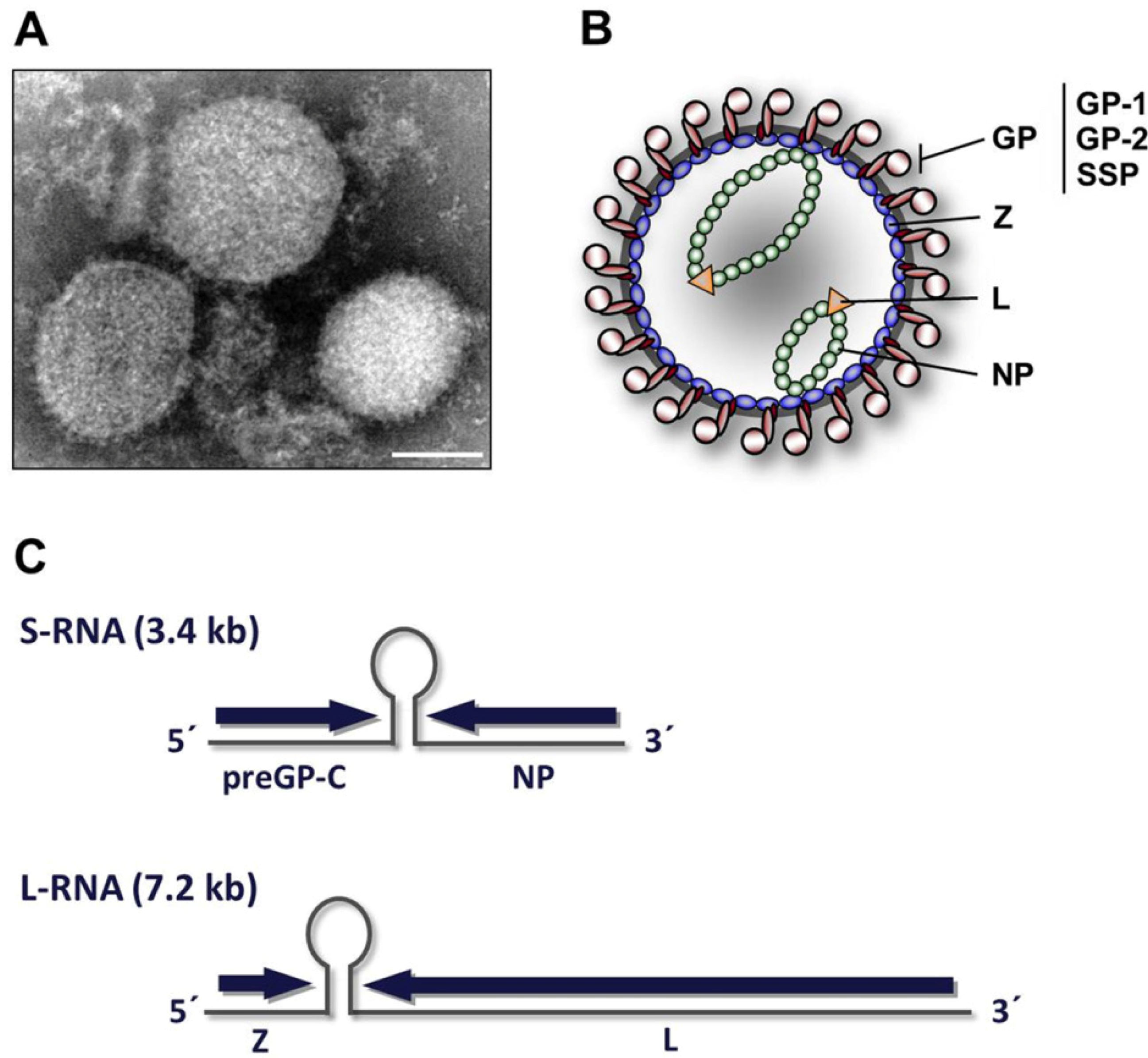
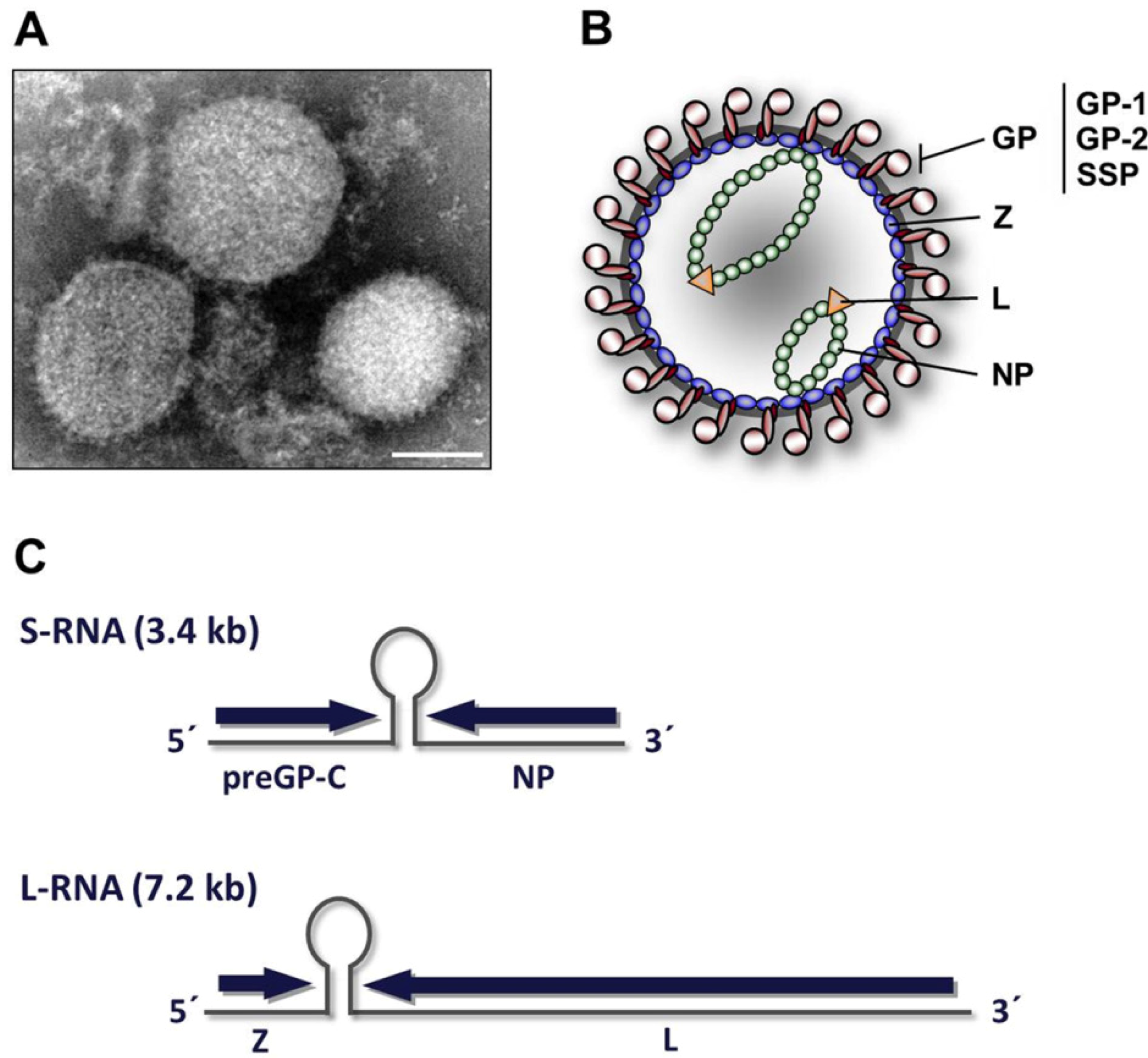
2. Virion architecture, gene organization and virus replication
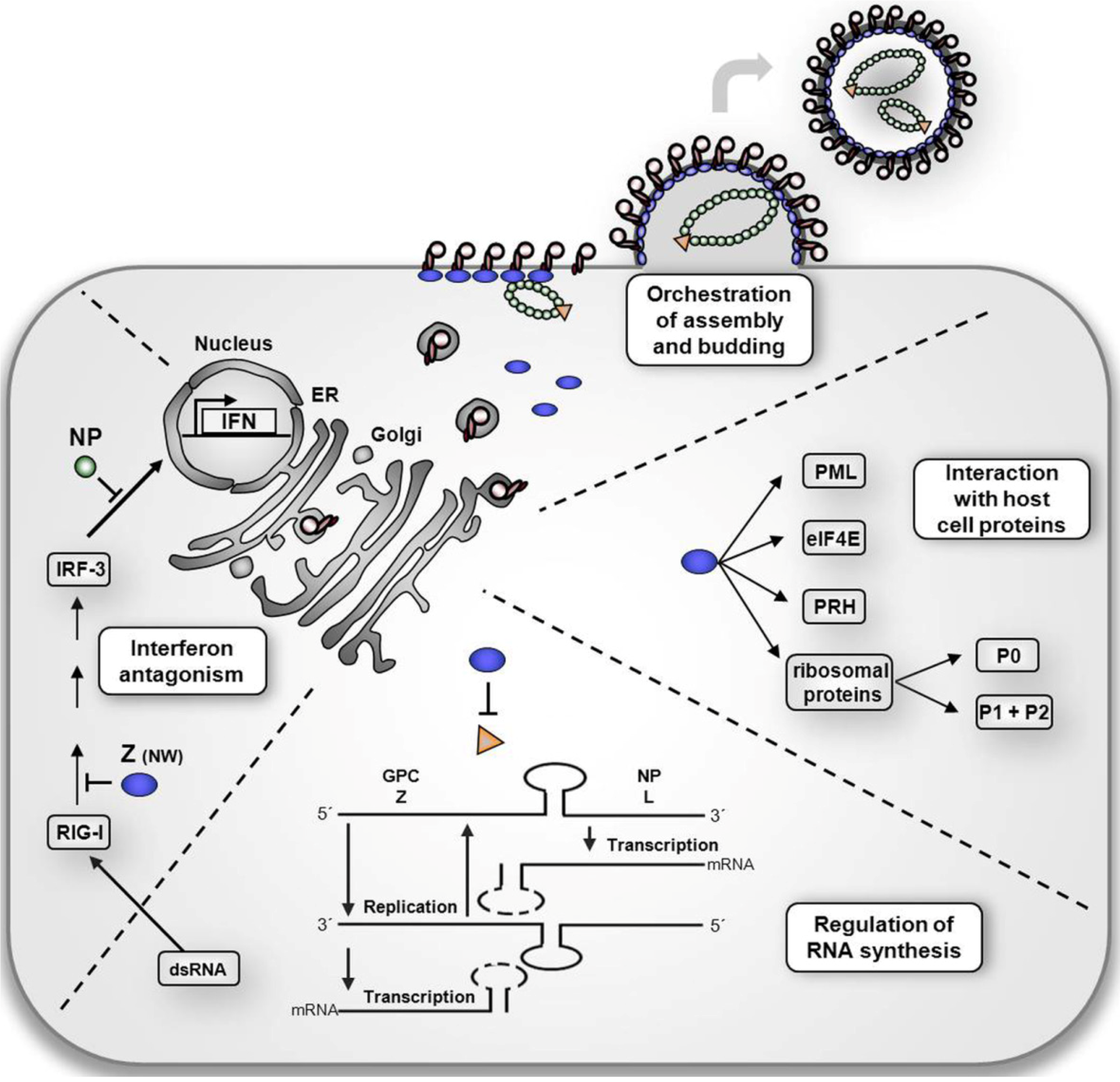
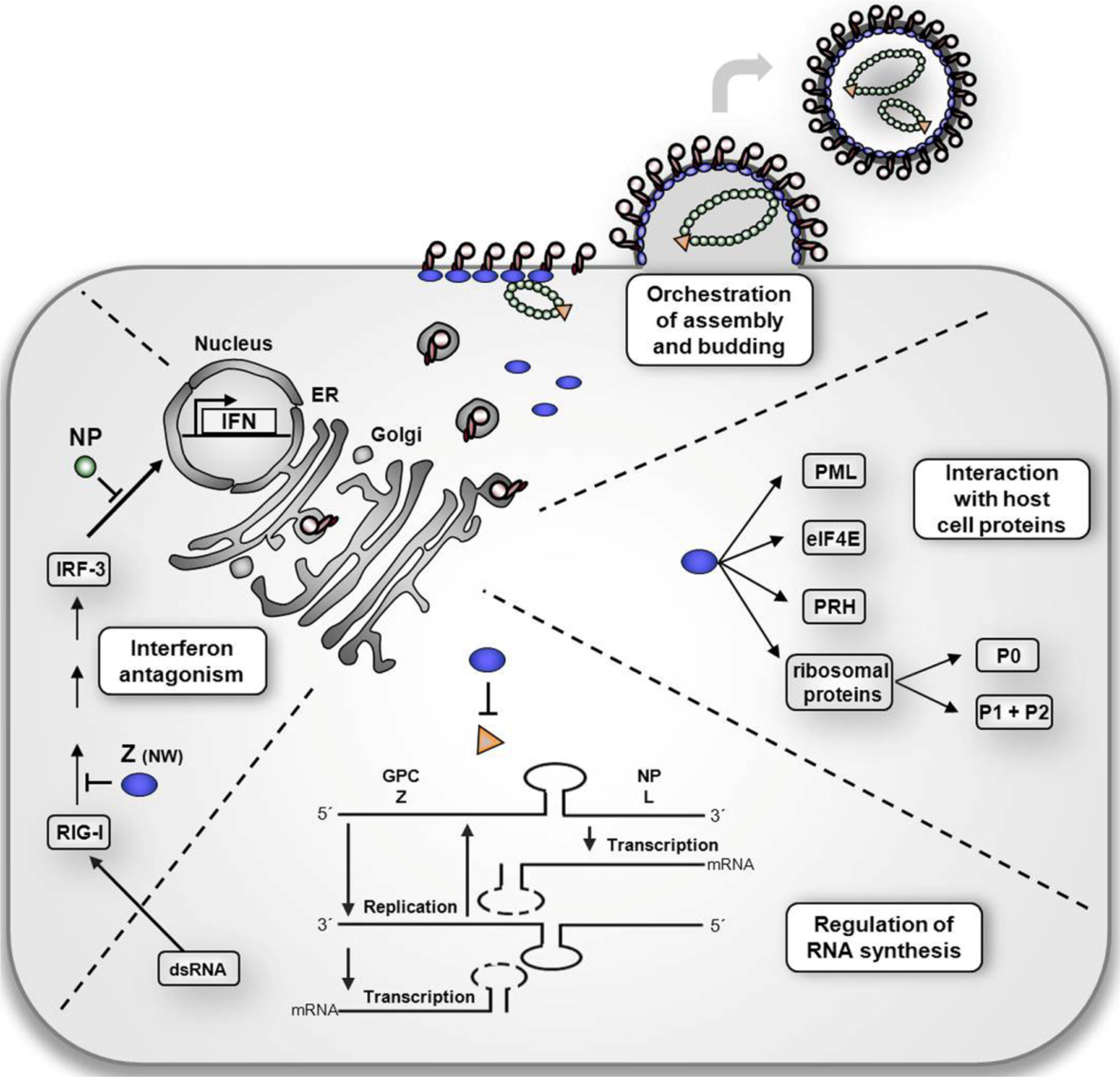
3. The multiple roles of Z protein in the arenavirus life cycle

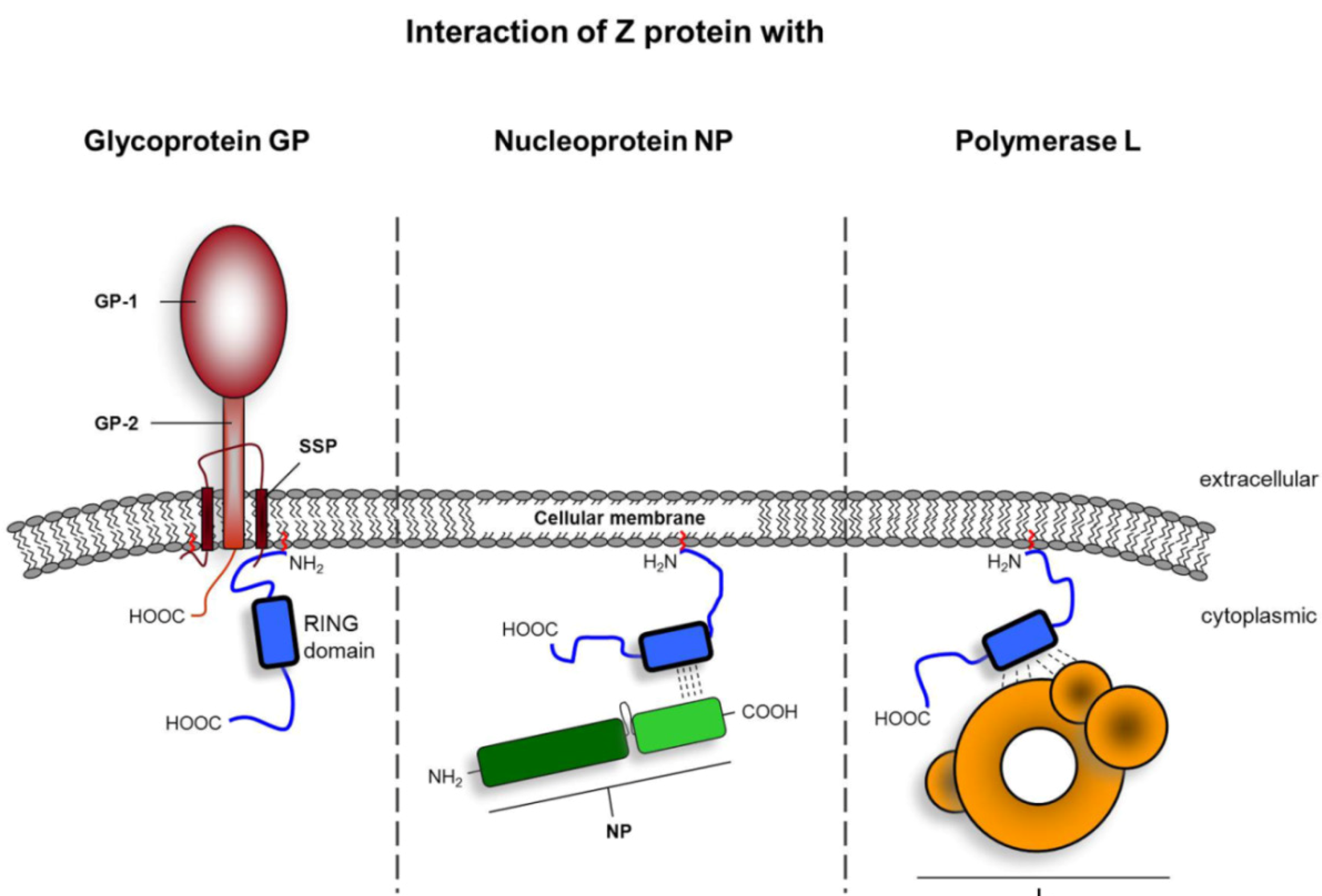
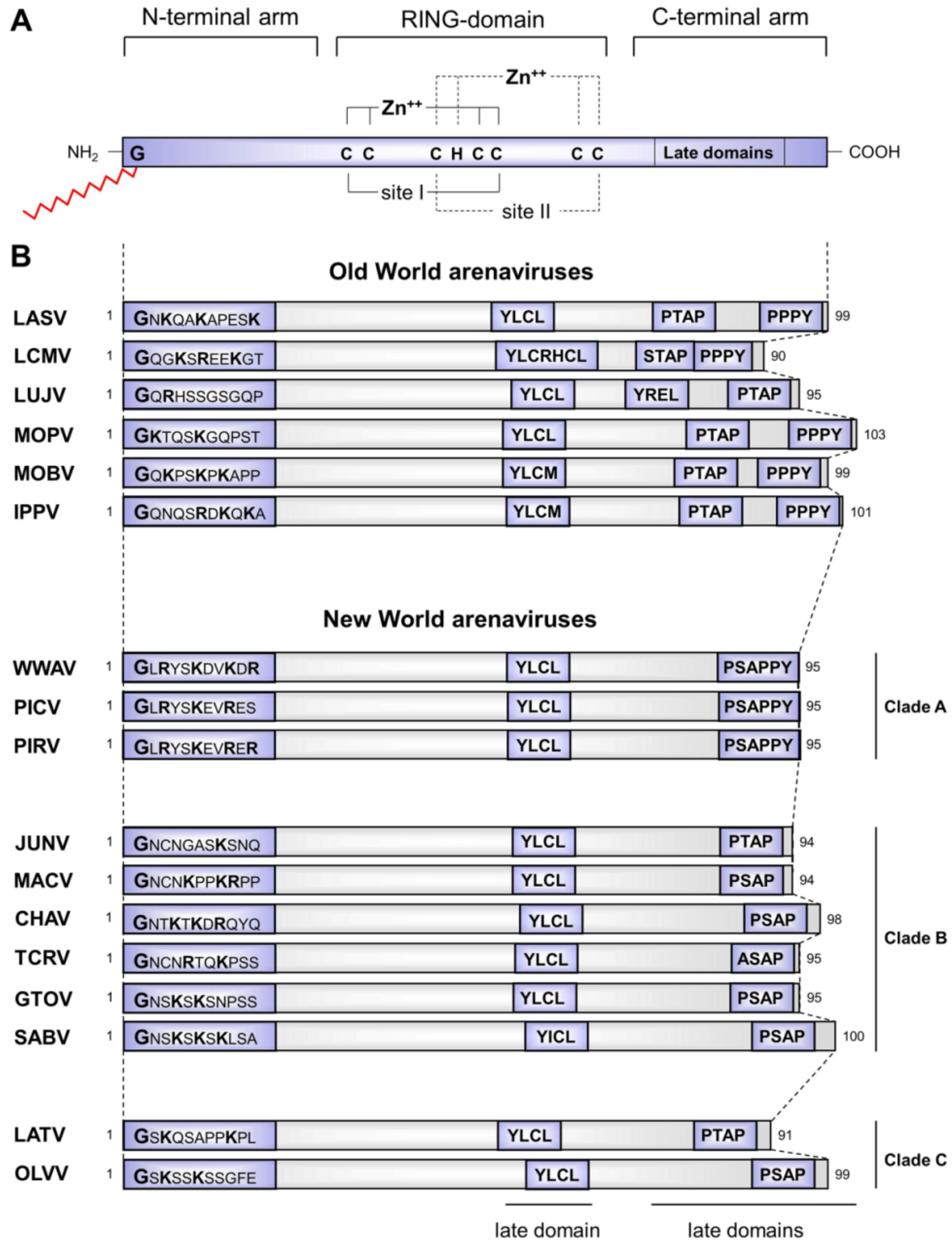
3.1 Structural aspects of the distinct functional properties of Z proteins
3.2 Z protein as a regulator of virus replication and transcription
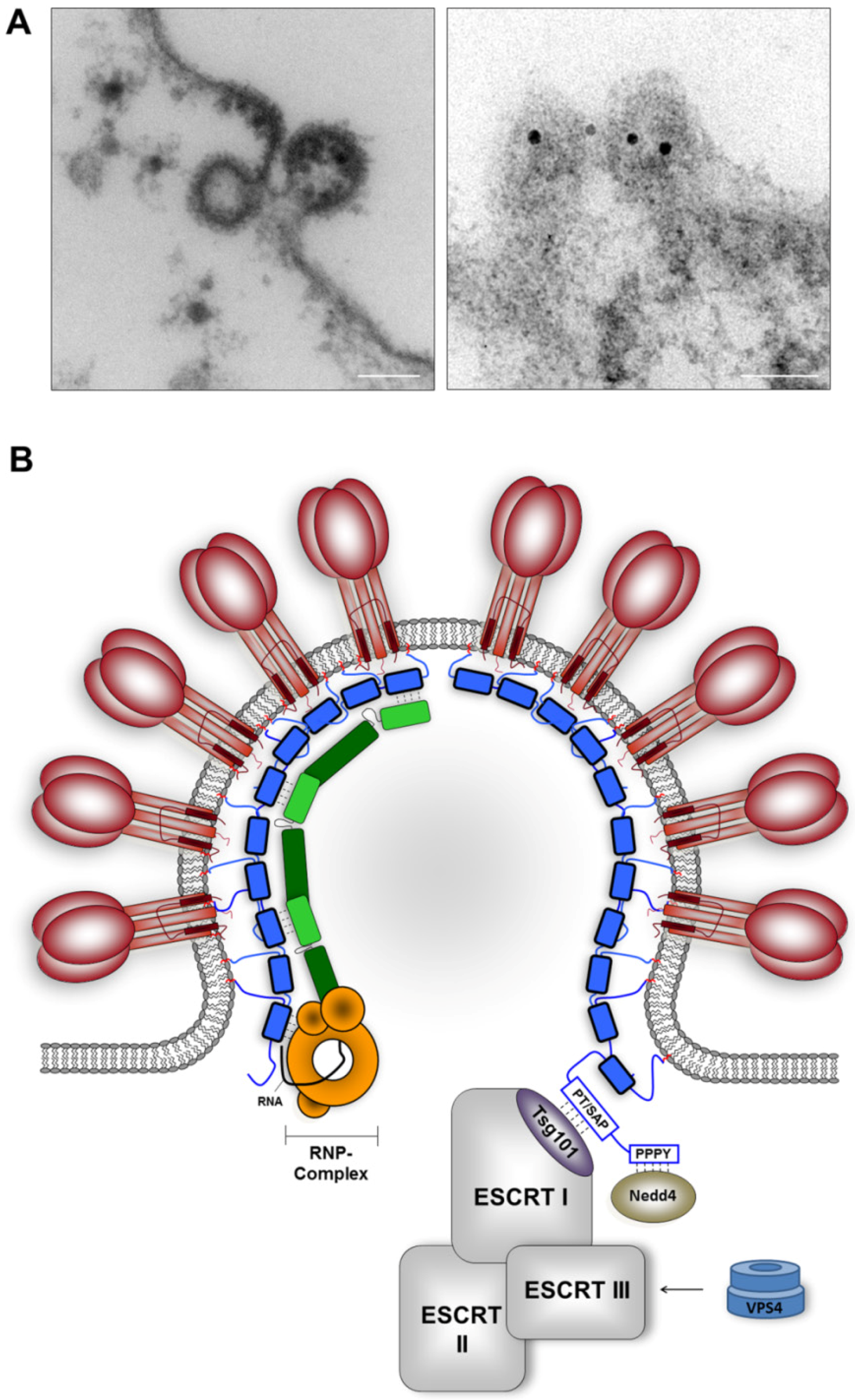
3.3 The multifunctional activities of the Z protein in virus assembly and budding
3.3.1 The role of the Z protein in glycoprotein GP incorporation
3.3.2 The role of Z protein in RNP packaging
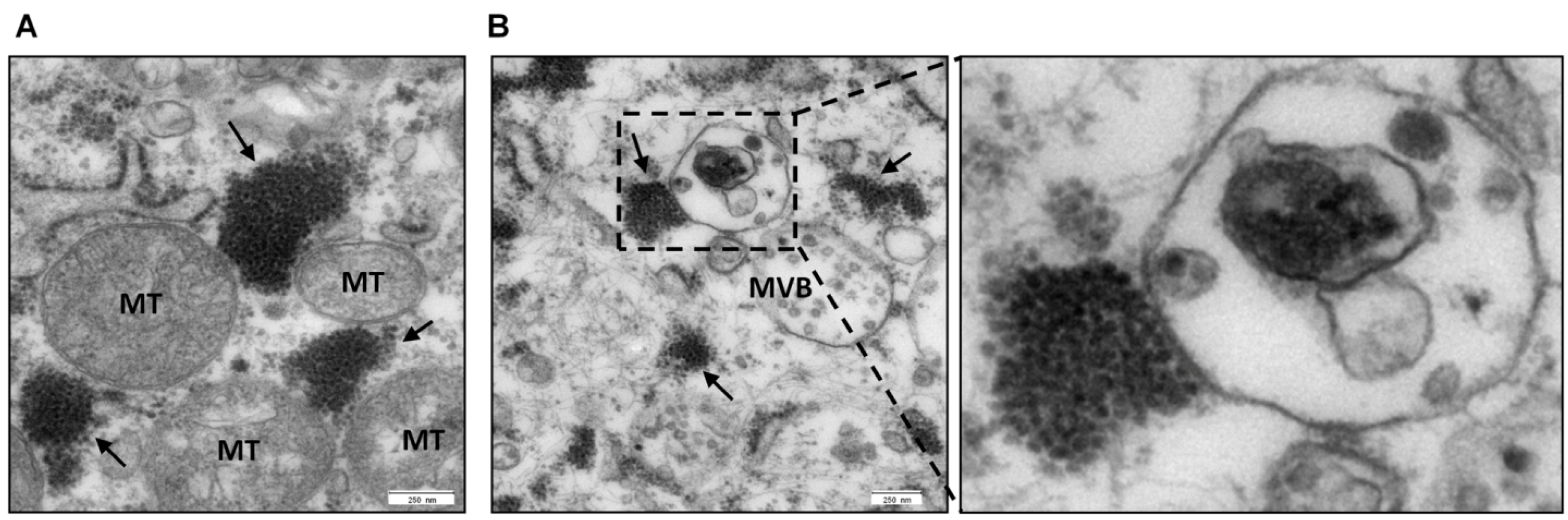
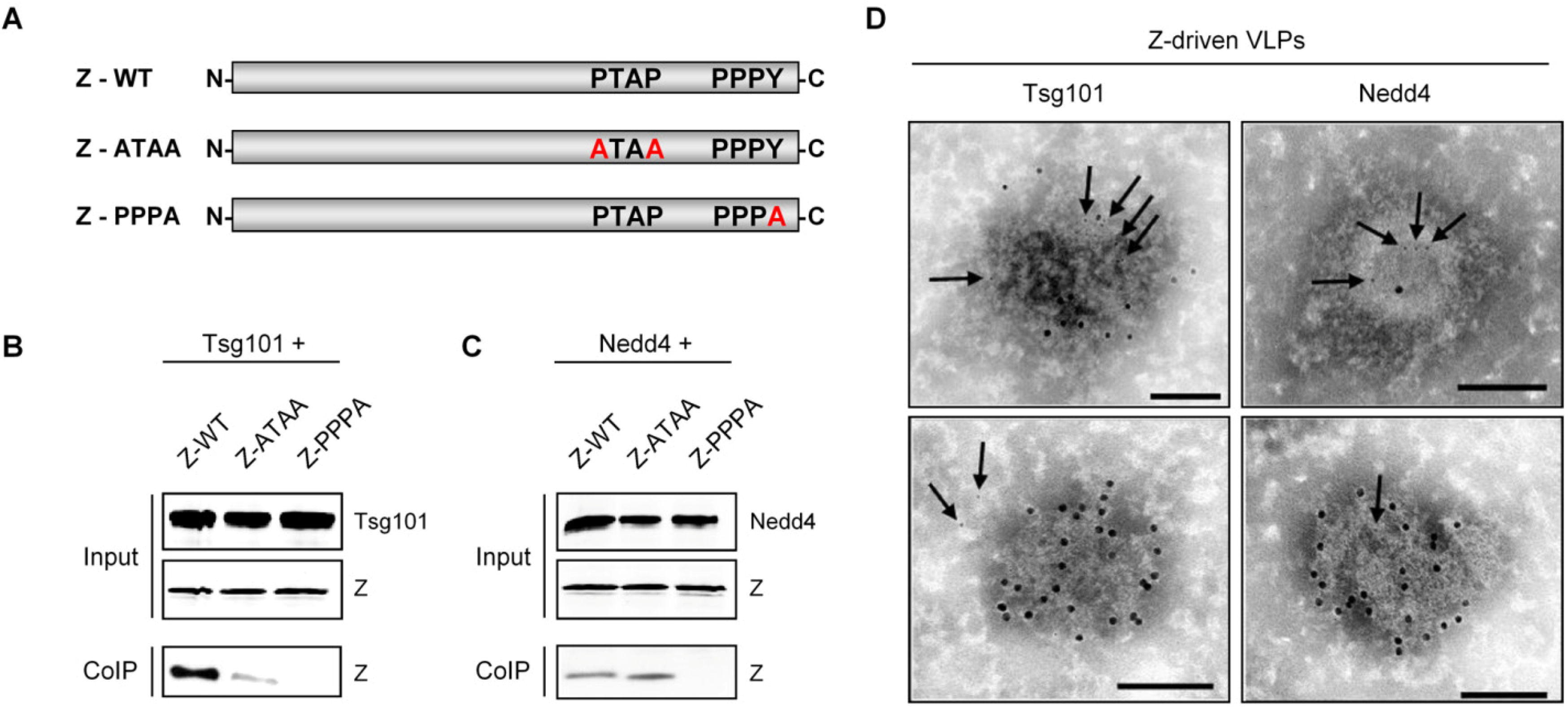
3.3.3 Z-mediated recruitment of host cell proteins essential for virus budding

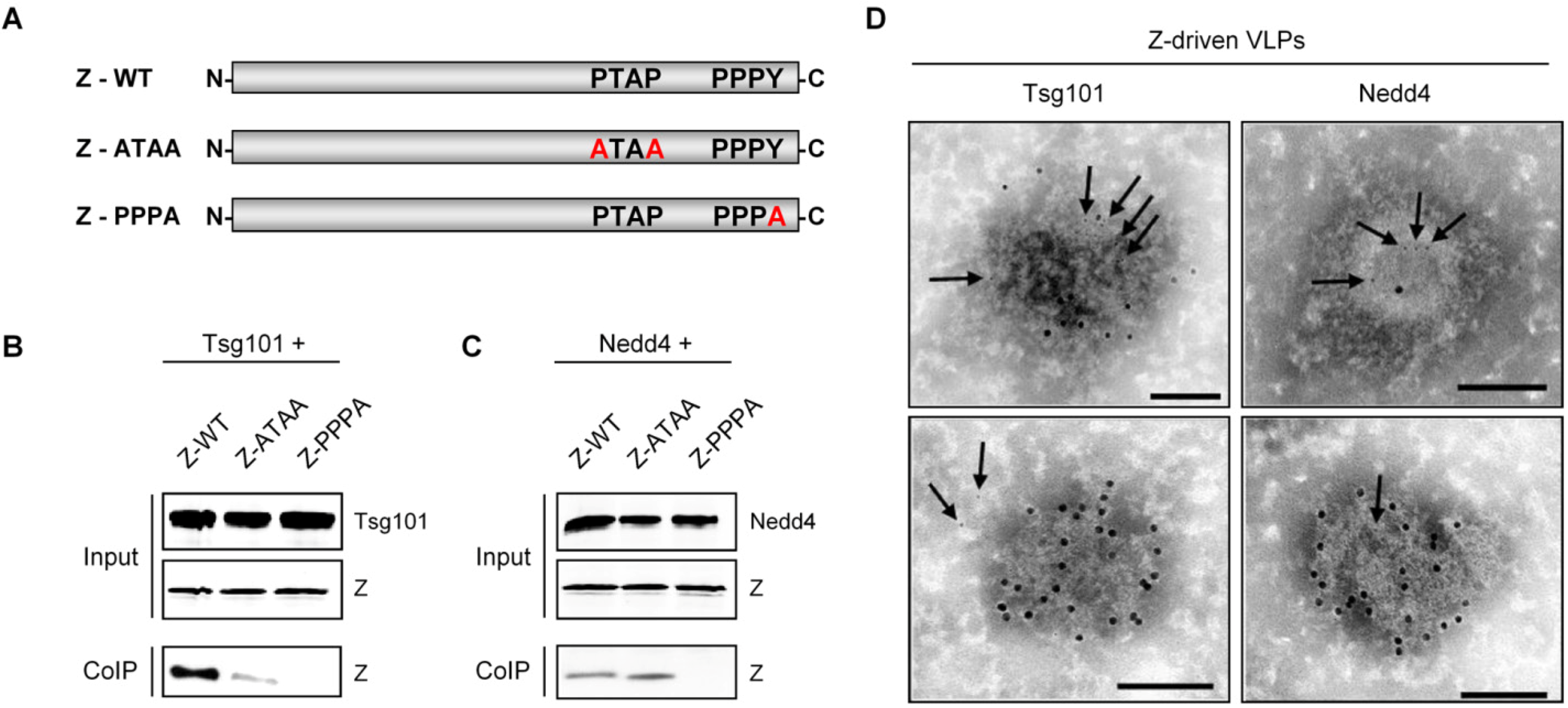
3.4 Z-mediated regulation of host cell functions
5. Concluding remarks and future outlook

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inter-arenaviral protein interactions | ||||||
| Interaction partner | Function | Domain/motifof Z protein | Shown for | Citations | ||
| GP | Z-GP interaction through association of Z with SSP of GP, recruitment of GP into Z-driven VLPs | N-terminal myristoylation | LASV LCMV | [72] | ||
| [106] | ||||||
| L | Inhibitory effect on transcription and replication by locking a polymerase-template complex | RING domain conserved W36 residue in LCMV Z | LCMV TCRV | [85] | ||
| [90] | ||||||
| [91] | ||||||
| Z-L interaction | RING domain | TCRV | [85] | |||
| NP | Z-NP interaction, recruitment of NP into VLPs | RING domain Residue L79 | JUNV | [118] | ||
| Recruitment of NP into VLPs, enhancement of TCRV Z-mediated budding | ASAP motif | TCRV | [117] | |||
| Recruitment of NP into VLPs, enhancement of TCRV Z-mediated budding | YLCL motif | TCRV | [117] | |||
| NP incorporation into Z-induced VLPs | YLCL motif | MOPV | [124] | |||
| Z | Association with membranes, intracellular targeting | N-terminal myristoylation | LASV LCMV | [71] | ||
| [70] | ||||||
| Self-association, multimerization | RING domain, N-terminal myristoylation | LCMV JUNV TCRV | [77] | |||
| [73] | ||||||
| Virus-host cell protein interactions | ||||||
| Interaction partner | Function | Domain/motifof Z protein | Shown for | Citations | ||
| Alix/AiP1 | NP-Z interaction | YLCL motif | MOPV | [124] | ||
| eIF-4E | Repression of eIF-4E dependent translation | RING domain | LCMV | [168] | ||
| Nedd4 | Exploitation of the VPS pathway for virus release | PPPY motif | LASV | Present study | ||
| P0 | unknown | unknown | LCMV | [166] | ||
| PML | Relocalization of PML from the nucleus to the cytoplasm and therefore evasion of host cell apoptosis | unknown | LCMV | [166] | ||
| PRH | Inhibition of the antiproliferative effect of PRH, favoring viral replication and leading to pathogenesis | unknown | LCMV | [170] | ||
| RIG-I | Inhibition of type-I IFN induction | unknown | JUNV GTOV MACV SABV | [181] | ||
| Tsg101 | Exploitation of the VPS pathway for virus release | PTAP and PPPY motifs | LCMV LASV | [130] | ||
| [129] | ||||||
| Present study | ||||||
Acknowledgments
Conflict of Interest
List of abbreviations
| BSL-4 | biosafety level 4 |
| eIF4E | eukaryotic translation initiation factor 4E |
| ESCRT | endosomal sorting complex required for transport |
| GP | glycoprotein |
| GP-C | glycoprotein precursor |
| IFN | interferon |
| IKKε | IκB kinase epsilon |
| IRF-3 | interferon regulatory factor 3 |
| JUNV | Junin virus |
| kDA | kilo Dalton |
| L | L protein, arenavirus polymerase |
| LASV | Lassa virus |
| LCMV | lymphocytic choriomeningitis virus |
| NF-ĸB | nuclear factor kappa light chain enhancer of activated B cells |
| Nedd4 | neural precursor cell expressed developmentally down-regulated protein 4 |
| NP | nucleoprotein |
| P0 | ribosomal protein P0 |
| PML | promyelocytic leukemia protein |
| PRH | proline-rich homeodomain protein |
| RIG-I | retinoic acid inducible gene I |
| RING | really interesting new gene |
| RNA | ribonucleic acid |
| RNAi | RNA interference |
| SSP | stable signal peptide |
| TCRV | Tacaribe virus |
| Tsg101 | tumor susceptibility gene 101 |
| VLP | virus-like particle |
| Z protein | zinc-binding matrix protein |
References and Notes
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; Nichol, S.T.; Lipkin, W.I. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog 2009, 5, e1000455. [Google Scholar] [CrossRef]
- Buckley, S.M.; Casals, J.; Downs, W.G. Isolation and antigenic characterization of Lassa virus. Nature 1970, 227, 174. [Google Scholar]
- Smadel, J.E.; Wall, M.J. Identification of the Virus of Lymphocytic Choriomeningitis. J Bacteriol 1941, 41, 421–430. [Google Scholar]
- Emonet, S.; Lemasson, J.J.; Gonzalez, J.P.; de Lamballerie, X.; Charrel, R.N. Phylogeny and evolution of old world arenaviruses. Virology 2006, 350, 251–257. [Google Scholar] [CrossRef]
- Lecompte, E.; ter Meulen, J.; Emonet, S.; Daffis, S.; Charrel, R.N. Genetic identification of Kodoko virus, a novel arenavirus of the African pigmy mouse (Mus Nannomys minutoides) in West Africa. Virology 2007, 364, 178–183. [Google Scholar] [CrossRef]
- Gunther, S.; Hoofd, G.; Charrel, R.; Roser, C.; Becker-Ziaja, B.; Lloyd, G.; Sabuni, C.; Verhagen, R.; van der Groen, G.; Kennis, J.; Katakweba, A.; Machang'u, R.; Makundi, R.; Leirs, H. Mopeia virus-related arenavirus in natal multimammate mice, Morogoro, Tanzania. Emerg Infect Dis 2009, 15, 2008–2012. [Google Scholar] [CrossRef]
- Delgado, S.; Erickson, B.R.; Agudo, R.; Blair, P.J.; Vallejo, E.; Albarino, C.G.; Vargas, J.; Comer, J.A.; Rollin, P.E.; Ksiazek, T.G.; Olson, J.G.; Nichol, S.T. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLoS Pathog 2008, 4, e1000047. [Google Scholar] [CrossRef]
- Charrel, R.N.; Feldmann, H.; Fulhorst, C.F.; Khelifa, R.; de Chesse, R.; de Lamballerie, X. Phylogeny of New World arenaviruses based on the complete coding sequences of the small genomic segment identified an evolutionary lineage produced by intrasegmental recombination. Biochem Biophys Res Commun 2002, 296, 1118–1124. [Google Scholar] [CrossRef]
- Bowen, M.D.; Peters, C.J.; Nichol, S.T. The phylogeny of New World (Tacaribe complex) arenaviruses. Virology 1996, 219, 285–290. [Google Scholar] [CrossRef]
- Downs, W.G.; Anderson, C.R.; Spence, L.; Aitken, T.H.; Greenhall, A.H. Tacaribe virus, a new agent isolated from Artibeus bats and mosquitoes in Trinidad, West Indies. Am J Trop Med Hyg 1963, 12, 640–646. [Google Scholar]
- Cogswell-Hawkinson, A.; Bowen, R.; James, S.; Gardiner, D.; Calisher, C.H.; Adams, R.; Schountz, T. Tacaribe Virus Causes Fatal Infection of An Ostensible Host, the Jamaican Fruit Bat. J Virol 2012.
- Ter Meulen, J.; Lukashevich, I.; Sidibe, K.; Inapogui, A.; Marx, M.; Dorlemann, A.; Yansane, M.L.; Koulemou, K.; Chang-Claude, J.; Schmitz, H. Hunting of peridomestic rodents and consumption of their meat as possible risk factors for rodent-to-human transmission of Lassa virus in the Republic of Guinea. Am J Trop Med Hyg 1996, 55, 661–666. [Google Scholar]
- Kerneis, S.; Koivogui, L.; Magassouba, N.; Koulemou, K.; Lewis, R.; Aplogan, A.; Grais, R.F.; Guerin, P.J.; Fichet-Calvet, E. Prevalence and risk factors of Lassa seropositivity in inhabitants of the forest region of Guinea: a cross-sectional study. PLoS Negl Trop Dis 2009, 3, e548. [Google Scholar] [CrossRef] [Green Version]
- Fisher-Hoch, S.P.; Tomori, O.; Nasidi, A.; Perez-Oronoz, G.I.; Fakile, Y.; Hutwagner, L.; McCormick, J.B. Review of cases of nosocomial Lassa fever in Nigeria: the high price of poor medical practice. BMJ 1995, 311, 857–859. [Google Scholar]
- Monath, T.P.; Mertens, P.E.; Patton, R.; Moser, C.R.; Baum, J.J.; Pinneo, L.; Gary, G.W.; Kissling, R.E. A hospital epidemic of Lassa fever in Zorzor, Liberia, March-April 1972. Am J Trop Med Hyg 1973, 22, 773–779. [Google Scholar]
- Carey, D.E.; Kemp, G.E.; White, H.A.; Pinneo, L.; Addy, R.F.; Fom, A.L.; Stroh, G.; Casals, J.; Henderson, B.E. Lassa fever. Epidemiological aspects of the 1970 epidemic, Jos, Nigeria. Trans R Soc Trop Med Hyg 1972, 66, 402–408. [Google Scholar] [CrossRef]
- Bowen, G.S.; Tomori, O.; Wulff, H.; Casals, J.; Noonan, A.; Downs, W.G. Lassa fever in Onitsha, East Central State, Nigeria in 1974. Bull World Health Organ 1975, 52, 599–604. [Google Scholar]
- Webb, P.A.; McCormick, J.B.; King, I.J.; Bosman, I.; Johnson, K.M.; Elliott, L.H.; Kono, G.K.; O'Sullivan, R. Lassa fever in children in Sierra Leone, West Africa. Trans R Soc Trop Med Hyg 1986, 80, 577–582. [Google Scholar] [CrossRef]
- McCormick, J.B. Epidemiology and control of Lassa fever. Curr Top Microbiol Immunol 1987, 134, 69–78. [Google Scholar]
- Omilabu, S.A.; Badaru, S.O.; Okokhere, P.; Asogun, D.; Drosten, C.; Emmerich, P.; Becker-Ziaja, B.; Schmitz, H.; Gunther, S. Lassa fever, Nigeria, 2003 and 2004. Emerg Infect Dis 2005, 11, 1642–1644. [Google Scholar] [CrossRef]
- Ehichioya, D.U.; Hass, M.; Olschlager, S.; Becker-Ziaja, B.; Onyebuchi Chukwu, C.O.; Coker, J.; Nasidi, A.; Ogugua, O.O.; Gunther, S.; Omilabu, S.A. Lassa fever, Nigeria, 2005-2008. Emerg Infect Dis 2010, 16, 1040–1041. [Google Scholar] [CrossRef]
- Fichet-Calvet, E.; Rogers, D.J. Risk maps of Lassa fever in West Africa. PLoS Negl Trop Dis 2009, 3, e388. [Google Scholar] [CrossRef]
- Georges, A.J.; Gonzalez, J.P.; Abdul-Wahid, S.; Saluzzo, J.F.; Meunier, D.M.; McCormick, J.B. Antibodies to Lassa and Lassa-like viruses in man and mammals in the Central African Republic. Trans R Soc Trop Med Hyg 1985, 79, 78–79. [Google Scholar] [CrossRef]
- Safronetz, D.; Lopez, J.E.; Sogoba, N.; Traore, S.F.; Raffel, S.J.; Fischer, E.R.; Ebihara, H.; Branco, L.; Garry, R.F.; Schwan, T.G.; Feldmann, H. Detection of Lassa virus, Mali. Emerg Infect Dis 2010, 16, 1123–1126. [Google Scholar]
- Saluzzo, J.F.; Adam, F.; McCormick, J.B.; Digoutte, J.P. Lassa fever virus in Senegal. J Infect Dis 1988, 157, 605. [Google Scholar] [CrossRef]
- Akoua-Koffi, C.; Ter Meulen, J.; Legros, D.; Akran, V.; Aidara, M.; Nahounou, N.; Dogbo, P.; Ehouman, A. [Detection of anti-Lassa antibodies in the Western Forest area of the Ivory Coast]. Med Trop (Mars) 2006, 66, 465–468. [Google Scholar]
- Zweighaft, R.M.; Fraser, D.W.; Hattwick, M.A.; Winkler, W.G.; Jordan, W.C.; Alter, M.; Wolfe, M.; Wulff, H.; Johnson, K.M. Lassa fever: response to an imported case. N Engl J Med 1977, 297, 803–807. [Google Scholar]
- Atkin, S.; Anaraki, S.; Gothard, P.; Walsh, A.; Brown, D.; Gopal, R.; Hand, J.; Morgan, D. The first case of Lassa fever imported from Mali to the United Kingdom, February 2009. Euro Surveill 2009, 14. [Google Scholar]
- ter Meulen, J.; Lenz, O.; Koivogui, L.; Magassouba, N.; Kaushik, S.K.; Lewis, R.; Aldis, W. Short communication: Lassa fever in Sierra Leone: UN peacekeepers are at risk. Trop Med Int Health 2001, 6, 83–84. [Google Scholar] [CrossRef]
- Amorosa, V.; MacNeil, A.; McConnell, R.; Patel, A.; Dillon, K.E.; Hamilton, K.; Erickson, B.R.; Campbell, S.; Knust, B.; Cannon, D.; Miller, D.; Manning, C.; Rollin, P.E.; Nichol, S.T. Imported Lassa fever, Pennsylvania, USA, 2010. Emerg Infect Dis 2010, 16, 1598–1600. [Google Scholar]
- Haas, W.H.; Breuer, T.; Pfaff, G.; Schmitz, H.; Kohler, P.; Asper, M.; Emmerich, P.; Drosten, C.; Golnitz, U.; Fleischer, K.; Günther, S. Imported Lassa fever in Germany: surveillance and management of contact persons. Clin Infect Dis 2003, 36, 1254–1258. [Google Scholar] [CrossRef]
- Schmitz, H.; Kohler, B.; Laue, T.; Drosten, C.; Veldkamp, P.J.; Gunther, S.; Emmerich, P.; Geisen, H.P.; Fleischer, K.; Beersma, M.F.; Hoerauf, A. Monitoring of clinical and laboratory data in two cases of imported Lassa fever. Microbes Infect 2002, 4, 43–50. [Google Scholar] [CrossRef]
- Günther, S.; Emmerich, P.; Laue, T.; Kuhle, O.; Asper, M.; Jung, A.; Grewing, T.; ter Meulen, J.; Schmitz, H. Imported lassa fever in Germany: molecular characterization of a new lassa virus strain. Emerg Infect Dis 2000, 6, 466–476. [Google Scholar] [CrossRef]
- Mahdy, M.S.; Chiang, W.; McLaughlin, B.; Derksen, K.; Truxton, B.H.; Neg, K. Lassa fever: the first confirmed case imported into Canada. Can Dis Wkly Rep 1989, 15, 193–198. [Google Scholar]
- Hirabayashi, Y.; Oka, S.; Goto, H.; Shimada, K.; Kurata, T.; Fisher-Hoch, S.P.; McCormick, J.B. [The first imported case of Lassa fever in Japan]. Nihon Rinsho 1989, 47, 71–75. [Google Scholar]
- Hirabayashi, Y.; Oka, S.; Goto, H.; Shimada, K.; Kurata, T.; Fisher-Hoch, S.P.; McCormick, J.B. An imported case of Lassa fever with late appearance of polyserositis. J Infect Dis 1988, 158, 872–875. [Google Scholar] [CrossRef]
- Kitching, A.; Addiman, S.; Cathcart, S.; Bischop, L.; Krahe, D.; Nicholas, M.; Coakley, J.; Lloyd, G.; Brooks, T.; Morgan, D.; Turbitt, D. A fatal case of Lassa fever in London, January 2009. Euro Surveill 2009, 14. [Google Scholar]
- Maiztegui, J.I.; McKee, K.T., Jr.; Barrera Oro, J.G.; Harrison, L.H.; Gibbs, P.H.; Feuillade, M.R.; Enria, D.A.; Briggiler, A.M.; Levis, S.C.; Ambrosio, A.M.; Halsey, N.A.; Peters, C.J. Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever. AHF Study Group. J Infect Dis 1998, 177, 277–283. [Google Scholar]
- Geisbert, T.W.; Jones, S.; Fritz, E.A.; Shurtleff, A.C.; Geisbert, J.B.; Liebscher, R.; Grolla, A.; Stroher, U.; Fernando, L.; Daddario, K.M.; Guttieri, M.C.; Mothe, B.R.; Larsen, T.; Hensley, L.E.; Jahrling, P.B.; Feldmann, H. Development of a new vaccine for the prevention of Lassa fever. PLoS Med 2005, 2, e183. [Google Scholar]
- Fisher-Hoch, S.P.; McCormick, J.B. Lassa fever vaccine. Expert Rev Vaccines 2004, 3, 189–197. [Google Scholar] [CrossRef]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliott, L.H.; Belmont-Williams, R. Lassa fever. Effective therapy with ribavirin. N Engl J Med 1986, 314, 20–26. [Google Scholar]
- Clegg, J.C.; Wilson, S.M.; Oram, J.D. Nucleotide sequence of the S RNA of Lassa virus (Nigerian strain) and comparative analysis of arenavirus gene products. Virus Res 1991, 18, 151–164. [Google Scholar] [CrossRef]
- Riviere, Y.; Ahmed, R.; Southern, P.J.; Buchmeier, M.J.; Dutko, F.J.; Oldstone, M.B. The S RNA segment of lymphocytic choriomeningitis virus codes for the nucleoprotein and glycoproteins 1 and 2. J Virol 1985, 53, 966–968. [Google Scholar]
- Auperin, D.D.; Galinski, M.; Bishop, D.H. The sequences of the N protein gene and intergenic region of the S RNA of pichinde arenavirus. Virology 1984, 134, 208–219. [Google Scholar] [CrossRef]
- Auperin, D.D.; Sasso, D.R.; McCormick, J.B. Nucleotide sequence of the glycoprotein gene and intergenic region of the Lassa virus S genome RNA. Virology 1986, 154, 155–167. [Google Scholar] [CrossRef]
- Djavani, M.; Lukashevich, I.S.; Sanchez, A.; Nichol, S.T.; Salvato, M.S. Completion of the Lassa fever virus sequence and identification of a RING finger open reading frame at the L RNA 5' End. Virology 1997, 235, 414–418. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Djavani, M.; Shapiro, K.; Sanchez, A.; Ravkov, E.; Nichol, S.T.; Salvato, M.S. The Lassa fever virus L gene: nucleotide sequence, comparison, and precipitation of a predicted 250 kDa protein with monospecific antiserum. J Gen Virol 1997, 78 ( Pt 3), 547–551. [Google Scholar]
- Salvato, M.S.; Shimomaye, E.M. The completed sequence of lymphocytic choriomeningitis virus reveals a unique RNA structure and a gene for a zinc finger protein. Virology 1989, 173, 1–10. [Google Scholar] [CrossRef]
- Singh, M.K.; Fuller-Pace, F.V.; Buchmeier, M.J.; Southern, P.J. Analysis of the genomic L RNA segment from lymphocytic choriomeningitis virus. Virology 1987, 161, 448–456. [Google Scholar] [CrossRef]
- Schlie, K.; Maisa, A.; Lennartz, F.; Stroher, U.; Garten, W.; Strecker, T. Characterization of Lassa virus glycoprotein oligomerization and influence of cholesterol on virus replication. J Virol 2010, 84, 983–992. [Google Scholar] [CrossRef]
- Neuman, B.W.; Adair, B.D.; Burns, J.W.; Milligan, R.A.; Buchmeier, M.J.; Yeager, M. Complementarity in the supramolecular design of arenaviruses and retroviruses revealed by electron cryomicroscopy and image analysis. J Virol 2005, 79, 3822–3830. [Google Scholar]
- Strecker, T.; Eichler, R.; Meulen, J.; Weissenhorn, W.; Klenk, H.D.; Garten, W.; Lenz, O. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles. J Virol 2003, 77, 10700–10705. [Google Scholar]
- Salvato, M.S.; Schweighofer, K.J.; Burns, J.; Shimomaye, E.M. Biochemical and immunological evidence that the 11 kDa zinc-binding protein of lymphocytic choriomeningitis virus is a structural component of the virus. Virus Res 1992, 22, 185–198. [Google Scholar] [CrossRef]
- Eichler, R.; Strecker, T.; Kolesnikova, L.; ter Meulen, J.; Weissenhorn, W.; Becker, S.; Klenk, H.D.; Garten, W.; Lenz, O. Characterization of the Lassa virus matrix protein Z: electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP). Virus Res 2004, 100, 249–255. [Google Scholar] [CrossRef]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar]
- Spiropoulou, C.F.; Kunz, S.; Rollin, P.E.; Campbell, K.P.; Oldstone, M.B. New World arenavirus clade C, but not clade A and B viruses, utilizes alpha-dystroglycan as its major receptor. J Virol 2002, 76, 5140–5146. [Google Scholar] [CrossRef]
- Shimojima, M.; Stroher, U.; Ebihara, H.; Feldmann, H.; Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J Virol 2012, 86, 2067–2078. [Google Scholar] [CrossRef]
- Shimojima, M.; Kawaoka, Y. Cell Surface Molecules Involved in Infection Mediated by Lymphocytic Choriomeningitis Virus Glycoprotein. J Vet Med Sci 2012.
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; Farzan, M.; Choe, H. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar]
- Martinez, M.G.; Cordo, S.M.; Candurra, N.A. Characterization of Junin arenavirus cell entry. J Gen Virol 2007, 88, 1776–1784. [Google Scholar] [CrossRef]
- Borrow, P.; Oldstone, M.B. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology 1994, 198, 1–9. [Google Scholar] [CrossRef]
- Quirin, K.; Eschli, B.; Scheu, I.; Poort, L.; Kartenbeck, J.; Helenius, A. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology 2008, 378, 21–33. [Google Scholar] [CrossRef]
- Kunz, S. Receptor binding and cell entry of Old World arenaviruses reveal novel aspects of virus-host interaction. Virology 2009, 387, 245–249. [Google Scholar] [CrossRef]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.Y.; Kunz, S. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog 2011, 7, e1002232. [Google Scholar] [CrossRef]
- Klewitz, C.; Klenk, H.D.; ter Meulen, J. Amino acids from both N-terminal hydrophobic regions of the Lassa virus envelope glycoprotein GP-2 are critical for pH-dependent membrane fusion and infectivity. J Gen Virol 2007, 88, 2320–2328. [Google Scholar] [CrossRef]
- Meyer, B.J.; Southern, P.J. Concurrent sequence analysis of 5' and 3' RNA termini by intramolecular circularization reveals 5' nontemplated bases and 3' terminal heterogeneity for lymphocytic choriomeningitis virus mRNAs. J Virol 1993, 67, 2621–2627. [Google Scholar]
- Lee, K.J.; Novella, I.S.; Teng, M.N.; Oldstone, M.B.; de La Torre, J.C. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J Virol 2000, 74, 3470–3477. [Google Scholar]
- Lopez, N.; Jacamo, R.; Franze-Fernandez, M.T. Transcription and RNA replication of tacaribe virus genome and antigenome analogs require N and L proteins: Z protein is an inhibitor of these processes. J Virol 2001, 75, 12241–12251. [Google Scholar] [CrossRef]
- Farazi, T.A.; Waksman, G.; Gordon, J.I. The biology and enzymology of protein N-myristoylation. J Biol Chem 2001, 276, 39501–39504. [Google Scholar] [CrossRef]
- Perez, M.; Greenwald, D.L.; de la Torre, J.C. Myristoylation of the RING finger Z protein is essential for arenavirus budding. J Virol 2004, 78, 11443–11448. [Google Scholar] [CrossRef]
- Strecker, T.; Maisa, A.; Daffis, S.; Eichler, R.; Lenz, O.; Garten, W. The role of myristoylation in the membrane association of the Lassa virus matrix protein Z. Virol J 2006, 3, 93. [Google Scholar] [CrossRef]
- Capul, A.A.; Perez, M.; Burke, E.; Kunz, S.; Buchmeier, M.J.; de la Torre, J.C. Arenavirus Z-glycoprotein association requires Z myristoylation but not functional RING or late domains. J Virol 2007, 81, 9451–9460. [Google Scholar]
- Loureiro, M.E.; Wilda, M.; Levingston Macleod, J.M.; D'Antuono, A.; Foscaldi, S.; Marino Buslje, C.; Lopez, N. Molecular determinants of arenavirus Z protein homo-oligomerization and L polymerase binding. J Virol 2011, 85, 12304–12314. [Google Scholar] [CrossRef]
- Cordo, S.M.; Candurra, N.A.; Damonte, E.B. Myristic acid analogs are inhibitors of Junin virus replication. Microbes Infect 1999, 1, 609–614. [Google Scholar] [CrossRef]
- Silverman, L.; Resh, M.D. Lysine residues form an integral component of a novel NH2-terminal membrane targeting motif for myristylated pp60v-src. J Cell Biol 1992, 119, 415–425. [Google Scholar] [CrossRef]
- Volpon, L.; Osborne, M.J.; Capul, A.A.; de la Torre, J.C.; Borden, K.L. Structural characterization of the Z RING-eIF4E complex reveals a distinct mode of control for eIF4E. Proc Natl Acad Sci U S A 2010, 107, 5441–5446. [Google Scholar]
- Kentsis, A.; Gordon, R.E.; Borden, K.L. Self-assembly properties of a model RING domain. Proc Natl Acad Sci U S A 2002, 99, 667–672. [Google Scholar] [CrossRef]
- Kentsis, A.; Gordon, R.E.; Borden, K.L. Control of biochemical reactions through supramolecular RING domain self-assembly. Proc Natl Acad Sci U S A 2002, 99, 15404–15409. [Google Scholar] [CrossRef]
- Kentsis, A.; Dwyer, E.C.; Perez, J.M.; Sharma, M.; Chen, A.; Pan, Z.Q.; Borden, K.L. The RING domains of the promyelocytic leukemia protein PML and the arenaviral protein Z repress translation by directly inhibiting translation initiation factor eIF4E. J Mol Biol 2001, 312, 609–623. [Google Scholar] [CrossRef]
- Freed, E.O. Viral late domains. J Virol 2002, 76, 4679–4687. [Google Scholar] [CrossRef]
- May, E.R.; Armen, R.S.; Mannan, A.M.; Brooks, C.L., 3rd. The flexible C-terminal arm of the Lassa arenavirus Z-protein mediates interactions with multiple binding partners. Proteins 2010, 78, 2251–2264. [Google Scholar] [CrossRef]
- Garcin, D.; Rochat, S.; Kolakofsky, D. The Tacaribe arenavirus small zinc finger protein is required for both mRNA synthesis and genome replication. J Virol 1993, 67, 807–812. [Google Scholar]
- Cornu, T.I.; de la Torre, J.C. RING finger Z protein of lymphocytic choriomeningitis virus (LCMV) inhibits transcription and RNA replication of an LCMV S-segment minigenome. J Virol 2001, 75, 9415–9426. [Google Scholar] [CrossRef]
- Cornu, T.I.; Feldmann, H.; de la Torre, J.C. Cells expressing the RING finger Z protein are resistant to arenavirus infection. J Virol 2004, 78, 2979–2983. [Google Scholar] [CrossRef]
- Jacamo, R.; Lopez, N.; Wilda, M.; Franze-Fernandez, M.T. Tacaribe virus Z protein interacts with the L polymerase protein to inhibit viral RNA synthesis. J Virol 2003, 77, 10383–10393. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Schenk, A.D.; Rahmeh, A.A.; Radoshitzky, S.R.; Bavari, S.; Walz, T.; Whelan, S.P. Assembly of a functional Machupo virus polymerase complex. Proc Natl Acad Sci U S A 2010, 107, 20069–20074. [Google Scholar]
- Wilda, M.; Lopez, N.; Casabona, J.C.; Franze-Fernandez, M.T. Mapping of the tacaribe arenavirus Z-protein binding sites on the L protein identified both amino acids within the putative polymerase domain and a region at the N terminus of L that are critically involved in binding. J Virol 2008, 82, 11454–11460. [Google Scholar] [CrossRef]
- Brunotte, L.; Lelke, M.; Hass, M.; Kleinsteuber, K.; Becker-Ziaja, B.; Gunther, S. Domain structure of Lassa virus L protein. J Virol 2011, 85, 324–333. [Google Scholar] [CrossRef]
- Capul, A.A.; de la Torre, J.C.; Buchmeier, M.J. Conserved residues in Lassa fever virus Z protein modulate viral infectivity at the level of the ribonucleoprotein. J Virol 2011, 85, 3172–3178. [Google Scholar] [CrossRef]
- Cornu, T.I.; de la Torre, J.C. Characterization of the arenavirus RING finger Z protein regions required for Z-mediated inhibition of viral RNA synthesis. J Virol 2002, 76, 6678–6688. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Whelan, S.P. Arenavirus Z protein controls viral RNA synthesis by locking a polymerase-promoter complex. Proc Natl Acad Sci U S A 2011, 108, 19743–19748. [Google Scholar] [CrossRef]
- Lenard, J. Negative-strand virus M and retrovirus MA proteins: all in a family? Virology 1996, 216, 289–298. [Google Scholar] [CrossRef]
- Mebatsion, T.; Weiland, F.; Conzelmann, K.K. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J Virol 1999, 73, 242–250. [Google Scholar]
- Newcomb, W.W.; Brown, J.C. Role of the vesicular stomatitis virus matrix protein in maintaining the viral nucleocapsid in the condensed form found in native virions. J Virol 1981, 39, 295–299. [Google Scholar]
- Eichler, R.; Lenz, O.; Strecker, T.; Garten, W. Signal peptide of Lassa virus glycoprotein GP-C exhibits an unusual length. FEBS Lett 2003, 538, 203–206. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; Oldstone, M.B. Protein structure of lymphocytic choriomeningitis virus: evidence for a cell-associated precursor of the virion glycopeptides. Virology 1979, 99, 111–120. [Google Scholar] [CrossRef]
- Beyer, W.R.; Popplau, D.; Garten, W.; von Laer, D.; Lenz, O. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. J Virol 2003, 77, 2866–2872. [Google Scholar] [CrossRef]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc Natl Acad Sci U S A 2001, 98, 12701–12705. [Google Scholar] [CrossRef]
- Rojek, J.M.; Lee, A.M.; Nguyen, N.; Spiropoulou, C.F.; Kunz, S. Site 1 protease is required for proteolytic processing of the glycoproteins of the South American hemorrhagic fever viruses Junin, Machupo, and Guanarito. J Virol 2008, 82, 6045–6051. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; Southern, P.J.; Parekh, B.S.; Wooddell, M.K.; Oldstone, M.B. Site-specific antibodies define a cleavage site conserved among arenavirus GP-C glycoproteins. J Virol 1987, 61, 982–985. [Google Scholar]
- Eichler, R.; Lenz, O.; Strecker, T.; Eickmann, M.; Klenk, H.D.; Garten, W. Identification of Lassa virus glycoprotein signal peptide as a trans-acting maturation factor. EMBO Rep 2003, 4, 1084–1088. [Google Scholar] [CrossRef]
- York, J.; Nunberg, J.H. A novel zinc-binding domain is essential for formation of the functional Junin virus envelope glycoprotein complex. J Virol 2007, 81, 13385–13391. [Google Scholar] [CrossRef]
- Briknarova, K.; Thomas, C.J.; York, J.; Nunberg, J.H. Structure of a zinc-binding domain in the Junin virus envelope glycoprotein. J Biol Chem 2011, 286, 1528–1536. [Google Scholar]
- Eichler, R.; Lenz, O.; Strecker, T.; Eickmann, M.; Klenk, H.D.; Garten, W. Lassa virus glycoprotein signal peptide displays a novel topology with an extended endoplasmic reticulum luminal region. J Biol Chem 2004, 279, 12293–12299. [Google Scholar]
- Agnihothram, S.S.; York, J.; Trahey, M.; Nunberg, J.H. Bitopic membrane topology of the stable signal peptide in the tripartite Junin virus GP-C envelope glycoprotein complex. J Virol 2007, 81, 4331–4337. [Google Scholar] [CrossRef]
- Schlie, K.; Maisa, A.; Freiberg, F.; Groseth, A.; Strecker, T.; Garten, W. Viral protein determinants of Lassa virus entry and release from polarized epithelial cells. J Virol 2010, 84, 3178–3188. [Google Scholar] [CrossRef]
- Tanaka, T.; Ames, J.B.; Harvey, T.S.; Stryer, L.; Ikura, M. Sequestration of the membrane-targeting myristoyl group of recoverin in the calcium-free state. Nature 1995, 376, 444–447. [Google Scholar]
- Agnihothram, S.S.; Dancho, B.; Grant, K.W.; Grimes, M.L.; Lyles, D.S.; Nunberg, J.H. Assembly of arenavirus envelope glycoprotein GPC in detergent-soluble membrane microdomains. J Virol 2009, 83, 9890–9900. [Google Scholar] [CrossRef]
- Mittler, E.; Kolesnikova, L.; Strecker, T.; Garten, W.; Becker, S. Role of the transmembrane domain of marburg virus surface protein GP in assembly of the viral envelope. J Virol 2007, 81, 3942–3948. [Google Scholar] [CrossRef]
- Kolesnikova, L.; Strecker, T.; Morita, E.; Zielecki, F.; Mittler, E.; Crump, C.; Becker, S. Vacuolar protein sorting pathway contributes to the release of Marburg virus. J Virol 2009, 83, 2327–2337. [Google Scholar] [CrossRef]
- Baird, N.L.; York, J.; Nunberg, J.H. Arenavirus infection induces discrete cytosolic structures for RNA replication. J Virol 2012.
- Murphy, F.A.; Whitfield, S.G. Morphology and morphogenesis of arenaviruses. Bull World Health Organ 1975, 52, 409–419. [Google Scholar]
- Noda, T.; Ebihara, H.; Muramoto, Y.; Fujii, K.; Takada, A.; Sagara, H.; Kim, J.H.; Kida, H.; Feldmann, H.; Kawaoka, Y. Assembly and budding of Ebolavirus. PLoS Pathog 2006, 2, e99. [Google Scholar] [CrossRef]
- Shtanko, O.; Imai, M.; Goto, H.; Lukashevich, I.S.; Neumann, G.; Watanabe, T.; Kawaoka, Y. A role for the C terminus of Mopeia virus nucleoprotein in its incorporation into Z protein-induced virus-like particles. J Virol 2010, 84, 5415–5422. [Google Scholar] [CrossRef]
- Ortiz-Riano, E.; Cheng, B.Y.; de la Torre, J.C.; Martinez-Sobrido, L. The C-terminal region of lymphocytic choriomeningitis virus nucleoprotein contains distinct and segregable functional domains involved in NP-Z interaction and counteraction of the type I interferon response. J Virol 2011, 85, 13038–13048. [Google Scholar] [CrossRef]
- Levingston Macleod, J.M.; D'Antuono, A.; Loureiro, M.E.; Casabona, J.C.; Gomez, G.A.; Lopez, N. Identification of two functional domains within the arenavirus nucleoprotein. J Virol 2011, 85, 2012–2023. [Google Scholar]
- Groseth, A.; Wolff, S.; Strecker, T.; Hoenen, T.; Becker, S. Efficient budding of the tacaribe virus matrix protein z requires the nucleoprotein. J Virol 2010, 84, 3603–3611. [Google Scholar] [CrossRef]
- Casabona, J.C.; Levingston Macleod, J.M.; Loureiro, M.E.; Gomez, G.A.; Lopez, N. The RING domain and the L79 residue of Z protein are involved in both the rescue of nucleocapsids and the incorporation of glycoproteins into infectious chimeric arenavirus-like particles. J Virol 2009, 83, 7029–7039. [Google Scholar] [CrossRef]
- Brunotte, L.; Kerber, R.; Shang, W.; Hauer, F.; Hass, M.; Gabriel, M.; Lelke, M.; Busch, C.; Stark, H.; Svergun, D.I.; Betzel, C.; Perbandt, M.; Gunther, S. Structure of the Lassa virus nucleoprotein revealed by X-ray crystallography, small-angle X-ray scattering, and electron microscopy. J Biol Chem 2011, 286, 38748–38756. [Google Scholar]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar]
- Hastie, K.M.; Liu, T.; Li, S.; King, L.B.; Ngo, N.; Zandonatti, M.A.; Woods, V.L., Jr.; de la Torre, J.C.; Saphire, E.O. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc Natl Acad Sci U S A 2011, 108, 19365–19370. [Google Scholar]
- Ortiz-Riano, E.; Cheng, B.Y.; de la Torre, J.C.; Martinez-Sobrido, L. Self-association of lymphocytic choriomeningitis virus nucleoprotein is mediated by its N-terminal region and is not required for its anti-interferon function. J Virol 2012, 86, 3307–3317. [Google Scholar] [CrossRef]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3' to 5' exonuclease activity essential for immune suppression. Proc Natl Acad Sci U S A 2011, 108, 2396–2401. [Google Scholar]
- Shtanko, O.; Watanabe, S.; Jasenosky, L.D.; Watanabe, T.; Kawaoka, Y. ALIX/AIP1 is required for NP incorporation into Mopeia virus Z-induced virus-like particles. J Virol 2011, 85, 3631–3641. [Google Scholar] [CrossRef]
- Garoff, H.; Hewson, R.; Opstelten, D.J. Virus maturation by budding. Microbiol Mol Biol Rev 1998, 62, 1171–1190. [Google Scholar]
- Neumann, P.; Lieber, D.; Meyer, S.; Dautel, P.; Kerth, A.; Kraus, I.; Garten, W.; Stubbs, M.T. Crystal structure of the Borna disease virus matrix protein (BDV-M) reveals ssRNA binding properties. Proc Natl Acad Sci U S A 2009, 106, 3710–3715. [Google Scholar]
- Alfadhli, A.; McNett, H.; Tsagli, S.; Bachinger, H.P.; Peyton, D.H.; Barklis, E. HIV-1 matrix protein binding to RNA. J Mol Biol 2011, 410, 653–666. [Google Scholar] [CrossRef]
- Saurin, A.J.; Borden, K.L.; Boddy, M.N.; Freemont, P.S. Does this have a familiar RING? Trends Biochem Sci 1996, 21, 208–214. [Google Scholar]
- Urata, S.; Noda, T.; Kawaoka, Y.; Yokosawa, H.; Yasuda, J. Cellular factors required for Lassa virus budding. J Virol 2006, 80, 4191–4195. [Google Scholar] [CrossRef]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc Natl Acad Sci U S A 2003, 100, 12978–12983. [Google Scholar] [CrossRef]
- Bieniasz, P.D. Late budding domains and host proteins in enveloped virus release. Virology 2006, 344, 55–63. [Google Scholar] [CrossRef]
- Hurley, J.H. ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol 2008, 20, 4–11. [Google Scholar] [CrossRef]
- Hurley, J.H. The ESCRT complexes. Crit Rev Biochem Mol Biol 2010, 45, 463–487. [Google Scholar] [CrossRef]
- Wollert, T.; Wunder, C.; Lippincott-Schwartz, J.; Hurley, J.H. Membrane scission by the ESCRT-III complex. Nature 2009, 458, 172–177. [Google Scholar]
- Babst, M. A protein's final ESCRT. Traffic 2005, 6, 2–9. [Google Scholar] [CrossRef]
- Gruenberg, J.; Stenmark, H. The biogenesis of multivesicular endosomes. Nature reviews 2004, 5, 317–323. [Google Scholar]
- Hurley, J.H.; Emr, S.D. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct 2006, 35, 277–298. [Google Scholar] [CrossRef]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; Myszka, D.G.; Sundquist, W.I. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc Natl Acad Sci U S A 2001, 98, 7724–7729. [Google Scholar]
- Martin-Serrano, J.; Yarovoy, A.; Perez-Caballero, D.; Bieniasz, P.D. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci U S A 2003, 100, 12414–12419. [Google Scholar]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Gottlinger, H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- von Schwedler, U.K.; Stuchell, M.; Muller, B.; Ward, D.M.; Chung, H.Y.; Morita, E.; Wang, H.E.; Davis, T.; He, G.P.; Cimbora, D.M.; Scott, A.; Krausslich, H.G.; Kaplan, J.; Morham, S.G.; Sundquist, W.I. The protein network of HIV budding. Cell 2003, 114, 701–713. [Google Scholar] [CrossRef]
- Chen, H.I.; Sudol, M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc Natl Acad Sci U S A 1995, 92, 7819–7823. [Google Scholar] [CrossRef]
- Harty, R.N.; Brown, M.E.; Wang, G.; Huibregtse, J.; Hayes, F.P. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc Natl Acad Sci U S A 2000, 97, 13871–13876. [Google Scholar] [CrossRef]
- Ingham, R.J.; Gish, G.; Pawson, T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 2004, 23, 1972–1984. [Google Scholar] [CrossRef]
- Kikonyogo, A.; Bouamr, F.; Vana, M.L.; Xiang, Y.; Aiyar, A.; Carter, C.; Leis, J. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc Natl Acad Sci U S A 2001, 98, 11199–11204. [Google Scholar]
- Vana, M.L.; Tang, Y.; Chen, A.; Medina, G.; Carter, C.; Leis, J. Role of Nedd4 and ubiquitination of Rous sarcoma virus Gag in budding of virus-like particles from cells. J Virol 2004, 78, 13943–13953. [Google Scholar] [CrossRef]
- Blot, V.; Perugi, F.; Gay, B.; Prevost, M.C.; Briant, L.; Tangy, F.; Abriel, H.; Staub, O.; Dokhelar, M.C.; Pique, C. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J Cell Sci 2004, 117, 2357–2367. [Google Scholar] [CrossRef]
- Sakurai, A.; Yasuda, J.; Takano, H.; Tanaka, Y.; Hatakeyama, M.; Shida, H. Regulation of human T-cell leukemia virus type 1 (HTLV-1) budding by ubiquitin ligase Nedd4. Microbes Infect 2004, 6, 150–156. [Google Scholar] [CrossRef]
- Urata, S.; Noda, T.; Kawaoka, Y.; Morikawa, S.; Yokosawa, H.; Yasuda, J. Interaction of Tsg101 with Marburg virus VP40 depends on the PPPY motif, but not the PT/SAP motif as in the case of Ebola virus, and Tsg101 plays a critical role in the budding of Marburg virus-like particles induced by VP40, NP, and GP. J Virol 2007, 81, 4895–4899. [Google Scholar] [CrossRef]
- Bouamr, F.; Melillo, J.A.; Wang, M.Q.; Nagashima, K.; de Los Santos, M.; Rein, A.; Goff, S.P. PPPYVEPTAP motif is the late domain of human T-cell leukemia virus type 1 Gag and mediates its functional interaction with cellular proteins Nedd4 and Tsg101 [corrected]. J Virol 2003, 77, 11882–11895. [Google Scholar]
- Shields, S.B.; Piper, R.C. How ubiquitin functions with ESCRTs. Traffic 2011, 12, 1306–1317. [Google Scholar] [CrossRef]
- Martin-Serrano, J. The role of ubiquitin in retroviral egress. Traffic 2007, 8, 1297–1303. [Google Scholar] [CrossRef]
- Kamynina, E.; Tauxe, C.; Staub, O. Distinct characteristics of two human Nedd4 proteins with respect to epithelial Na(+) channel regulation. Am J Physiol Renal Physiol 2001, 281, F469–477. [Google Scholar]
- Pornillos, O.; Alam, S.L.; Rich, R.L.; Myszka, D.G.; Davis, D.R.; Sundquist, W.I. Structure and functional interactions of the Tsg101 UEV domain. Embo J 2002, 21, 2397–2406. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Schubert, H.L.; Kelly, B.N.; Hill, G.C.; Holton, J.M.; Hill, C.P. Ubiquitin recognition by the human TSG101 protein. Mol Cell 2004, 13, 783–789. [Google Scholar] [CrossRef]
- Urata, S.; Yasuda, J.; de la Torre, J.C. The z protein of the new world arenavirus tacaribe virus has bona fide budding activity that does not depend on known late domain motifs. J Virol 2009, 83, 12651–12655. [Google Scholar] [CrossRef]
- Obiang, L.; Raux, H.; Ouldali, M.; Blondel, D.; Gaudin, Y. Phenotypes of vesicular stomatitis virus mutants with mutations in the PSAP motif of the matrix protein. J Gen Virol 2012, 93, 857–865. [Google Scholar] [CrossRef]
- Reineke, E.L.; Kao, H.Y. PML: An emerging tumor suppressor and a target with therapeutic potential. Cancer Ther 2009, 7, 219–226. [Google Scholar]
- Borden, K.L.; Campbell Dwyer, E.J.; Salvato, M.S. An arenavirus RING (zinc-binding) protein binds the oncoprotein promyelocyte leukemia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J Virol 1998, 72, 758–766. [Google Scholar]
- Borden, K.L.; CampbellDwyer, E.J.; Salvato, M.S. The promyelocytic leukemia protein PML has a pro-apoptotic activity mediated through its RING domain. FEBS Lett 1997, 418, 30–34. [Google Scholar] [CrossRef]
- Bernardi, R.; Papa, A.; Pandolfi, P.P. Regulation of apoptosis by PML and the PML-NBs. Oncogene 2008, 27, 6299–6312. [Google Scholar] [CrossRef]
- Regad, T.; Chelbi-Alix, M.K. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 2001, 20, 7274–7286. [Google Scholar] [CrossRef]
- Regad, T.; Saib, A.; Lallemand-Breitenbach, V.; Pandolfi, P.P.; de The, H.; Chelbi-Alix, M.K. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J 2001, 20, 3495–3505. [Google Scholar] [CrossRef]
- Borden, K.L.; Campbelldwyer, E.J.; Carlile, G.W.; Djavani, M.; Salvato, M.S. Two RING finger proteins, the oncoprotein PML and the arenavirus Z protein, colocalize with the nuclear fraction of the ribosomal P proteins. J Virol 1998, 72, 3819–3826. [Google Scholar]
- Pestova, T.V.; Kolupaeva, V.G.; Lomakin, I.B.; Pilipenko, E.V.; Shatsky, I.N.; Agol, V.I.; Hellen, C.U. Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci U S A 2001, 98, 7029–7036. [Google Scholar]
- Campbell Dwyer, E.J.; Lai, H.; MacDonald, R.C.; Salvato, M.S.; Borden, K.L. The lymphocytic choriomeningitis virus RING protein Z associates with eukaryotic initiation factor 4E and selectively represses translation in a RING-dependent manner. J Virol 2000, 74, 3293–3300. [Google Scholar]
- Soufi, A.; Jayaraman, P.S. PRH/Hex: an oligomeric transcription factor and multifunctional regulator of cell fate. Biochem J 2008, 412, 399–413. [Google Scholar] [CrossRef]
- Djavani, M.; Topisirovic, I.; Zapata, J.C.; Sadowska, M.; Yang, Y.; Rodas, J.; Lukashevich, I.S.; Bogue, C.W.; Pauza, C.D.; Borden, K.L.; Salvato, M.S. The proline-rich homeodomain (PRH/HEX) protein is down-regulated in liver during infection with lymphocytic choriomeningitis virus. J Virol 2005, 79, 2461–2473. [Google Scholar]
- Lukashevich, I.S.; Rodas, J.D.; Tikhonov, II; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. LCMV-mediated hepatitis in rhesus macaques: WE but not ARM strain activates hepatocytes and induces liver regeneration. Arch Virol 2004, 149, 2319–2336. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Djavani, M.; Rodas, J.D.; Zapata, J.C.; Usborne, A.; Emerson, C.; Mitchen, J.; Jahrling, P.B.; Salvato, M.S. Hemorrhagic fever occurs after intravenous, but not after intragastric, inoculation of rhesus macaques with lymphocytic choriomeningitis virus. J Med Virol 2002, 67, 171–186. [Google Scholar] [CrossRef]
- Topcu, Z.; Mack, D.L.; Hromas, R.A.; Borden, K.L. The promyelocytic leukemia protein PML interacts with the proline-rich homeodomain protein PRH: a RING may link hematopoiesis and growth control. Oncogene 1999, 18, 7091–7100. [Google Scholar] [CrossRef]
- Zinkernagel, R.M.; Haenseler, E.; Leist, T.; Cerny, A.; Hengartner, H.; Althage, A. T cell-mediated hepatitis in mice infected with lymphocytic choriomeningitis virus. Liver cell destruction by H-2 class I-restricted virus-specific cytotoxic T cells as a physiological correlate of the 51Cr-release assay? J Exp Med 1986, 164, 1075–1092. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J Virol 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J Virol 2011, 85, 12093–12097. [Google Scholar] [CrossRef]
- Muller, S.; Geffers, R.; Gunther, S. Analysis of gene expression in Lassa virus-infected HuH-7 cells. J Gen Virol 2007, 88, 1568–1575. [Google Scholar] [CrossRef]
- Groseth, A.; Hoenen, T.; Weber, M.; Wolff, S.; Herwig, A.; Kaufmann, A.; Becker, S. Tacaribe virus but not junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl Trop Dis 2011, 5, e1137. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Zuniga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martinez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus Nucleoprotein Targets Interferon Regulatory Factor-Activating Kinase IKK{varepsilon}. J Virol 2012, 86, 7728–7738. [Google Scholar] [CrossRef]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J Virol 2010, 84, 1785–1791. [Google Scholar] [CrossRef]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J Virol 2010, 84, 9452–9462. [Google Scholar]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol 2009, 83, 1837–1844. [Google Scholar] [CrossRef]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of Lassa and Marburg virus production by tetherin. J Virol 2009, 83, 2382–2385. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Dong, L.; Chi, X.; Clester, J.C.; Retterer, C.; Spurgers, K.; Kuhn, J.H.; Sandwick, S.; Ruthel, G.; Kota, K.; Boltz, D.; Warren, T.; Kranzusch, P.J.; Whelan, S.P.; Bavari, S. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J Virol 2010, 84, 10569–10580. [Google Scholar]
- Johnson, K.M.; McCormick, J.B.; Webb, P.A.; Smith, E.S.; Elliott, L.H.; King, I.J. Clinical virology of Lassa fever in hospitalized patients. J Infect Dis 1987, 155, 456–464. [Google Scholar] [CrossRef]
- Sanchez, A.B.; Perez, M.; Cornu, T.; de la Torre, J.C. RNA interference-mediated virus clearance from cells both acutely and chronically infected with the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 2005, 79, 11071–11081. [Google Scholar] [CrossRef]
- Muller, S.; Gunther, S. Broad-spectrum antiviral activity of small interfering RNA targeting the conserved RNA termini of Lassa virus. Antimicrob Agents Chemother 2007, 51, 2215–2218. [Google Scholar] [CrossRef]
- Artuso, M.C.; Ellenberg, P.C.; Scolaro, L.A.; Damonte, E.B.; Garcia, C.C. Inhibition of Junin virus replication by small interfering RNAs. Antiviral Res 2009, 84, 31–37. [Google Scholar] [CrossRef]
- Garcia, C.C.; Candurra, N.A.; Damonte, E.B. Antiviral and virucidal activities against arenaviruses of zinc-finger active compounds. Antivir Chem Chemother 2000, 11, 231–237. [Google Scholar]
- Garcia, C.C.; Ellenberg, P.C.; Artuso, M.C.; Scolaro, L.A.; Damonte, E.B. Characterization of Junin virus particles inactivated by a zinc finger-reactive compound. Virus Res 2009, 143, 106–113. [Google Scholar] [CrossRef]
- Garcia, C.C.; Djavani, M.; Topisirovic, I.; Borden, K.L.; Salvato, M.S.; Damonte, E.B. Arenavirus Z protein as an antiviral target: virus inactivation and protein oligomerization by zinc finger-reactive compounds. J Gen Virol 2006, 87, 1217–1228. [Google Scholar] [CrossRef]
- Garcia, C.C.; Topisirovic, I.; Djavani, M.; Borden, K.L.; Damonte, E.B.; Salvato, M.S. An antiviral disulfide compound blocks interaction between arenavirus Z protein and cellular promyelocytic leukemia protein. Biochem Biophys Res Commun 2010, 393, 625–630. [Google Scholar] [CrossRef]
- Timmins, J.; Schoehn, G.; Ricard-Blum, S.; Scianimanico, S.; Vernet, T.; Ruigrok, R.W.; Weissenhorn, W. Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J Mol Biol 2003, 326, 493–502. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fehling, S.K.; Lennartz, F.; Strecker, T. Multifunctional Nature of the Arenavirus RING Finger Protein Z. Viruses 2012, 4, 2973-3011. https://doi.org/10.3390/v4112973
Fehling SK, Lennartz F, Strecker T. Multifunctional Nature of the Arenavirus RING Finger Protein Z. Viruses. 2012; 4(11):2973-3011. https://doi.org/10.3390/v4112973
Chicago/Turabian StyleFehling, Sarah Katharina, Frank Lennartz, and Thomas Strecker. 2012. "Multifunctional Nature of the Arenavirus RING Finger Protein Z" Viruses 4, no. 11: 2973-3011. https://doi.org/10.3390/v4112973
APA StyleFehling, S. K., Lennartz, F., & Strecker, T. (2012). Multifunctional Nature of the Arenavirus RING Finger Protein Z. Viruses, 4(11), 2973-3011. https://doi.org/10.3390/v4112973