Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
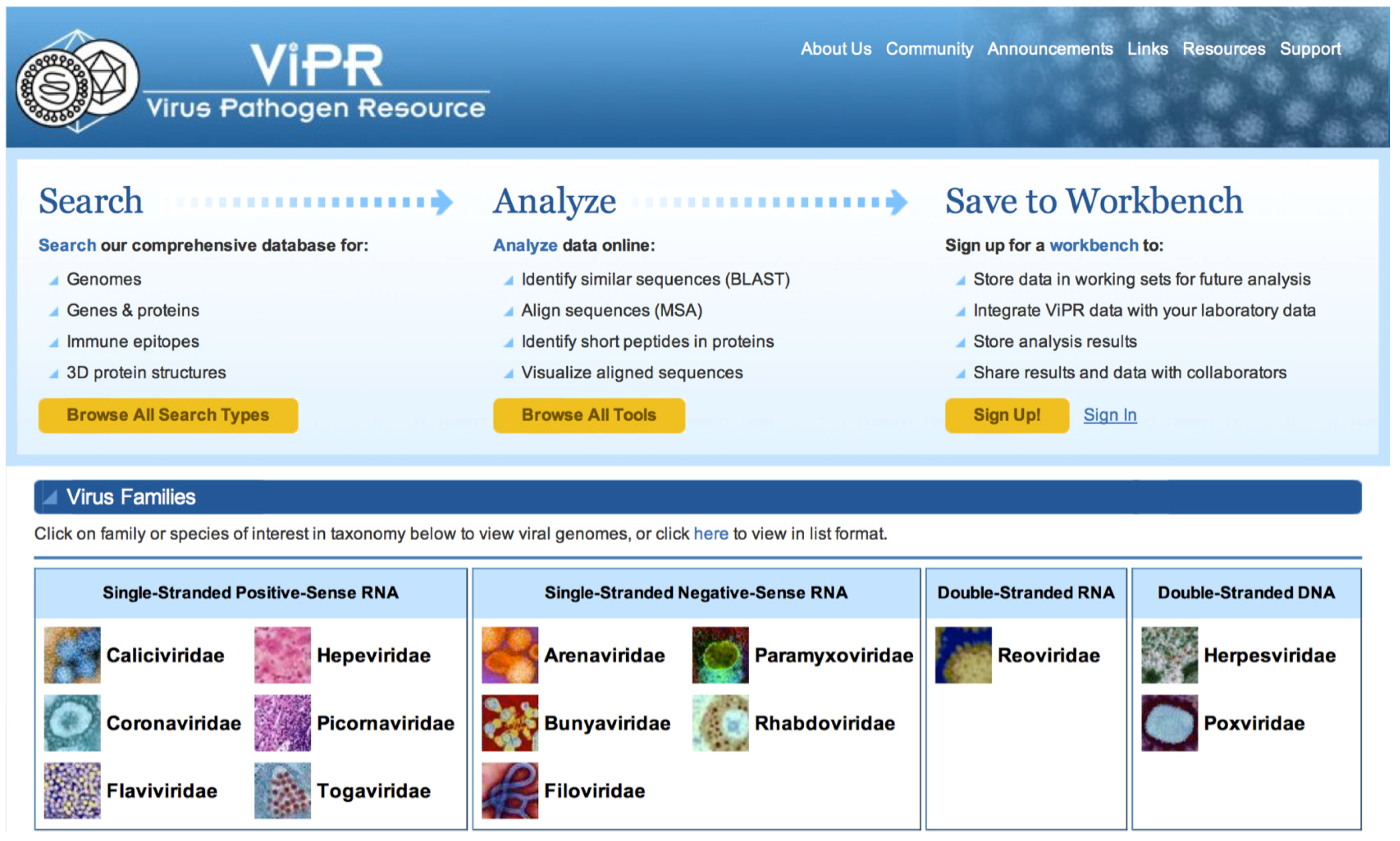
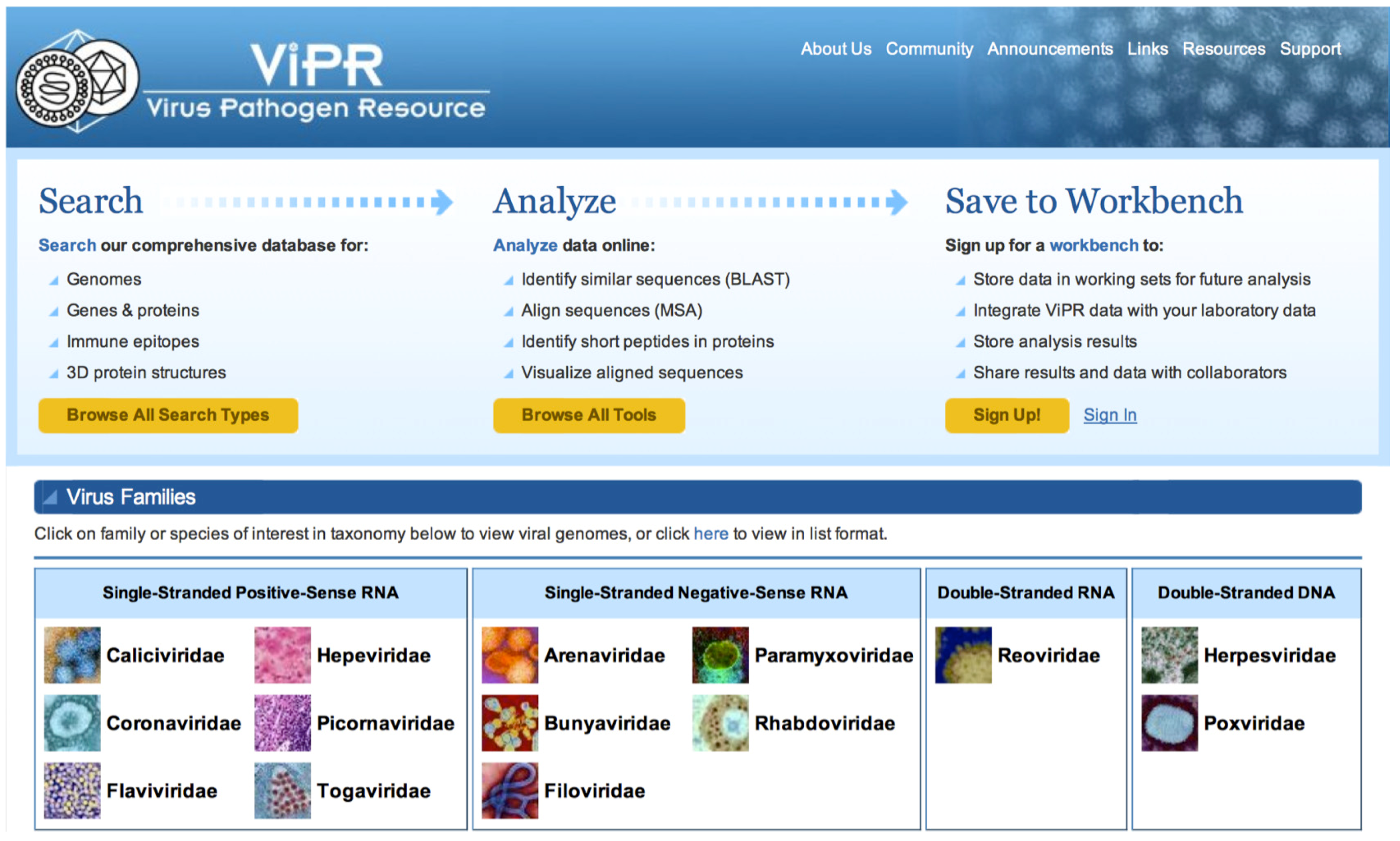
2.1. ViPR Overview
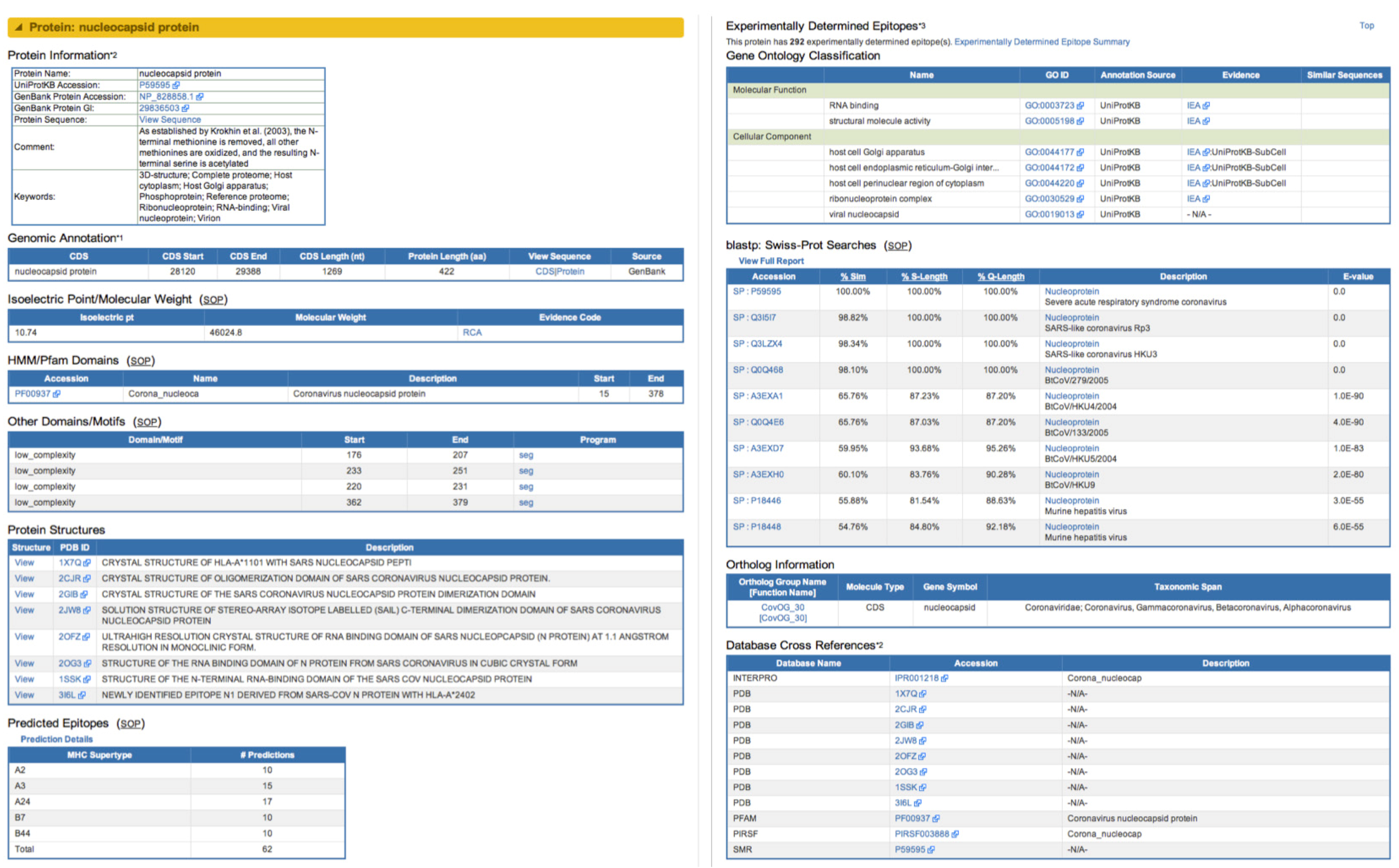
2.2. Data Contained in ViPR
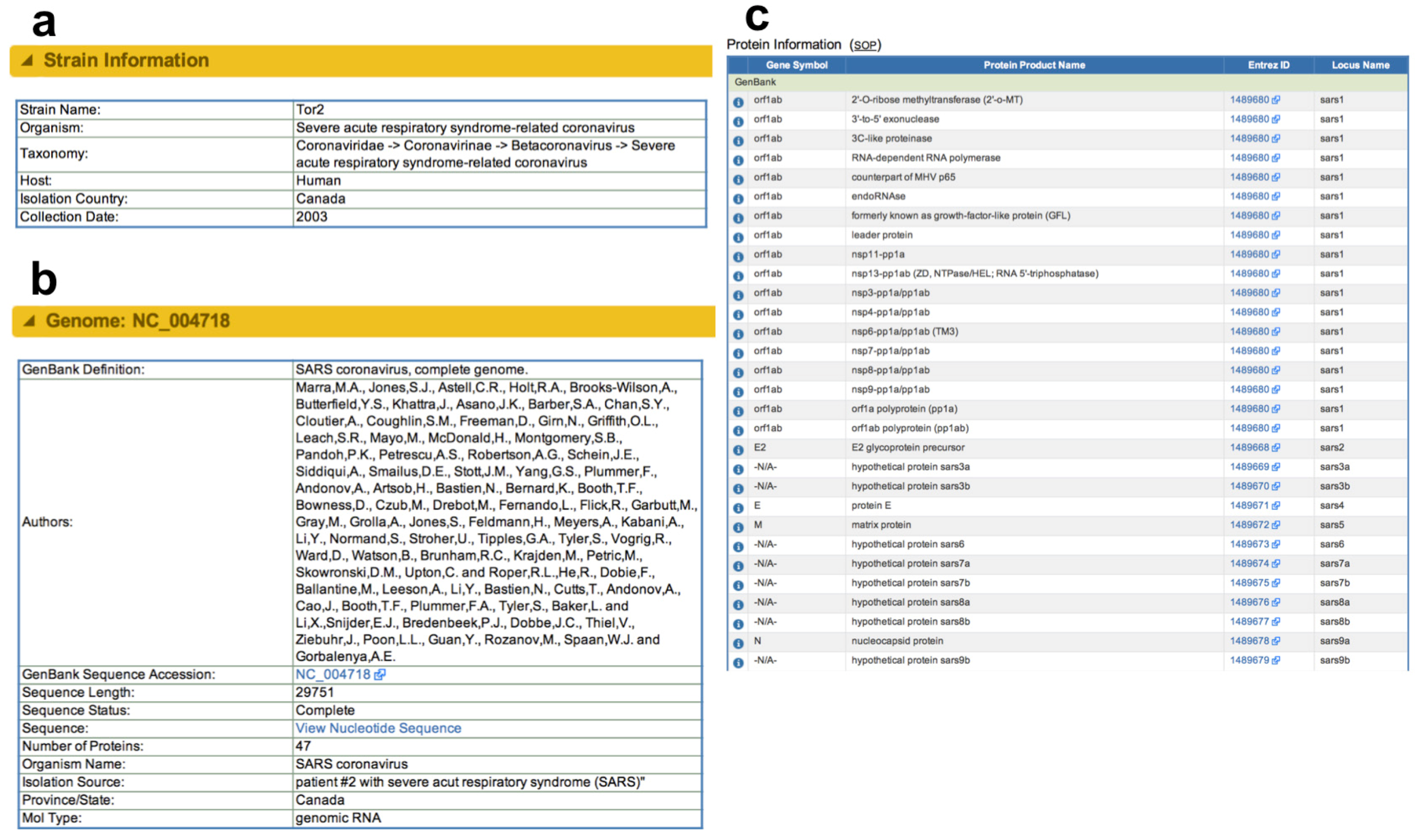
2.2.1. Data from Public Archives
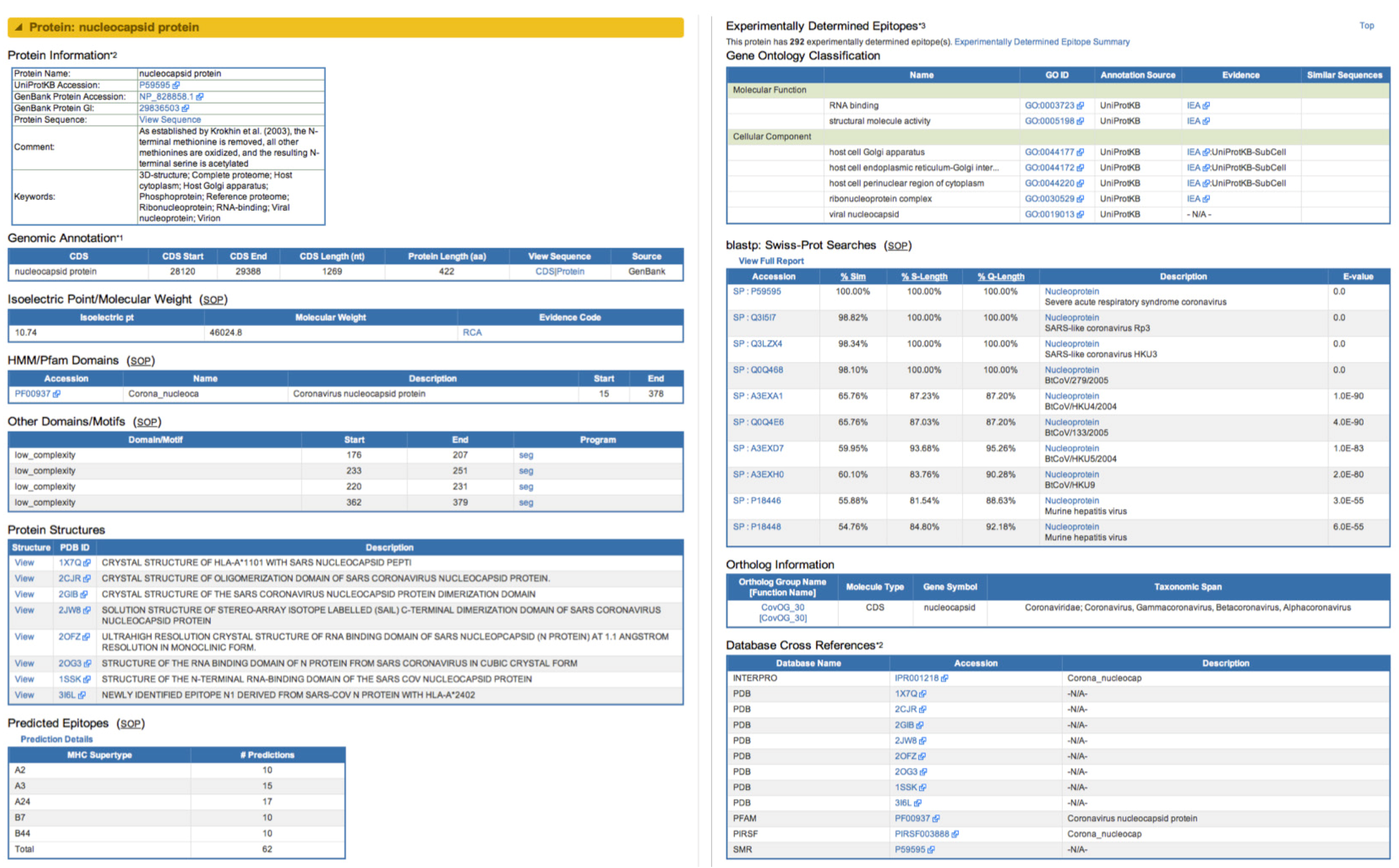
2.2.2. Novel Derived and Predicted Data
2.2.3. Data from Direct Submission
2.2.4. Search Capabilities in ViPR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Source | Data Type | Number of Records* |
|---|---|---|
| Imported from Public Archives | NCBI Virus Species | 384 |
| Virus Strains | 8,635 | |
| Genome Sequences | 10,729 | |
| Complete Genomes | 615 | |
| Unique UniProt Proteins | 11,171 | |
| Genes/Proteins | 20,531 | |
| Unique Protein Annotations | 2,040 | |
| Unique Gene Ontology Identifiers | 134 | |
| 3D Protein Structures | 197 | |
| Immune Epitopes | 2,253 | |
| ViPR-Generated | Pfam Domains | 8,232 |
| Other Domains/Motifs | 5,297 | |
| PubMed References | 3,969 | |
| Predicted Ortholog Groups | 50 | |
| Predicted Mature Peptides | 5,413 | |
| Predicted CD8 Epitopes | 5,116 | |
| Nearest BLASTp Hits | 18,977 | |
| Direct Submission | Host Factor Experiments | 8 |
| Biosets | 77 |

2.3. Analytical and Visualization Capabilities in ViPR
| Tool Name | Function / Purpose |
|---|---|
| MUSCLE | Calculate a multiple sequence alignment (MSA) using either nucleotide or amino acid sequences |
| ReadSeq | Convert between various MSA formats |
| JalView | Visualize and modify nucleotide or amino acid MSA |
| FastME, PhyML, RAxML | Infer phylogenetic trees for nucleotide or amino acid sequences using either similarity or maximum likelihood-based algorithms |
| modelCompare, ProtTest | Determine which evolutionary model to use when constructing maximum likelihood trees |
| Archaeopteryx | Visualize, manipulate and decorate phylogenetic trees |
| Jmol | 3D protein structure visualization / exploration |
| meta-CATS | Statistically compare groups of sequences to identify positions that significantly differ between them |
| BLAST | Identify similar nucleotide or amino acid sequences in a variety of custom ViPR databases |
| Sequence Variation Calculator | Compute the entropy present at each nucleotide or amino acid position at each position of user-defined groups of virus sequences |
| Short Peptide Identification Tool | Find short amino acid strings in target proteins using exact, fuzzy, or pattern matching |
| Genome Annotation Transfer Utility | Annotate a new genome sequence using an existing well-annotated reference genome |
| Primer3 | Design PCR primers to amplify specific virus sequence(s) based on the data within ViPR |
2.4. Workbench
2.5. Scientific Use Case
2.5.1. Searching for Relevant Sequence Records
2.5.2. Saving to the Workbench
2.5.3. Performing a Multiple Sequence Alignment
2.5.4. Viewing and Exploring a Phylogenetic Tree
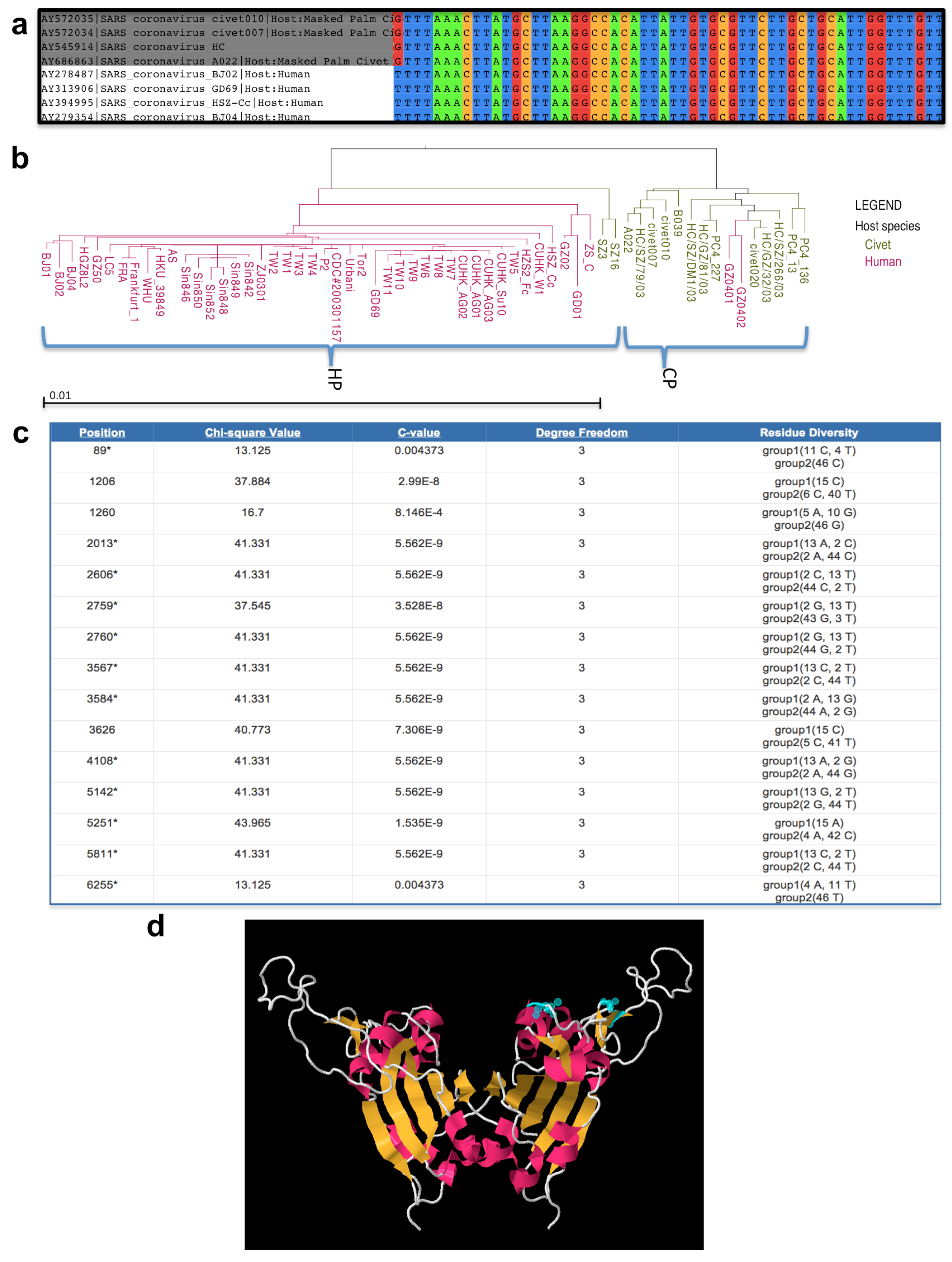
2.5.5. Metadata-driven Comparative Analysis Tool for Sequences
2.5.6. 3D Protein Structure Visualization and Exploration
2.5.7. Conclusions from Scientific Use Case

3. Conclusions
Acknowledgments
Conflict of Interest
Supplementary Files
References and Notes
- CDC, From the centers for disease control and prevention. Outbreak of west nile-like viral encephalitis--new york, 1999. MMWR Morb. Mortal. Wkly. Rep. 1999, 48, 845–849. [Google Scholar]
- Trifonov, V.; Khiabanian, H.; Rabadan, R. Geographic dependence, surveillance, and origins of the 2009 influenza a (h1n1) virus. N. Engl. J. Med. 2009, 361, 115–119. [Google Scholar] [CrossRef]
- Ha, D.Q.; Tien, N.T.; Huong, V.T.; Loan, H.T.; Thang, C.M. Dengue epidemic in southern vietnam, 1998. Emerg. Infect. Dis. 2000, 6, 422–425. [Google Scholar]
- CDC, From the centers for disease control and prevention. Severe acute respiratory syndrome--taiwan, 2003. Jama 2003, 289, 2930–2932. [Google Scholar] [CrossRef]
- Skowronski, D.M.; Astell, C.; Brunham, R.C.; Low, D.E.; Petric, M.; Roper, R.L.; Talbot, P.J.; Tam, T.; Babiuk, L. Severe acute respiratory syndrome (sars): A year in review. Ann. Rev. Med. 2005, 56, 357–381. [Google Scholar] [CrossRef]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nature Med. 2004, 10, S88–97. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef]
- Shi, Z.; Hu, Z. A review of studies on animal reservoirs of the sars coronavirus. Virus Res. 2008, 133, 74–87. [Google Scholar] [CrossRef]
- Song, H.D.; Tu, C.C.; Zhang, G.W.; Wang, S.Y.; Zheng, K.; Lei, L.C.; Chen, Q.X.; Gao, Y.W.; Zhou, H.Q.; Xiang, H.; et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA 2005, 102, 2430–2435. [Google Scholar]
- Marra, M.A.; Jones, S.J.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The genome sequence of the sars-associated coronavirus. Science 2003, 300, 1399–1404. [Google Scholar]
- Peiris, J.S.; Lai, S.T.; Poon, L.L.; Guan, Y.; Yam, L.Y.; Lim, W.; Nicholls, J.; Yee, W.K.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar]
- Fouchier, R.A.; Kuiken, T.; Schutten, M.; van Amerongen, G.; van Doornum, G.J.; van den Hoogen, B.G.; Peiris, M.; Lim, W.; Stohr, K.; Osterhaus, A.D. Aetiology: Koch's postulates fulfilled for sars virus. Nature 2003, 423, 240. [Google Scholar]
- Twu, S.J.; Chen, T.J.; Chen, C.J.; Olsen, S.J.; Lee, L.T.; Fisk, T.; Hsu, K.H.; Chang, S.C.; Chen, K.T.; Chiang, I.H.; et al. Control measures for severe acute respiratory syndrome (sars) in taiwan. Emerg. Infect. Dis. 2003, 9, 718–720. [Google Scholar] [CrossRef]
- Aderem, A.; Adkins, J.N.; Ansong, C.; Galagan, J.; Kaiser, S.; Korth, M.J.; Law, G.L.; McDermott, J.G.; Proll, S.C.; Rosenberger, C.; et al. A systems biology approach to infectious disease research: Innovating the pathogen-host research paradigm. mBio 2011, 2, e00325–00310. [Google Scholar]
- Greene, J.M.; Collins, F.; Lefkowitz, E.J.; Roos, D.; Scheuermann, R.H.; Sobral, B.; Stevens, R.; White, O.; Di Francesco, V. National institute of allergy and infectious diseases bioinformatics resource centers: New assets for pathogen informatics. Infect. Immun. 2007, 75, 3212–3219. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. Vipr: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–598. [Google Scholar] [CrossRef]
- Squires, R.B.; Noronha, J.; Hunt, V.; Garcia-Sastre, A.; Macken, C.; Baumgarth, N.; Suarez, D.; Pickett, B.E.; Zhang, Y.; Larsen, C.N.; et al. Influenza research database: An integrated bioinformatics resource for influenza research and surveillance. Influenza and other respiratory viruses 2012.
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. Genbank. Nucleic Acids Res. 2011, 39, D32–37. [Google Scholar] [CrossRef]
- Reorganizing the protein space at the universal protein resource (uniprot). Nucleic Acids Res. 2012, 40, D71–75. [CrossRef]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlic, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The rcsb protein data bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, D392–401. [Google Scholar]
- Kim, Y.; Ponomarenko, J.; Zhu, Z.; Tamang, D.; Wang, P.; Greenbaum, J.; Lundegaard, C.; Sette, A.; Lund, O.; Bourne, P.E.; et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012, 40, W525–530. [Google Scholar] [CrossRef]
- The gene ontology: Enhancements for 2011. Nucleic Acids Res. 2012, 40, D559–564. [CrossRef]
- Zdobnov, E.M.; Apweiler, R. Interproscan--an integration platform for the signature-recognition methods in interpro. Bioinform. 2001, 17, 847–848. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic t-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. Orthomcl: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. Ncbi reference sequences (refseq): Current status, new features and genome annotation policy. Nucleic Acids Res. 2012, 40, D130–135. [Google Scholar] [CrossRef]
- Noronha, J.M.; Liu, M.; Squires, R.B.; Pickett, B.E.; Hale, B.G.; Air, G.M.; Galloway, S.E.; Takimoto, T.; Schmolke, M.; Hunt, V.; et al. Influenza virus sequence feature variant type analysis: Evidence of a role for ns1 in influenza virus host range restriction. J. Virol. 2012, 86, 5857–5866. [Google Scholar] [CrossRef]
- Karp, D.R.; Marthandan, N.; Marsh, S.G.; Ahn, C.; Arnett, F.C.; Deluca, D.S.; Diehl, A.D.; Dunivin, R.; Eilbeck, K.; Feolo, M.; et al. Novel sequence feature variant type analysis of the hla genetic association in systemic sclerosis. Hum. Mol. Gen. 2010, 19, 707–719. [Google Scholar]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinform. 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS One 2010, 5, e11147. [Google Scholar]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview version 2--a multiple sequence alignment editor and analysis workbench. Bioinform. 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Gilbert, D. Sequence file format conversion with command-line readseq. Current protocols in bioinformatics / editoral board, Andreas D. Baxevanis ... [et al.], 2003; Appendix 1, Appendix 1E. [Google Scholar]
- Desper, R.; Gascuel, O. Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J. Comput. Biol. 2002, 9, 687–705. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Stamatakis, A.; Ludwig, T.; Meier, H. Raxml-iii: A fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinform. 2005, 21, 456–463. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Posada, D. Prottest: Selection of best-fit models of protein evolution. Bioinform. 2005, 21, 2104–2105. [Google Scholar] [CrossRef]
- Zmasek, C.M.; Eddy, S.R. Atv: Display and manipulation of annotated phylogenetic trees. Bioinform. 2001, 17, 383–384. [Google Scholar] [CrossRef]
- Hanson, R. Jmol - a paradigm shift in crystallographic visualization. J. Appl. Crystall. 2010, 43, 1250–1260. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome annotation transfer utility (gatu): Rapid annotation of viral genomes using a closely related reference genome. BMC Genomics 2006, 7, 150. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012. [Google Scholar]
- Han, M.V.; Zmasek, C.M. Phyloxml: Xml for evolutionary biology and comparative genomics. BMC Bioinform. 2009, 10, 356. [Google Scholar] [CrossRef]
- Li, W.; Wong, S.K.; Li, F.; Kuhn, J.H.; Huang, I.C.; Choe, H.; Farzan, M. Animal origins of the severe acute respiratory syndrome coronavirus: Insight from ace2-s-protein interactions. J. Virol. 2006, 80, 4211–4219. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Wen, K.; Mou, Z.; Zou, L.; Che, X.; Ni, B.; Wu, Y. Antibody binding site mapping of sars-cov spike protein receptor-binding domain by a combination of yeast surface display and phage peptide library screening. Viral Immun. 2009, 22, 407–415. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Liu, S.; Kou, Z.; Li, W.; Farzan, M.; Jiang, S. Receptor-binding domain of sars-cov spike protein induces highly potent neutralizing antibodies: Implication for developing subunit vaccine. Biochem. Biophys. Res. Comm. 2004, 324, 773–781. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community. Viruses 2012, 4, 3209-3226. https://doi.org/10.3390/v4113209
Pickett BE, Greer DS, Zhang Y, Stewart L, Zhou L, Sun G, Gu Z, Kumar S, Zaremba S, Larsen CN, et al. Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community. Viruses. 2012; 4(11):3209-3226. https://doi.org/10.3390/v4113209
Chicago/Turabian StylePickett, Brett E., Douglas S. Greer, Yun Zhang, Lucy Stewart, Liwei Zhou, Guangyu Sun, Zhiping Gu, Sanjeev Kumar, Sam Zaremba, Christopher N. Larsen, and et al. 2012. "Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community" Viruses 4, no. 11: 3209-3226. https://doi.org/10.3390/v4113209
APA StylePickett, B. E., Greer, D. S., Zhang, Y., Stewart, L., Zhou, L., Sun, G., Gu, Z., Kumar, S., Zaremba, S., Larsen, C. N., Jen, W., Klem, E. B., & Scheuermann, R. H. (2012). Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community. Viruses, 4(11), 3209-3226. https://doi.org/10.3390/v4113209