Human Metapneumovirus Antagonism of Innate Immune Responses



Abstract
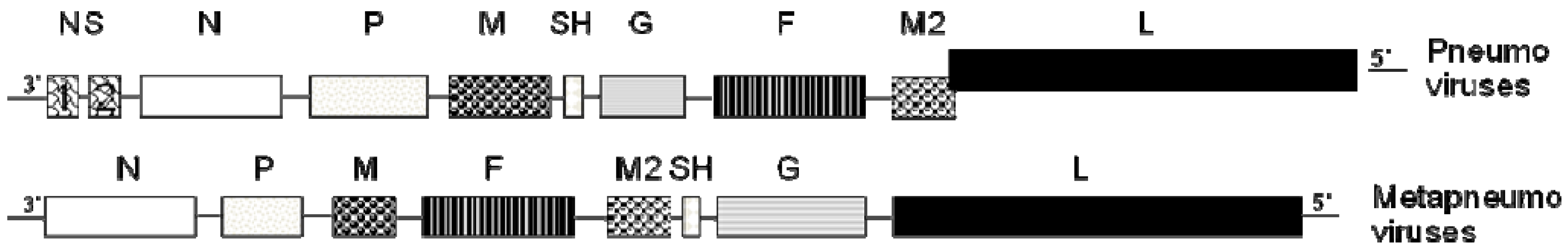
:1. Human Metapneumovirus (hMPV): A Recently Discovered Human Viral Pathogen

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Genus | Virus |
|---|---|---|
| Paramyxovirinae | ||
| Henipavirus | Hendravirus | |
| Nipah virus | ||
| Morbillivirus | Measles virus (MeV) | |
| Respirovirus | Sendai virus (SeV) | |
| Human parainfluenza virus type 1 (hPIV1) | ||
| Human parainfluenza virus type 3 (hPIV3) | ||
| Bovine parainfluenza virus type 3 (BPIV3) | ||
| Rubulavirus | Parainfluenza virus type 5 (PIV5) | |
| Mumps virus (MuV) | ||
| Human parainfluenza virus type 2 (hPIV2) | ||
| Pneumovirinae | ||
| Pneumovirus | Human respiratory syncytial virus (hRSV) | |
| Bovine respiratory syncytial virus (BRSV) | ||
| Metapneumovirus | Avian pneumovirus (APV) | |
| Human metapneumovirus (hMPV) |

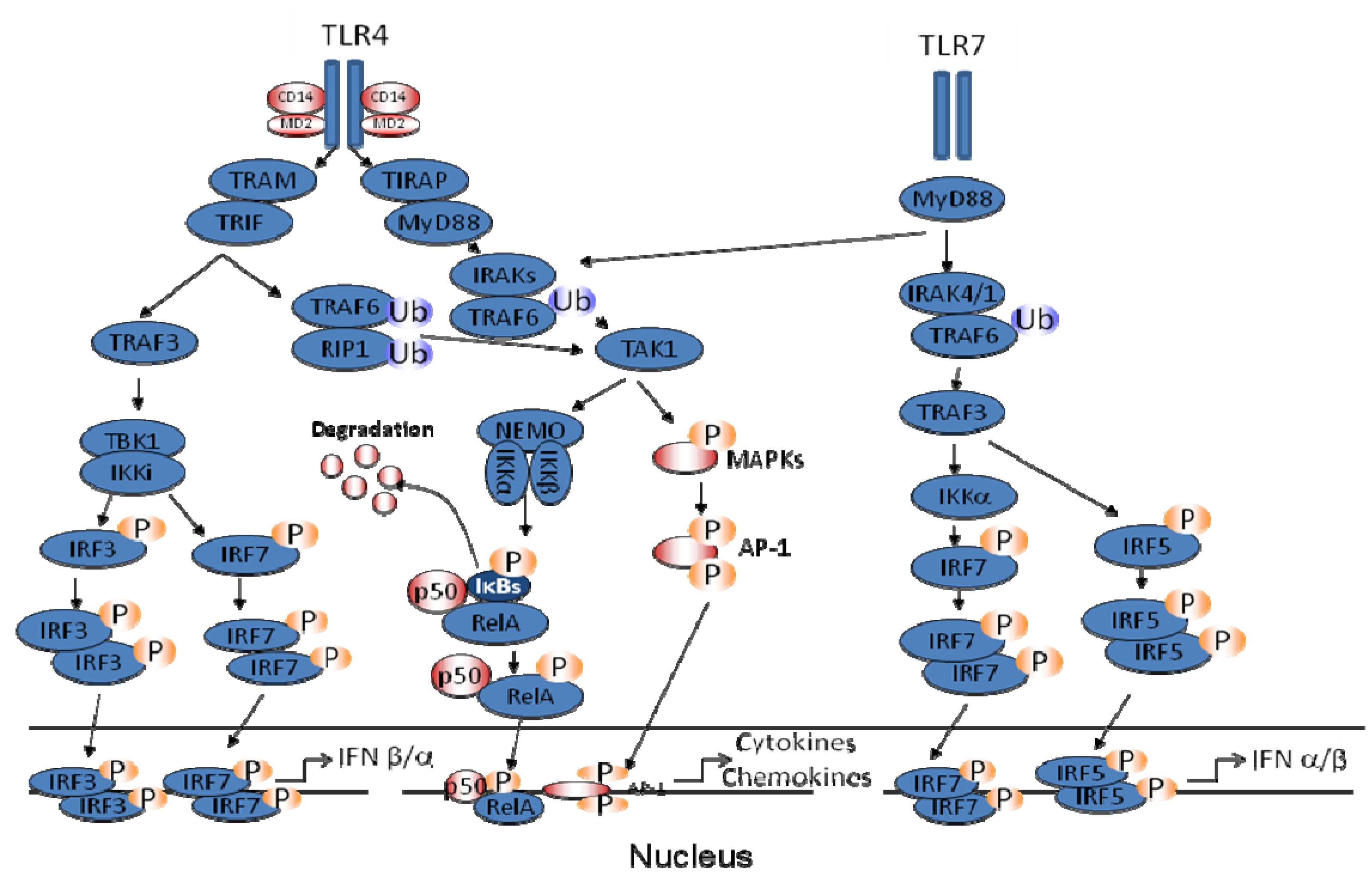
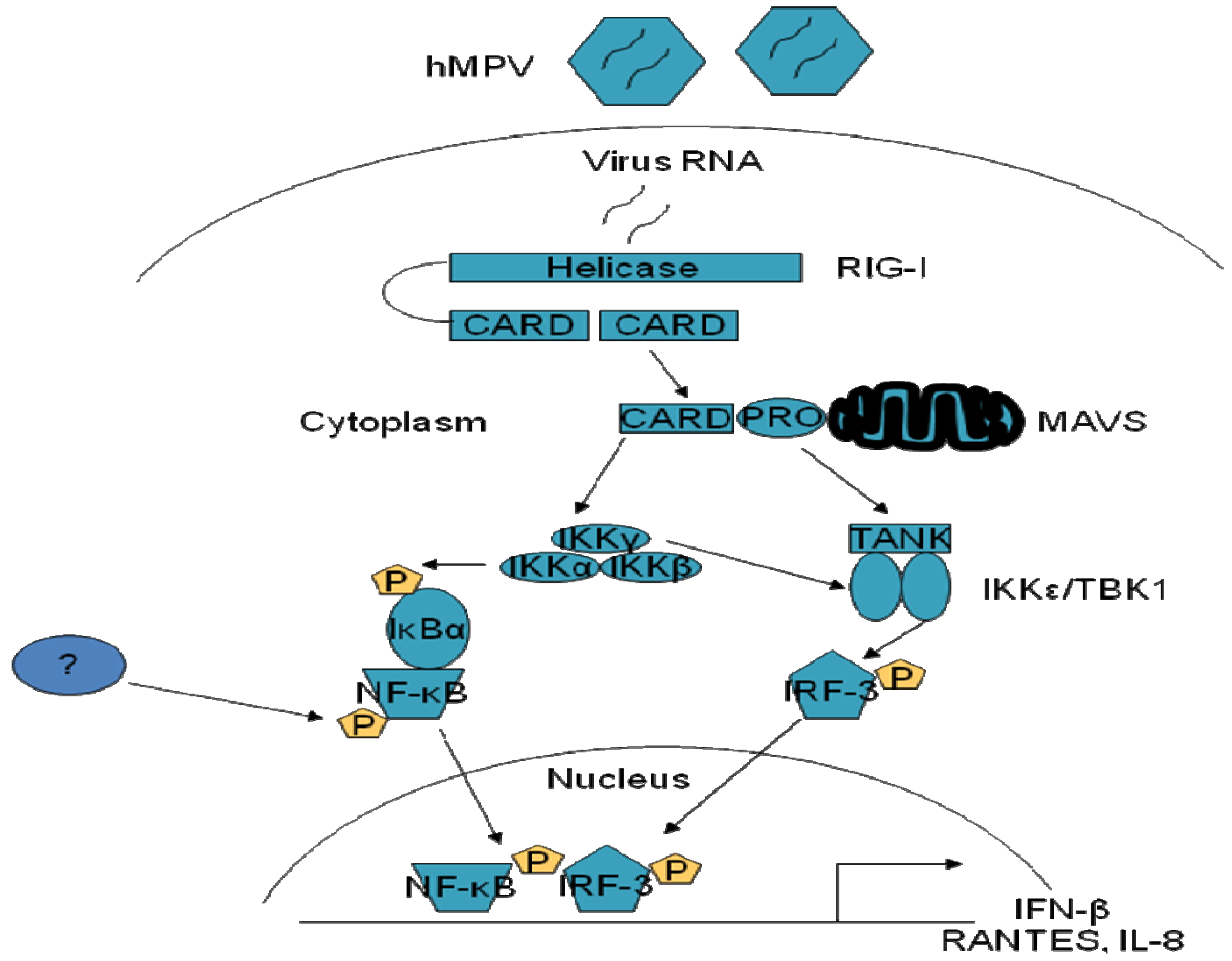
2. Pattern Recognition Receptors (PRRs) in hMPV-Induced Signaling


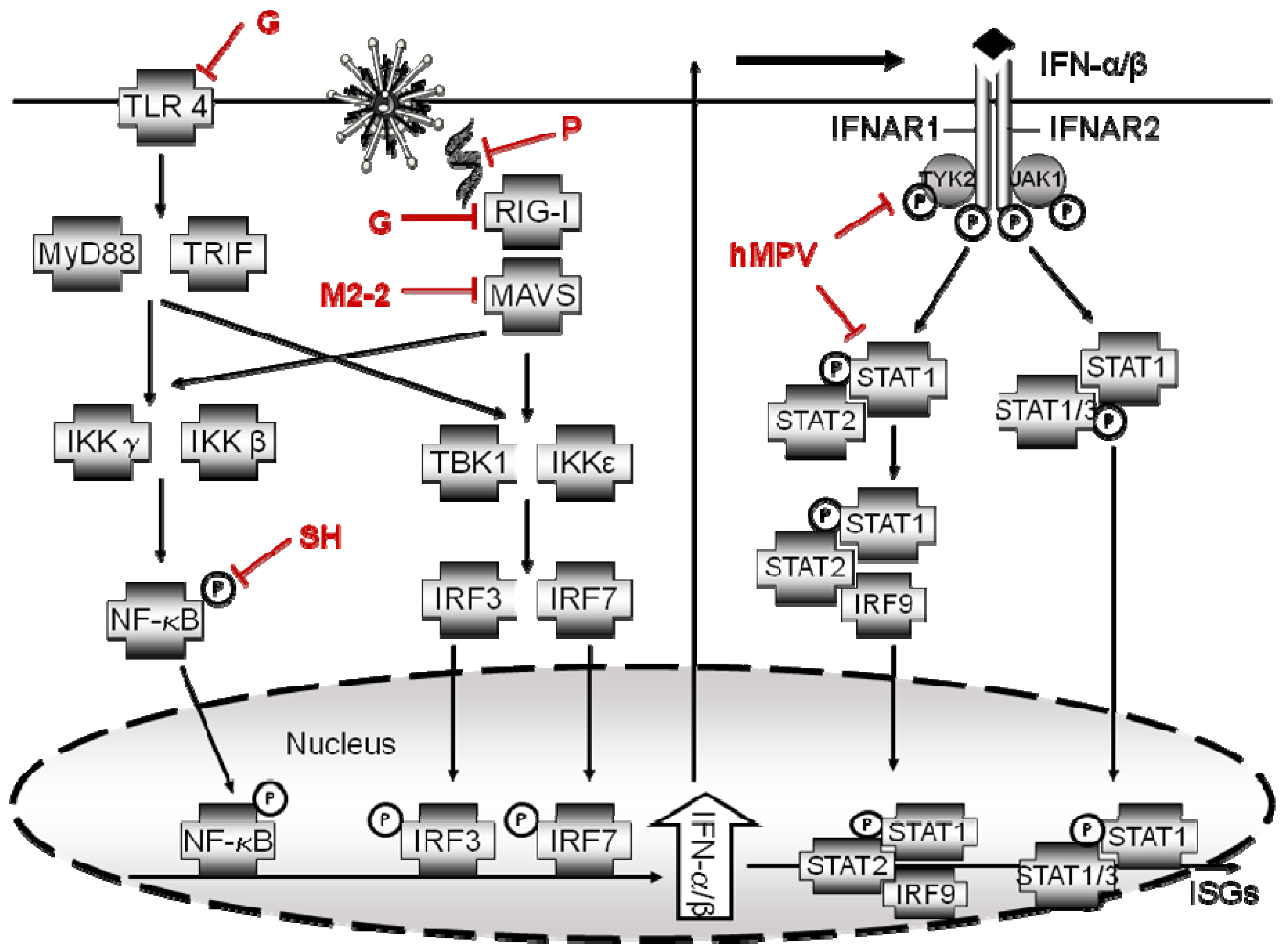
3. Inhibition of TLR Signaling
4. Interferon Signaling Antagonism
5. hMPV Proteins Identified as Antagonist of Host Innate Signaling Pathways

Acknowledgments
Conflict of Interest
References
- Lamb, R.A.; Kolakofsky, D. Paramyxoviridae: The Viruses and Their Replication. In Fundamental Virology, 4th; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2001; pp. 689–724. [Google Scholar]
- Principi, N.; Bosis, S.; Esposito, S. Human metapneumovirus in paediatric patients. Clin. Microbiol. Infect. 2006, 12, 301–308. [Google Scholar] [CrossRef]
- van den Hoogen, B.G.; de Jong, J.C.; Groen, J.; Kuiken, T.; de Groot, R.; Fouchier, R.A.; Osterhaus, A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001, 7, 719–724. [Google Scholar]
- Kahn, J.S. Epidemiology of human metapneumovirus. Clin. Microbiol. Rev. 2006, 19, 546–557. [Google Scholar] [CrossRef]
- Williams, J.V.; Harris, P.A.; Tollefson, S.J.; Halburnt-Rush, L.L.; Pingsterhaus, J.M.; Edwards, K.M.; Wright, P.F.; Crowe, J.E., Jr. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N. Engl. J. Med. 2004, 350, 443–450. [Google Scholar] [CrossRef]
- Crowe, J.E., Jr. Human metapneumovirus as a major cause of human respiratory tract disease. Pediatr. Infect. Dis. J. 2004, 23, 215–221. [Google Scholar] [CrossRef]
- Boivin, G.; Abed, L.; Pelletier, G.; Ruel, L.; Moisan, D.; Cote', S.; Peret, T.C.; Erdman, D.D.; Anderson, L.J. Virological features and clinical manifestations associated with the human metapneumovirus, a newly discovered paramyxovirus. J. Infect. Dis. 2002, 186, 1330–1334. [Google Scholar]
- Esper, F.; Boucher, D.; Weibel, C.; Martinello, R.A.; Kahn, J.S. Human metapneumovirus infection in the United States: clinical manifestations associated with a newly emerging respiratory infection in children. Pediatrics 2003, 111, 1407–1410. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Casola, A.; Garofalo, R.P. Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J. Virol. 2005, 79, 14992–14997. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Casola, A.; Suarez, G.; Yu, X.; Spetch, L.; Peeples, M.E.; Garofalo, R.P. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2006, 34, 320–329. [Google Scholar]
- Bao, X.; Liu, T.; Spetch, L.; Kolli, D.; Garofalo, R.P.; Casola, A. Airway epithelial cell response to human metapneumovirus infection. Virology 2007, 386, 91–101. [Google Scholar]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death. Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar]
- Groskreutz, D.J.; Monick, M.M.; Powers, L.S.; Yarovinsky, T.O.; Look, D.C.; Hunninghake, G.W. Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J. Immunol. 2006, 176, 1733–1740. [Google Scholar]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic Acid-inducible gene I mediates early antiviral response and toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef]
- Liao, S.; Bao, X.; Liu, T.; Lai, S.; Li, K.; Garofalo, R.P.; Casola, A. Role of retinoic acid inducible gene-I in human metapneumovirus-induced cellular signalling. J. Gen. Virol. 2008, 89, 1978–1986. [Google Scholar] [CrossRef]
- Kolli, D.; Bao, X.; Liu, T.; Hong, C.; Wang, T.; Garofalo, R.P.; Casola, A. Human metapneumovirus glycoprotein G inhibits TLR4-dependent signaling in monocyte-derived dendritic cells. J. Immunol. 2011, 187, 47–54. [Google Scholar] [CrossRef]
- Beutler, B.; Hoebe, K.; Georgel, P.; Tabeta, K.; Du, X. Genetic analysis of innate immunity: Identification and function of the TIR adapter proteins. Adv. Exp. Med. Biol. 2005, 560, 29–39. [Google Scholar]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef]
- Rassa, J.C.; Meyers, J.L.; Zhang, Y.; Kudaravalli, R.; Ross, S.R. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2281–2286. [Google Scholar] [CrossRef]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; Finberg, R.W. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol 2000, 1, 398–401. [Google Scholar] [CrossRef]
- Lizundia, R.; Sauter, K.S.; Taylor, G.; Werling, D. Host species-specific usage of the TLR4-LPS receptor complex. Innate. Immun. 2008, 14, 223–231. [Google Scholar] [CrossRef]
- Haeberle, H.; Takizawa, R.; Casola, A.; Brasier, A.R.; Dieterich, H.-J.; van Rooijen, N.; Gatalica, Z.; Garofalo, R.P. Respiratory syncytial virus-induced activation of NF-kB in the lung involves alveolar macrophages and Toll-like receptor 4-dependent pathways. J. Infect. Dis. 2002, 186, 1199–1206. [Google Scholar] [CrossRef]
- Cyr, S.L.; Angers, I.; Guillot, L.; Stoica-Popescu, I.; Lussier, M.; Qureshi, S.; Burt, D.S.; Ward, B.J. TLR4 and MyD88 control protection and pulmonary granulocytic recruitment in a murine intranasal RSV immunization and challenge model. Vaccine 2009, 27, 421–430. [Google Scholar] [CrossRef]
- Velayutham, T.S.; Kolli, D.; Ivanciuc, D.; Garofalo, R.P.; Casola, A. Critical role of Toll-like receptor 4 in human metapneumovirus innate immune responses and disease pathogenesis. J. Infect. Dis. 2012. manuscript in submission. [Google Scholar]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Jurk, M.; Heil, F.; Vollmer, J.; Schetter, C.; Krieg, A.M.; Wagner, H.; Lipford, G.; Bauer, S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 2002, 3, 499. [Google Scholar]
- Krug, A.; French, A.R.; Barchet, W.; Fischer, J.A.; Dzionek, A.; Pingel, J.T.; Orihuela, M.M.; Akira, S.; Yokoyama, W.M.; Colonna, M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004, 21, 107–119. [Google Scholar] [CrossRef]
- Krug, A.; Luker, G.D.; Barchet, W.; Leib, D.A.; Akira, S.; Colonna, M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103, 1433–1437. [Google Scholar]
- Goutagny, N.; Jiang, Z.; Tian, J.; Parroche, P.; Schickli, J.; Monks, B.G.; Ulbrandt, N.; Ji, H.; Kiener, P.A.; Coyle, A.J.; Fitzgerald, K.A. Cell type-specific recognition of human metapneumoviruses (HMPVs) by retinoic acid-inducible gene I (RIG-I) and TLR7 and viral interference of RIG-I ligand recognition by HMPV-B1 phosphoprotein. J. Immunol. 2010, 184, 1168–1179. [Google Scholar]
- Davidson, S.; Kaiko, G.; Loh, Z.; Lalwani, A.; Zhang, V.; Spann, K.; Foo, S.Y.; Hansbro, N.; Uematsu, S.; Akira, S.; Matthaei, K.I.; Rosenberg, H.F.; Foster, P.S.; Phipps, S. Plasmacytoid Dendritic Cells Promote Host Defense against Acute Pneumovirus Infection via the TLR7ΓÇôMyD88-Dependent Signaling Pathway. The Journal of Immunology 2011, 186, 5938–5948. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; Akira, S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 17264–17269. [Google Scholar]
- Breiman, A.; Grandvaux, N.; Lin, R.; Ottone, C.; Akira, S.; Yoneyama, M.; Fujita, T.; Hiscott, J.; Meurs, E.F. Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKepsilon. J. Virol. 2005, 79, 3969–3978. [Google Scholar] [CrossRef]
- tenOever, B.R.; Sharma, S.; Zou, W.; Sun, Q.; Grandvaux, N.; Julkunen, I.; Hemmi, H.; Yamamoto, M.; Akira, S.; Yeh, W.C.; Lin, R.; Hiscott, J. Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol. 2004, 78, 10636–10649. [Google Scholar]
- Siren, J.; Imaizumi, T.; Sarkar, D.; Pietila, T.; Noah, D.L.; Lin, R.; Hiscott, J.; Krug, R.M.; Fisher, P.B.; Julkunen, I.; Matikainen, S. Retinoic acid inducible gene-I and mda-5 are involved in influenza A virus-induced expression of antiviral cytokines. Microbes. Infect. 2006, 8, 2013–2020. [Google Scholar]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e, S. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science 2006, 314, 997–1001. [Google Scholar]
- Hiscott, J.; Lin, R.; Nakhaei, P.; Paz, S. MasterCARD: a priceless link to innate immunity. Trends Mol. Med. 2006, 12, 53–56. [Google Scholar] [CrossRef]
- Johnson, C.L.; Gale, M., Jr. CARD games between virus and host get a new player. Trends Immunol. 2006, 27, 1–4. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Tschopp, J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol. Cell 2006, 22, 561–569. [Google Scholar] [CrossRef]
- Liu, Y.J.; Kanzler, H.; Soumelis, V.; Gilliet, M. Dendritic cell lineage, plasticity and cross-regulation. Nat. Immunol. 2001, 2, 585–589. [Google Scholar]
- Rinaldo, C.R., Jr.; Piazza, P. Virus infection of dendritic cells: portal for host invasion and host defense. Trends Microbiol. 2004, 12, 337–345. [Google Scholar] [CrossRef]
- Stumbles, P.A.; Upham, J.W.; Holt, P.G. Airway dendritic cells: co-ordinators of immunological homeostasis and immunity in the respiratory tract. APMIS 2003, 111, 741–755. [Google Scholar] [CrossRef]
- Rescigno, M.; Borrow, P. The host-pathogen interaction: new themes from dendritic cell biology. Cell 2001, 106, 267–270. [Google Scholar] [CrossRef]
- Tan, M.C.; Battini, L.; Tuyama, A.C.; Macip, S.; Melendi, G.A.; Horga, M.A.; Gusella, G.L. Characterization of human metapneumovirus infection of myeloid dendritic cells. Virology 2007, 357, 1–9. [Google Scholar] [CrossRef]
- Le, N.C.; Munir, S.; Losq, S.; Winter, C.C.; McCarty, T.; Stephany, D.A.; Holmes, K.L.; Bukreyev, A.; Rabin, R.L.; Collins, P.L.; Buchholz, U.J. Infection and maturation of monocyte-derived human dendritic cells by human respiratory syncytial virus, human metapneumovirus, and human parainfluenza virus type 3. Virology 2009, 385, 169–182. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Baron, S.; Poast, J.S.; Adegboyega, P.A.; Casola, A.; Garofalo, R.P. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J. Virol. 2005, 79, 10190–10199. [Google Scholar] [CrossRef]
- Horvath, C.M. Silencing STATs: lessons from paramyxovirus interferon evasion. Cytokine Growth Factor Rev. 2004, 15, 117–127. [Google Scholar]
- Horvath, C.M. Weapons of STAT destruction. Interferon evasion by paramyxovirus V protein. Eur. J. Biochem. 2004, 271, 4621–4628. [Google Scholar] [CrossRef]
- Bao, X.; Liu, T.; Spetch, L.; Kolli, D.; Garofalo, R.P.; Casola, A. Airway epithelial cell response to human metapneumovirus infection. Virology 2007, 368, 91–101. [Google Scholar]
- Colamonici, O.R.; Uyttendaele, H.; Domanski, P.; Yan, H.; Krolewski, J.J. p135tyk2, an interferon-alpha-activated tyrosine kinase, is physically associated with an interferon-alpha receptor. J. Biol. Chem. 1994, 269, 3518–3522. [Google Scholar]
- Colamonici, O.; Yan, H.; Domanski, P.; Handa, R.; Smalley, D.; Mullersman, J.; Witte, M.; Krishnan, K.; Krolewski, J. Direct binding to and tyrosine phosphorylation of the alpha subunit of the type I interferon receptor by p135tyk2 tyrosine kinase. Mol. Cell Biol. 1994, 14, 8133–8142. [Google Scholar]
- Leung, S.; Qureshi, S.A.; Kerr, I.M.; Darnell, J.E., Jr.; Stark, G.R. Role of STAT2 in the alpha interferon signaling pathway. Mol. Cell Biol. 1995, 15, 1312–1317. [Google Scholar]
- Qureshi, S.A.; Salditt-Georgieff, M.; Darnell, J.E., Jr. Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 3829–3833. [Google Scholar] [CrossRef]
- Uze, G.; Schreiber, G.; Piehler, J.; Pellegrini, S. The receptor of the type I interferon family. Curr. Top. Microbiol. Immunol. 2007, 316, 71–95. [Google Scholar]
- Li, X.; Leung, S.; Burns, C.; Stark, G.R. Cooperative binding of Stat1-2 heterodimers and ISGF3 to tandem DNA elements. Biochimie 1998, 80, 703–710. [Google Scholar] [CrossRef]
- Hata, N.; Sato, M.; Takaoka, A.; Asagiri, M.; Tanaka, N.; Taniguchi, T. Constitutive IFN-alpha/beta signal for efficient IFN-alpha/beta gene induction by virus. Biochem. Biophys. Res. Commun. 2001, 285, 518–525. [Google Scholar] [CrossRef]
- Trinchieri, G. Type I interferon: friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le, B.A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef]
- Christophi, G.P.; Hudson, C.A.; Panos, M.; Gruber, R.C.; Massa, P.T. Modulation of macrophage infiltration and inflammatory activity by the phosphatase SHP-1 in virus-induced demyelinating disease. J. Virol. 2009, 83, 522–539. [Google Scholar] [CrossRef]
- Yasukawa, H.; Misawa, H.; Sakamoto, H.; Masuhara, M.; Sasaki, A.; Wakioka, T.; Ohtsuka, S.; Imaizumi, T.; Matsuda, T.; Ihle, J.N.; Yoshimura, A. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999, 18, 1309–1320. [Google Scholar] [CrossRef]
- Ramaswamy, M.; Shi, L.; Monick, M.M.; Hunninghake, G.W.; Look, D.C. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2004, 30, 893–900. [Google Scholar]
- Didcock, L.; Young, D.F.; Goodbourn, S.; Randall, R.E. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 1999, 73, 9928–9933. [Google Scholar]
- Kubota, T.; Yokosawa, N.; Yokota, S.; Fujii, N. C terminal CYS-RICH region of mumps virus structural V protein correlates with block of interferon alpha and gamma signal transduction pathway through decrease of STAT 1-alpha. Biochem. Biophys. Res. Commun. 2001, 283, 255–259. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Cruz, C.D.; Horvath, C.M. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J. Virol. 2004, 78, 5358–5367. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Parisien, J.P.; Horvath, C.M. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 2002, 76, 11476–11483. [Google Scholar]
- Takeuchi, K.; Kadota, S.I.; Takeda, M.; Miyajima, N.; Nagata, K. Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett. 2003, 545, 177–182. [Google Scholar] [CrossRef]
- Dinwiddie, D.L.; Harrod, K.S. Human Metapneumovirus Inhibits IFN-{alpha} Signaling Through Inhibition of STAT1 Phosphorylation. Am. J. Respir. Cell Mol. Biol. 2008, 38, 661–670. [Google Scholar] [CrossRef]
- Ren, J.; Kolli, D.; Liu, T.; Xu, R.; Garofalo, R.P.; Casola, A.; Bao, X. Human metapneumovirus inhibits IFN-beta signaling by downregulating Jak1 and Tyk2 cellular levels. PLoS. ONE. 2011, 6, e24496. [Google Scholar]
- Bao, X.; Kolli, D.; Liu, T.; Shan, Y.; Garofalo, R.P.; Casola, A. Human metapneumovirus small hydrophobic protein inhibits NF-kappaB transcriptional activity. J. Virol. 2008, 82, 8224–8229. [Google Scholar] [CrossRef]
- Bao, X.; Liu, T.; Shan, Y.; Li, K.; Garofalo, R.P.; Casola, A. Human metapneumovirus glycoprotein G inhibits innate immune responses. PLoS. Pathog. 2008, 4, e1000077. [Google Scholar]
- Ren, J.; Wang, Q.; Kolli, D.; Prusak, D.J.; Tseng, C.T.; Chen, Z.J.; Li, K.; Wood, T.G.; Bao, X. Human metapneumovirus M2-2 protein inhibits innate cellular signaling by targeting MAVS. J. Virol. 2012, 86, 13049–13061. [Google Scholar]
- van den Hoogen, B.G.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Analysis of the genomic sequence of a human metapneumovirus. Virology 2002, 295, 119–132. [Google Scholar] [CrossRef]
- Biacchesi, S.; Skiadopoulos, M.H.; Boivin, G.; Hanson, C.T.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Genetic diversity between human metapneumovirus subgroups. Virology 2003, 315, 1–9. [Google Scholar] [CrossRef]
- Biacchesi, S.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Frequent frameshift and point mutations in the SH gene of human metapneumovirus passaged in vitro. J. Virol. 2007, 81, 6057–6067. [Google Scholar] [CrossRef]
- Biacchesi, S.; Skiadopoulos, M.H.; Yang, L.; Lamirande, E.W.; Tran, K.C.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Recombinant human Metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J. Virol. 2004, 78, 12877–12887. [Google Scholar]
- Biacchesi, S.; Pham, Q.N.; Skiadopoulos, M.H.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 2005, 79, 12608–12613. [Google Scholar] [CrossRef]
- Wilson, R.L.; Fuentes, S.M.; Wang, P.; Taddeo, E.C.; Klatt, A.; Henderson, A.J.; He, B. Function of small hydrophobic proteins of paramyxovirus. J. Virol. 2006, 80, 1700–1709. [Google Scholar]
- Fuentes, S.; Tran, K.C.; Luthra, P.; Teng, M.N.; He, B. Function of the Respiratory Syncytial Virus small hydrophobic protein. J. Virol. 2007, 81, 8361–8366. [Google Scholar]
- Li, Z.; Xu, J.; Patel, J.; Fuentes, S.; Lin, Y.; Anderson, D.; Sakamoto, K.; Wang, L.F.; He, B. Function of the small hydrophobic protein of J paramyxovirus. J. Virol. 2011, 85, 32–42. [Google Scholar]
- Xu, P.; Li, Z.; Sun, D.; Lin, Y.; Wu, J.; Rota, P.A.; He, B. Rescue of wild-type mumps virus from a strain associated with recent outbreaks helps to define the role of the SH ORF in the pathogenesis of mumps virus. Virology 2011, 417, 126–136. [Google Scholar]
- Karin, M.; Delhase, M. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin. Immunol 2000, 12, 85–98. [Google Scholar] [CrossRef]
- Schmidt, M.; Chiorini, J.A.; Afione, S.; Kotin, R. Adeno-associated virus type 2 Rep78 inhibition of PKA and PRKX: fine mapping and analysis of mechanism. J. Virol. 2002, 76, 1033–1042. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Smith, S.E.; Dutch, R.E. Characterization of human metapneumovirus F protein-promoted membrane fusion: critical roles for proteolytic processing and low pH. J. Virol. 2006, 80, 10931–10941. [Google Scholar] [CrossRef]
- Cox, R.G.; Livesay, S.B.; Johnson, M.; Ohi, M.D.; Williams, J.V. The Human Metapneumovirus Fusion Protein Mediates Entry Via an Interaction with RGD-binding Integrins. J. Virol. 2012, 86, 12148–12160. [Google Scholar]
- Kolli, D.; Bao, X.; Liu, T.; Hong, C.; Wang, T.; Garofalo, R.P.; Casola, A. Human metapneumovirus glycoprotein G inhibits TLR4-dependent signaling in monocyte-derived dendritic cells. J. Immunol. 2011, 187, 47–54. [Google Scholar]
- Bao, X.; Kolli, D.; Ren, J.; Liu, T.; Garofalo, R.P.; Casola, A. Human Metapneumovirus Glycoprotein G Targets RIG-I to Inhibit Airway Epithelial Cell Responses. J. Virol. 2012. manuscript in submission. [Google Scholar]
- Pulendran, B.; Palucka, K.; Banchereau, J. Sensing pathogens and tuning immune responses. Science 2001, 293, 253–256. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Delgado, M.F.; Coviello, S.; Monsalvo, A.C.; Melendi, G.A.; Hernandez, J.Z.; Batalle, J.P.; Diaz, L.; Trento, A.; Chang, H.Y.; Mitzner, W.; Ravetch, J.; Melero, J.A.; Irusta, P.M.; Polack, F.P. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat. Med. 2009, 15, 34–41. [Google Scholar]
- Polack, F.P.; Irusta, P.M.; Hoffman, S.J.; Schiatti, M.P.; Melendi, G.A.; Delgado, M.F.; Laham, F.R.; Thumar, B.; Hendry, R.M.; Melero, J.A.; Karron, R.A.; Collins, P.L.; Kleeberger, S.R. The cysteine-rich region of respiratory syncytial virus attachment protein inhibits innate immunity elicited by the virus and endotoxin. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 8996–9001. [Google Scholar]
- Arnold, R.; Konig, B.; Werchau, H.; Konig, W. Respiratory syncytial virus deficient in soluble G protein induced an increased proinflammatory response in human lung epithelial cells. Virology 2004, 330, 384–397. [Google Scholar] [CrossRef]
- Alff, P.J.; Gavrilovskaya, I.N.; Gorbunova, E.; Endriss, K.; Chong, Y.; Geimonen, E.; Sen, N.; Reich, N.C.; Mackow, E.R. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 2006, 80, 9676–9686. [Google Scholar] [CrossRef]
- Geimonen, E.; LaMonica, R.; Springer, K.; Farooqui, Y.; Gavrilovskaya, I.N.; Mackow, E.R. Hantavirus pulmonary syndrome-associated hantaviruses contain conserved and functional ITAM signaling elements. J. Virol. 2003, 77, 1638–1643. [Google Scholar]
- Buchholz, U.J.; Biacchesi, S.; Pham, Q.N.; Tran, K.C.; Yang, L.; Luongo, C.L.; Skiadopoulos, M.H.; Murphy, B.R.; Collins, P.L. Deletion of M2 gene open reading frames 1 and 2 of human metapneumovirus: Effects on RNA synthesis, attenuation, and immunogenicity. J. Virol. 2005, 79, 6588–6597. [Google Scholar]
- Herfst, S.; de Graaf, M.; Schickli, J.H.; Tang, R.S.; Kaur, J.; Yang, C.F.; Spaete, R.R.; Haller, A.A.; van den Hoogen, B.G.; Osterhaus, A.D.; Fouchier, R.A. Recovery of human metapneumovirus genetic lineages a and B from cloned cDNA. J. Virol. 2004, 78, 8264–8270. [Google Scholar]
- Sutherland, K.A.; Collins, P.L.; Peeples, M.E. Synergistic effects of gene-end signal mutations and the M2-1 protein on transcription termination by respiratory syncytial virus. Virology 2001, 288, 295–307. [Google Scholar] [CrossRef]
- Schickli, J.H.; Kaur, J.; MacPhail, M.; Guzzetta, J.M.; Spaete, R.R.; Tang, R.S. Deletion of human metapneumovirus M2-2 increases mutation frequency and attenuates growth in hamsters. Virol. J. 2008, 3, 5–69. [Google Scholar]
- Ren, J.; Wang, Q.; Kolli, D.; Prusak, D.J.; Tseng, C.T.; Li, K.; Wood, T.G.; Bao, X. Human metapneumovirus M2-2 protein inhibits the innate cellular signaling by targeting MAVS. J. Virol. 2012, 86, 13049–13061. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; Garcia-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The Influenza Virus Protein PB1-F2 Inhibits the Induction of Type I Interferon at the Level of the MAVS Adaptor Protein. PLoS. Pathog. 2011, 7, e1002067. [Google Scholar]
- Graef, K.M.; Vreede, F.T.; Lau, Y.F.; McCall, A.W.; Carr, S.M.; Subbarao, K.; Fodor, E. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 2010, 84, 8433–8445. [Google Scholar] [CrossRef]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.G.; Casola, A.; Bao, X. A Novel Mechanism for Inhibition of IRF-3-Dependent Gene Expression by Human Respiratory Syncytial Virus NS1 Protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar]
- Bastien, N.; Normand, S.; Taylor, T.; Ward, D.; Peret, T.C.; Boivin, G.; Anderson, L.J.; Li, Y. Sequence analysis of the N, P, M and F genes of Canadian human metapneumovirus strains. Virus Res. 2003, 93, 51–62. [Google Scholar] [CrossRef]
- Tedcastle, A.B.; Fenwick, F.; Ingram, R.E.; King, B.J.; Robinson, M.J.; Toms, G.L. The characterization of monoclonal antibodies to human metapneumovirus and the detection of multiple forms of the virus nucleoprotein and phosphoprotein. J. Med. Virolog. 2012, 84, 1061–1070. [Google Scholar] [CrossRef]
- Derdowski, A.; Peters, T.R.; Glover, N.; Qian, R.; Utley, T.J.; Burnett, A.; Williams, J.V.; Spearman, P.; Crowe, J.E., Jr. Human metapneumovirus nucleoprotein and phosphoprotein interact and provide the minimal requirements for inclusion body formation. J. Gen. Virol. 2008, 89, 2698–2708. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kolli, D.; Bao, X.; Casola, A. Human Metapneumovirus Antagonism of Innate Immune Responses. Viruses 2012, 4, 3551-3571. https://doi.org/10.3390/v4123551
Kolli D, Bao X, Casola A. Human Metapneumovirus Antagonism of Innate Immune Responses. Viruses. 2012; 4(12):3551-3571. https://doi.org/10.3390/v4123551
Chicago/Turabian StyleKolli, Deepthi, Xiaoyong Bao, and Antonella Casola. 2012. "Human Metapneumovirus Antagonism of Innate Immune Responses" Viruses 4, no. 12: 3551-3571. https://doi.org/10.3390/v4123551
APA StyleKolli, D., Bao, X., & Casola, A. (2012). Human Metapneumovirus Antagonism of Innate Immune Responses. Viruses, 4(12), 3551-3571. https://doi.org/10.3390/v4123551