Interplay between Interferon-Mediated Innate Immunity and Porcine Reproductive and Respiratory Syndrome Virus

Abstract
:
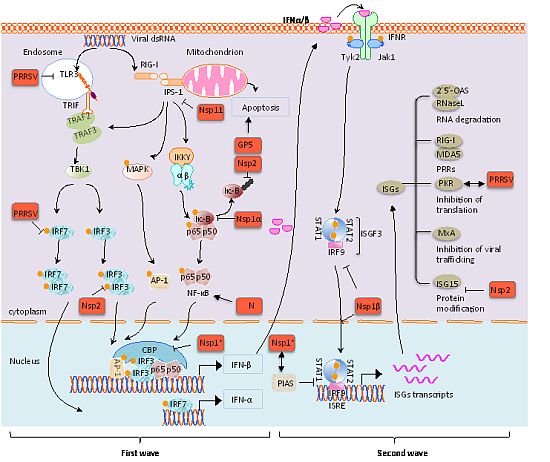{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
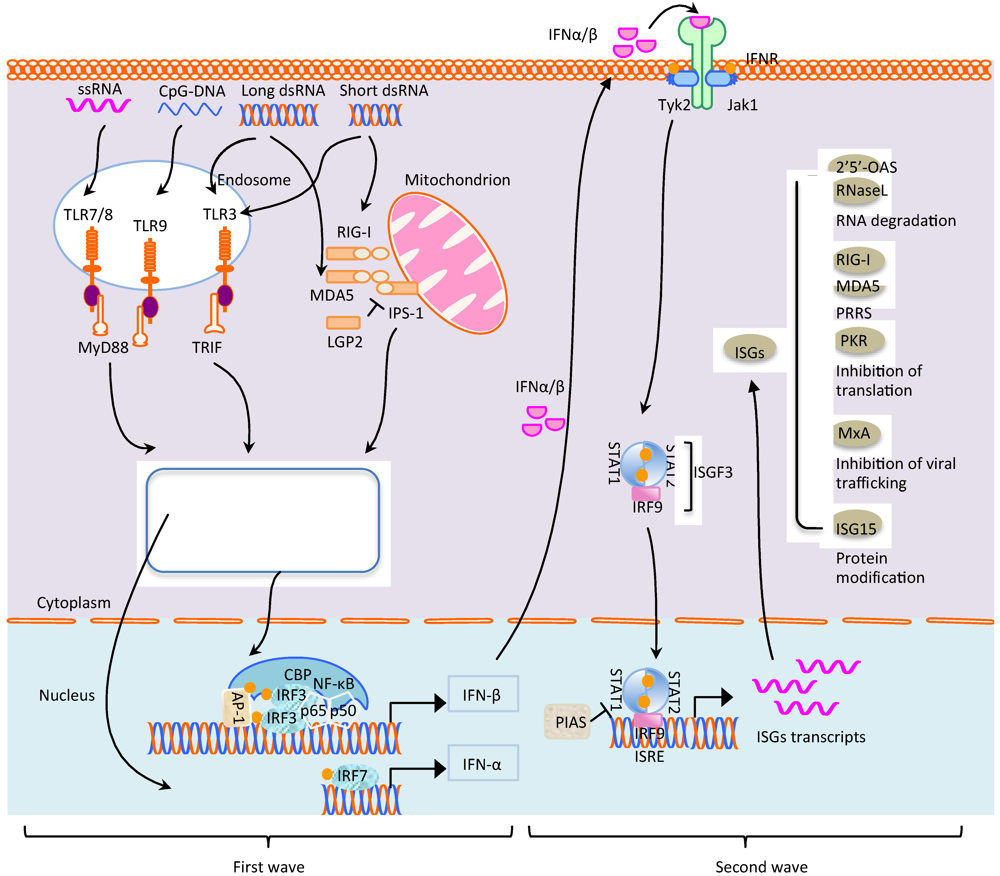
2. Signaling Pathways for Innate Immunity against Virus Infection

2.1. Toll-Like Receptor (TLR) Signaling
2.1.1. TLR3
2.1.2. TLR7/8/9
2.2. Cytoplasmic Receptors and Their Signaling
2.2.1. RIG-I/MDA5 Pathway
2.2.2. IPS-1 Signaling
2.2.3. RNA Recognition by RIG-I and MDA5
2.3. IFN Signaling Pathways
2.3.1. JAK-STAT Pathway
2.3.2. IFN Inducible Genes and Their Functions
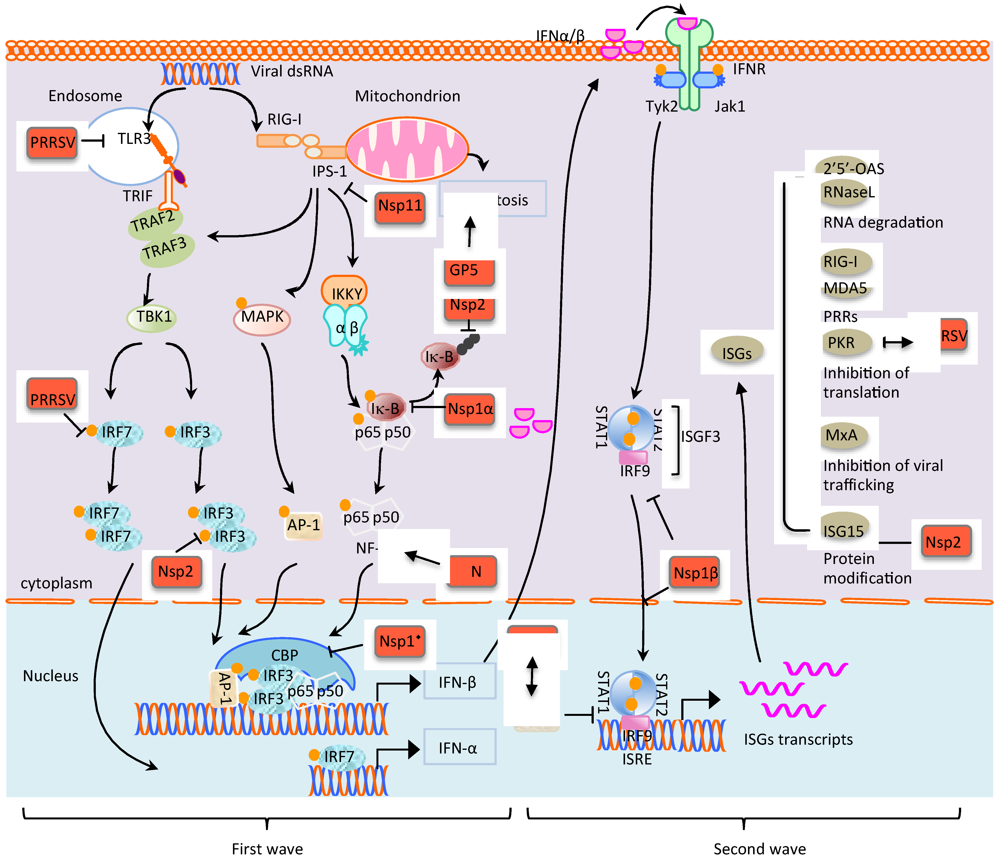
3. Modulation of Innate Immunity by PRRSV
3.1. TLRs and the Virus

3.2. PRRSV Targeting IFN Enhanceosome through IRF-Mediated RIG-I/MDA5 Pathways
3.3. Modulation of NF-κB Signaling by PRRSV
3.3.1. Up-regulation of NF-κB by N protein
3.3.2. Down-Regulation of NF-κB by Nsp1α and Nsp2
3.4. Inhibition of JAK-STAT Signaling and ISG Expressions by PRRSV
3.5. Apoptosis and the Virus
4. Conclusion and Future Prospects
Acknowledgments
Conflict of Interest
References and Notes
- Wensvoort, G.; de Kluyver, E.P.; Pol, J.M.; Wagenaar, F.; Moormann, R.J.; Hulst, M.M.; Bloemraad, R.; den Besten, A.; Zetstra, T.; Terpstra, C. Lelystad virus, the cause of porcine epidemic abortion and respiratory syndrome: A review of mystery swine disease research at Lelystad. Vet. Microbiol. 1992, 33, 185–193. [Google Scholar]
- Collins, J.E.; Benfield, D.A.; Christianson, W.T.; Harris, L.; Hennings, J.C.; Shaw, D.P.; Goyal, S.M.; McCullough, S.; Morrison, R.B.; Joo, H.S.; et al. Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J. Vet. Diagn. Invest. 1992, 4, 117–126. [Google Scholar] [CrossRef]
- Allende, R.; Lewis, T.L.; Lu, Z.; Rock, D.L.; Kutish, G.F.; Ali, A.; Doster, A.R.; Osorio, F.A. North American and European porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J. Gen. Virol. 1999, 80, 307–315. [Google Scholar]
- Nelsen, C.J.; Murtaugh, M.P.; Faaberg, K.S. Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J. Virol. 1999, 73, 270–280. [Google Scholar]
- Xiao, S.; Mo, D.; Wang, Q.; Jia, J.; Qin, L.; Yu, X.; Niu, Y.; Zhao, X.; Liu, X.; Chen, Y. Aberrant host immune response induced by highly virulent PRRSV identified by digital gene expression tag profiling. BMC Genom. 2010, 11, 544. [Google Scholar]
- Conzelmann, K.K.; Visser, N.; Van Woensel, P.; Thiel, H.J. Molecular characterization of porcine reproductive and respiratory syndrome virus, a member of the arterivirus group. Virology 1993, 193, 329–339. [Google Scholar]
- Meulenberg, J.J.; Hulst, M.M.; de Meijer, E.J.; Moonen, P.L.; den Besten, A.; de Kluyver, E.P.; Wensvoort, G.; Moormann, R.J. Lelystad virus, the causative agent of porcine epidemic abortion and respiratory syndrome (PEARS), is related to LDV and EAV. Virology 1993, 192, 62–72. [Google Scholar] [CrossRef]
- Snijder, E.J. The arterivirus replicase. The road from RNA to protein(s), and back again. Adv. Exp. Med. Biol. 1998, 440, 97–108. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar]
- Firth, A.E.; Zevenhoven-Dobbe, J.C.; Wills, N.M.; Go, Y.Y.; Balasuriya, U.B.; Atkins, J.F.; Snijder, E.J.; Posthuma, C.C. Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production. J. Gen. Virol. 2011, 92, 1097–1106. [Google Scholar]
- Johnson, C.R.; Griggs, T.F.; Gnanandarajah, J.; Murtaugh, M.P. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J. Gen. Virol. 2011, 92, 1107–1116. [Google Scholar]
- den Boon, J.A.; Snijder, E.J.; Chirnside, E.D.; de Vries, A.A.; Horzinek, M.C.; Spaan, W.J. Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J. Virol. 1991, 65, 2910–2920. [Google Scholar]
- Wootton, S.; Yoo, D.; Rogan, D. Full-length sequence of a Canadian porcine reproductive and respiratory syndrome virus (PRRSV) isolate. Arch. Virol. 2000, 145, 2297–2323. [Google Scholar]
- Fang, Y.; Snijder, E.J. The PRRSV replicase: Exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res. 2010, 154, 61–76. [Google Scholar]
- Dea, S.; Gagnon, C.A.; Mardassi, H.; Pirzadeh, B.; Rogan, D. Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: Comparison of the North American and European isolates. Arch. Virol. 2000, 145, 659–688. [Google Scholar]
- Meulenberg, J.J.; Bende, R.J.; Pol, J.M.; Wensvoort, G.; Moormann, R.J. Nucleocapsid protein N of Lelystad virus: Expression by recombinant baculovirus, immunological properties, and suitability for detection of serum antibodies. Clin. Diagn. Lab. Immunol. 1995, 2, 652–656. [Google Scholar]
- Mardassi, H.; Massie, B.; Dea, S. Intracellular synthesis, processing, and transport of proteins encoded by ORFs 5 to 7 of porcine reproductive and respiratory syndrome virus. Virology 1996, 221, 98–112. [Google Scholar] [CrossRef]
- Wissink, E.H.; Kroese, M.V.; van Wijk, H.A.; Rijsewijk, F.A.; Meulenberg, J.J.; Rottier, P.J. Envelope protein requirements for the assembly of infectious virions of porcine reproductive and respiratory syndrome virus. J. Virol. 2005, 79, 12495–12506. [Google Scholar]
- Wootton, S.K.; Yoo, D. Homo-oligomerization of the porcine reproductive and respiratory syndrome virus nucleocapsid protein and the role of disulfide linkages. J. Virol. 2003, 77, 4546–4557. [Google Scholar]
- Yoo, D.; Wootton, S.K.; Li, G.; Song, C.; Rowland, R.R. Colocalization and interaction of the porcine arterivirus nucleocapsid protein with the small nucleolar RNA-associated protein fibrillarin. J. Virol. 2003, 77, 12173–12183. [Google Scholar]
- Yoo, D.; Song, C.; Sun, Y.; Du, Y.; Kim, O.; Liu, H.C. Modulation of host cell responses and evasion strategies for porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 48–60. [Google Scholar]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasure. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar]
- Decker, T.; Muller, M.; Stockinger, S. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 2005, 5, 675–687. [Google Scholar]
- Takaoka, A.; Yanai, H. Interferon signalling network in innate defence. Cell. Microbiol. 2006, 8, 907–922. [Google Scholar]
- Sang, Y.; Rowland, R.R.; Hesse, R.A.; Blecha, F. Differential expression and activity of the porcine type I interferon family. Physiol. Genom. 2010, 42, 248–258. [Google Scholar]
- O'Neill, L.A.; Fitzgerald, K.A.; Bowie, A.G. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003, 24, 286–290. [Google Scholar]
- Zhang, S.Y.; Jouanguy, E.; Sancho-Shimizu, V.; von Bernuth, H.; Yang, K.; Abel, L.; Picard, C.; Puel, A.; Casanova, J.L. Human Toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol. Rev. 2007, 220, 225–236. [Google Scholar]
- Baccala, R.; Hoebe, K.; Kono, D.H.; Beutler, B.; Theofilopoulos, A.N. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 2007, 13, 543–551. [Google Scholar]
- Matsumoto, M.; Funami, K.; Tanabe, M.; Oshiumi, H.; Shingai, M.; Seto, Y.; Yamamoto, A.; Seya, T. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 2003, 171, 3154–3162. [Google Scholar]
- Kariko, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar]
- Okahira, S.; Nishikawa, F.; Nishikawa, S.; Akazawa, T.; Seya, T.; Matsumoto, M. Interferon-beta induction through toll-like receptor 3 depends on double-stranded RNA structure. DNA Cell Biol. 2005, 24, 614–623. [Google Scholar]
- Vercammen, E.; Staal, J.; Beyaert, R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin. Microbiol. Rev. 2008, 21, 13–25. [Google Scholar]
- Unterholzner, L.; Bowie, A.G. The interplay between viruses and innate immune signaling: Recent insights and therapeutic opportunities. Biochem. Pharmacol. 2008, 75, 589–602. [Google Scholar]
- Gantier, M.P.; Williams, B.R. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 2007, 18, 363–371. [Google Scholar]
- Vollmer, J.; Weeratna, R.; Payette, P.; Jurk, M.; Schetter, C.; Laucht, M.; Wader, T.; Tluk, S.; Liu, M.; Davis, H.L.; et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 2004, 34, 251–262. [Google Scholar] [CrossRef]
- O'Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar]
- Edwards, M.R.; Slater, L.; Johnston, S.L. Signalling pathways mediating type I interferon gene expression. Microb. Infect. 2007, 9, 1245–1251. [Google Scholar]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef]
- Komuro, A.; Horvath, C.M. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol. 2006, 80, 12332–12342. [Google Scholar]
- Rothenfusser, S.; Goutagny, N.; DiPerna, G.; Gong, M.; Monks, B.G.; Schoenemeyer, A.; Yamamoto, M.; Akira, S.; Fitzgerald, K.A. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 2005, 175, 5260–5268. [Google Scholar]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 8459–8464. [Google Scholar]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science 2006, 314, 997–1001. [Google Scholar]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5'-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar]
- Schindler, C.; Darnell, J.E., Jr. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annu. Rev. Biochem. 1995, 64, 621–651. [Google Scholar]
- Krebs, D.L.; Hilton, D.J. SOCS proteins: Negative regulators of cytokine signaling. Stem Cell. 2001, 19, 378–387. [Google Scholar]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar]
- Wilkinson, K.A.; Henley, J.M. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010, 428, 133–145. [Google Scholar]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar]
- Blomstrom, D.C.; Fahey, D.; Kutny, R.; Korant, B.D.; Knight, E., Jr. Molecular characterization of the interferon-induced 15-kDa protein. Molecular cloning and nucleotide and amino acid sequence. J. Biol. Chem. 1986, 261, 8811–8816. [Google Scholar]
- Narasimhan, J.; Potter, J.L.; Haas, A.L. Conjugation of the 15-kDa interferon-induced ubiquitin homolog is distinct from that of ubiquitin. J. Biol. Chem. 1996, 271, 324–330. [Google Scholar]
- Lu, G.; Reinert, J.T.; Pitha-Rowe, I.; Okumura, A.; Kellum, M.; Knobeloch, K.P.; Hassel, B.; Pitha, P.M. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell. Mol. Biol. (Noisy-le-grand) 2006, 52, 29–41. [Google Scholar]
- D'Cunha, J.; Ramanujam, S.; Wagner, R.J.; Witt, P.L.; Knight, E., Jr.; Borden, E.C. In vitro and in vivo secretion of human ISG15, an IFN-induced immunomodulatory cytokin. J. Immunol. 1996, 157, 4100–4108. [Google Scholar]
- Haller, O.; Arnheiter, H.; Lindenmann, J.; Gresser, I. Host gene influences sensitivity to interferon action selectively for influenza virus. Nature 1980, 283, 660–662. [Google Scholar]
- Haller, O.; Frese, M.; Rost, D.; Nuttall, P.A.; Kochs, G. Tick-borne thogoto virus infection in mice is inhibited by the orthomyxovirus resistance gene product Mx1. J. Virol. 1995, 69, 2596–2601. [Google Scholar]
- Kochs, G.; Haller, O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 2082–2086. [Google Scholar]
- Hovanessian, A.G.; Justesen, J. The human 2'-5'oligoadenylate synthetase family: Unique interferon-inducible enzymes catalyzing 2'-5' instead of 3'-5' phosphodiester bond formation. Biochimie 2007, 89, 779–788. [Google Scholar]
- Roberts, W.K.; Hovanessian, A.; Brown, R.E.; Clemens, M.J.; Kerr, I.M. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature 1976, 264, 477–480. [Google Scholar]
- Ank, N.; West, H.; Paludan, S.R. IFN-lambda: Novel antiviral cytokines. J. Interferon Cytokine Res. 2006, 26, 373–379. [Google Scholar]
- Nanduri, S.; Carpick, B.W.; Yang, Y.; Williams, B.R.; Qin, J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998, 17, 5458–5465. [Google Scholar]
- Nallagatla, S.R.; Hwang, J.; Toroney, R.; Zheng, X.; Cameron, C.E.; Bevilacqua, P.C. 5'-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 2007, 318, 1455–1458. [Google Scholar]
- Beyer, J.; Fichtner, D.; Schirrmeier, H.; Polster, U.; Weiland, E.; Wege, H. Porcine reproductive and respiratory syndrome virus (PRRSV): Kinetics of infection in lymphatic organs and lung. J. Vet. Med. B 2000, 47, 9–25. [Google Scholar]
- Rowland, R.R.; Lawson, S.; Rossow, K.; Benfield, D.A. Lymphoid tissue tropism of porcine reproductive and respiratory syndrome virus replication during persistent infection of pigs originally exposed to virus in utero. Vet. Microbiol. 2003, 96, 219–235. [Google Scholar]
- Swenson, S.L.; Hill, H.T.; Zimmerman, J.J.; Evans, L.E.; Landgraf, J.G.; Wills, R.W.; Sanderson, T.P.; McGinley, M.J.; Brevik, A.K.; Ciszewski, D.K.; et al. Excretion of porcine reproductive and respiratory syndrome virus in semen after experimentally induced infection in boars. J. Am. Vet. Med. Assoc. 1994, 204, 1943–1948. [Google Scholar]
- Albina, E.; Piriou, L.; Hutet, E.; Cariolet, R.; L'Hospitalier, R. Immune responses in pigs infected with porcine reproductive and respiratory syndrome virus (PRRSV). Vet. Immunol. Immunopathol. 1998, 61, 49–66. [Google Scholar]
- Van Reeth, K.; Labarque, G.; Nauwynck, H.; Pensaert, M. Differential production of proinflammatory cytokines in the pig lung during different respiratory virus infections: Correlations with pathogenicity. Res. Vet. Sci. 1999, 67, 47–52. [Google Scholar]
- Buddaert, W.; Van Reeth, K.; Pensaert, M. In vivo and in vitro interferon (IFN) studies with the porcine reproductive and respiratory syndrome virus (PRRSV). Adv. Exp. Med. Biol. 1998, 440, 461–467. [Google Scholar] [CrossRef]
- Miller, L.C.; Laegreid, W.W.; Bono, J.L.; Chitko-McKown, C.G.; Fox, J.M. Interferon type I response in porcine reproductive and respiratory syndrome virus-infected MARC-145 cells. Arch. Virol. 2004, 149, 2453–2463. [Google Scholar]
- Luo, R.; Xiao, S.; Jiang, Y.; Jin, H.; Wang, D.; Liu, M.; Chen, H.; Fang, L. Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway. Mol. Immunol. 2008, 45, 2839–2846. [Google Scholar]
- Ait-Ali, T.; Wilson, A.D.; Carre, W.; Westcott, D.G.; Frossard, J.P.; Mellencamp, M.A.; Mouzaki, D.; Matika, O.; Waddington, D.; Drew, T.W.; et al. Host inhibits replication of European porcine reproductive and respiratory syndrome virus in macrophages by altering differential regulation of type-I interferon transcriptional response. Immunogenetics 2011, 63, 437–448. [Google Scholar] [CrossRef]
- Tohno, M.; Shimosato, T.; Moue, M.; Aso, H.; Watanabe, K.; Kawai, Y.; Yamaguchi, T.; Saito, T.; Kitazawa, H. Toll-like receptor 2 and 9 are expressed and functional in gut-associated lymphoid tissues of presuckling newborn swine. Vet. Res. 2006, 37, 791–812. [Google Scholar]
- Sang, Y.; Ross, C.R.; Rowland, R.R.; Blecha, F. Toll-like receptor 3 activation decreases porcine arterivirus infection. Viral. Immunol. 2008, 21, 303–313. [Google Scholar]
- Sang, Y.; Yang, J.; Ross, C.R.; Rowland, R.R.; Blecha, F. Molecular identification and functional expression of porcine Toll-like receptor (TLR) 3 and TLR7. Vet. Immunol. Immunopathol. 2008, 125, 162–167. [Google Scholar]
- Chaung, H.C.; Chen, C.W.; Hsieh, B.L.; Chung, W.B. Toll-Like Receptor expressions in porcine alveolar macrophages and Dendritic Cells in responding to poly IC stimulation and porcine reproductive and respiratory syndrome virus (PRRSV) infection. Comp. Immunol. Microbiol. Infect. Dis. 2010, 33, 197–213. [Google Scholar]
- Miller, L.C.; Lager, K.M.; Kehrli, M.E., Jr. Role of Toll-like receptors in activation of porcine alveolar macrophages by porcine reproductive and respiratory syndrome virus. Clin. Vaccine Immunol. 2009, 16, 360–365. [Google Scholar]
- Kim, O.; Sun, Y.; Lai, F.W.; Song, C.; Yoo, D. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology 2010, 402, 315–326. [Google Scholar]
- Li, H.; Zheng, Z.; Zhou, P.; Zhang, B.; Shi, Z.; Hu, Q.; Wang, H. The cysteine protease domain of porcine reproductive and respiratory syndrome virus non-structural protein 2 antagonizes interferon regulatory factor 3 activation. J. Gen. Virol. 2010, 91, 2947–2958. [Google Scholar]
- Nedialkova, D.D.; Ulferts, R.; van den Born, E.; Lauber, C.; Gorbalenya, A.E.; Ziebuhr, J.; Snijder, E.J. Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. J. Virol. 2009, 83, 5671–5682. [Google Scholar]
- Lee, S.M.; Kleiboeker, S.B. Porcine arterivirus activates the NF-kappaB pathway through IkappaB degradation. Virology 2005, 342, 47–59. [Google Scholar]
- Calzada-Nova, G.; Schnitzlein, W.M.; Husmann, R.J.; Zuckermann, F.A. North American porcine reproductive and respiratory syndrome viruses inhibit type I interferon production by plasmacytoid dendritic cells. J. Virol. 2011, 85, 2703–2713. [Google Scholar]
- Song, C.; Krell, P.; Yoo, D. Nonstructural protein 1alpha subunit-based inhibition of NF-kappaB activation and suppression of interferon-beta production by porcine reproductive and respiratory syndrome virus. Virology 2010, 407, 268–280. [Google Scholar]
- Han, M.; Du, Y.; Song, C.; Yoo, D. The regulatory role of the zinc finger motif of PRRSV Nsp1α protein for IFN modilation. University of Illinois at Urbana-Champaign: Urbana, IL, USA, 2011; Unpublished work. [Google Scholar]
- Frias-Staheli, N.; Giannakopoulos, N.V.; Kikkert, M.; Taylor, S.L.; Bridgen, A.; Paragas, J.; Richt, J.A.; Rowland, R.R.; Schmaljohn, C.S.; Lenschow, D.J.; et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2007, 2, 404–416. [Google Scholar] [CrossRef]
- Sun, Z.; Chen, Z.; Lawson, S.R.; Fang, Y. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 2010, 84, 7832–7846. [Google Scholar]
- Patel, D.; Nan, Y.; Shen, M.; Ritthipichai, K.; Zhu, X.; Zhang, Y.J. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J. Virol. 2010, 84, 11045–11055. [Google Scholar]
- Chen, Z.; Lawson, S.; Sun, Z.; Zhou, X.; Guan, X.; Christopher-Hennings, J.; Nelson, E.A.; Fang, Y. Identification of two auto-cleavage products of nonstructural protein 1 (nsp1) in porcine reproductive and respiratory syndrome virus infected cells: nsp1 function as interferon antagonist. Virology 2010, 398, 87–97. [Google Scholar]
- Song, C.; Du, Y.; Kim, O.; Liu, H. C.; Yoo, D. Interaction of PRRSV Nsp1α and protein inhibitor of activated STAT1 (PIAS1) mediated sumolation of Nsp1α. University of Illinois at Urbana-Champaign: Urbana, IL, USA, 2010; Unpublished work. [Google Scholar]
- Rowland, R.R.; Robinson, B.; Stefanick, J.; Kim, T.S.; Guanghua, L.; Lawson, S.R.; Benfield, D.A. Inhibition of porcine reproductive and respiratory syndrome virus by interferon-gamma and recovery of virus replication with 2-aminopurine. Arch. Virol. 2001, 146, 539–555. [Google Scholar]
- Suarez, P.; Diaz-Guerra, M.; Prieto, C.; Esteban, M.; Castro, J.M.; Nieto, A.; Ortin, J. Open reading frame 5 of porcine reproductive and respiratory syndrome virus as a cause of virus-induced apoptosis. J. Virol. 1996, 70, 2876–2882. [Google Scholar]
- Sirinarumitr, T.; Zhang, Y.; Kluge, J.P.; Halbur, P.G.; Paul, P.S. A pneumo-virulent United States isolate of porcine reproductive and respiratory syndrome virus induces apoptosis in bystander cells both in vitro and in vivo. J. Gen. Virol. 1998, 79, 2989–2995. [Google Scholar]
- Sur, J.H.; Doster, A.R.; Osorio, F.A. Apoptosis induced in vivo during acute infection by porcine reproductive and respiratory syndrome virus. Vet. Pathol. 1998, 35, 506–514. [Google Scholar]
- Fernandez, A.; Suarez, P.; Castro, J.M.; Tabares, E.; Diaz-Guerra, M. Characterization of regions in the GP5 protein of porcine reproductive and respiratory syndrome virus required to induce apoptotic cell death. Virus Res. 2002, 83, 103–118. [Google Scholar]
- Miller, L.C.; Fox, J.M. Apoptosis and porcine reproductive and respiratory syndrome virus. Vet. Immunol. Immunopathol. 2004, 102, 131–142. [Google Scholar]
- Costers, S.; Lefebvre, D.J.; Delputte, P.L.; Nauwynck, H.J. Porcine reproductive and respiratory syndrome virus modulates apoptosis during replication in alveolar macrophages. Arch. Virol. 2008, 153, 1453–1465. [Google Scholar]
- Wang, X.; Eaton, M.; Mayer, M.; Li, H.; He, D.; Nelson, E.; Christopher-Hennings, J. Porcine reproductive and respiratory syndrome virus productively infects monocyte-derived dendritic cells and compromises their antigen-presenting ability. Arch. Virol. 2007, 152, 289–303. [Google Scholar]
- Lei, Y.; Moore, C.B.; Liesman, R.M.; O'Connor, B.P.; Bergstralh, D.T.; Chen, Z.J.; Pickles, R.J.; Ting, J.P. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One 2009, 4, e5466. [Google Scholar]
- Lee, S.M.; Schommer, S.K.; Kleiboeker, S.B. Porcine reproductive and respiratory syndrome virus field isolates differ in in vitro interferon phenotypes. Vet. Immunol. Immunopathol. 2004, 102, 217–231. [Google Scholar] [CrossRef]
- He, D.; Overend, C.; Ambrogio, J.; Maganti, R.J.; Grubman, M.J.; Garmendia, A.E. Marked differences between MARC-145 cells and swine alveolar macrophages in IFNbeta-induced activation of antiviral state against PRRSV. Vet. Immunol. Immunopathol. 2011, 139, 57–60. [Google Scholar]
- Ait-Ali, T.; Wilson, A.D.; Westcott, D.G.; Clapperton, M.; Waterfall, M.; Mellencamp, M.A.; Drew, T.W.; Bishop, S.C.; Archibald, A.L. Innate immune responses to replication of porcine reproductive and respiratory syndrome virus in isolated Swine alveolar macrophages. Viral. Immunol. 2007, 20, 105–118. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, Y.; Han, M.; Kim, C.; Calvert, J.G.; Yoo, D. Interplay between Interferon-Mediated Innate Immunity and Porcine Reproductive and Respiratory Syndrome Virus. Viruses 2012, 4, 424-446. https://doi.org/10.3390/v4040424
Sun Y, Han M, Kim C, Calvert JG, Yoo D. Interplay between Interferon-Mediated Innate Immunity and Porcine Reproductive and Respiratory Syndrome Virus. Viruses. 2012; 4(4):424-446. https://doi.org/10.3390/v4040424
Chicago/Turabian StyleSun, Yan, Mingyuan Han, Chiyong Kim, Jay G. Calvert, and Dongwan Yoo. 2012. "Interplay between Interferon-Mediated Innate Immunity and Porcine Reproductive and Respiratory Syndrome Virus" Viruses 4, no. 4: 424-446. https://doi.org/10.3390/v4040424
APA StyleSun, Y., Han, M., Kim, C., Calvert, J. G., & Yoo, D. (2012). Interplay between Interferon-Mediated Innate Immunity and Porcine Reproductive and Respiratory Syndrome Virus. Viruses, 4(4), 424-446. https://doi.org/10.3390/v4040424