Host Cell Factors as Antiviral Targets in Arenavirus Infection

 ,
, 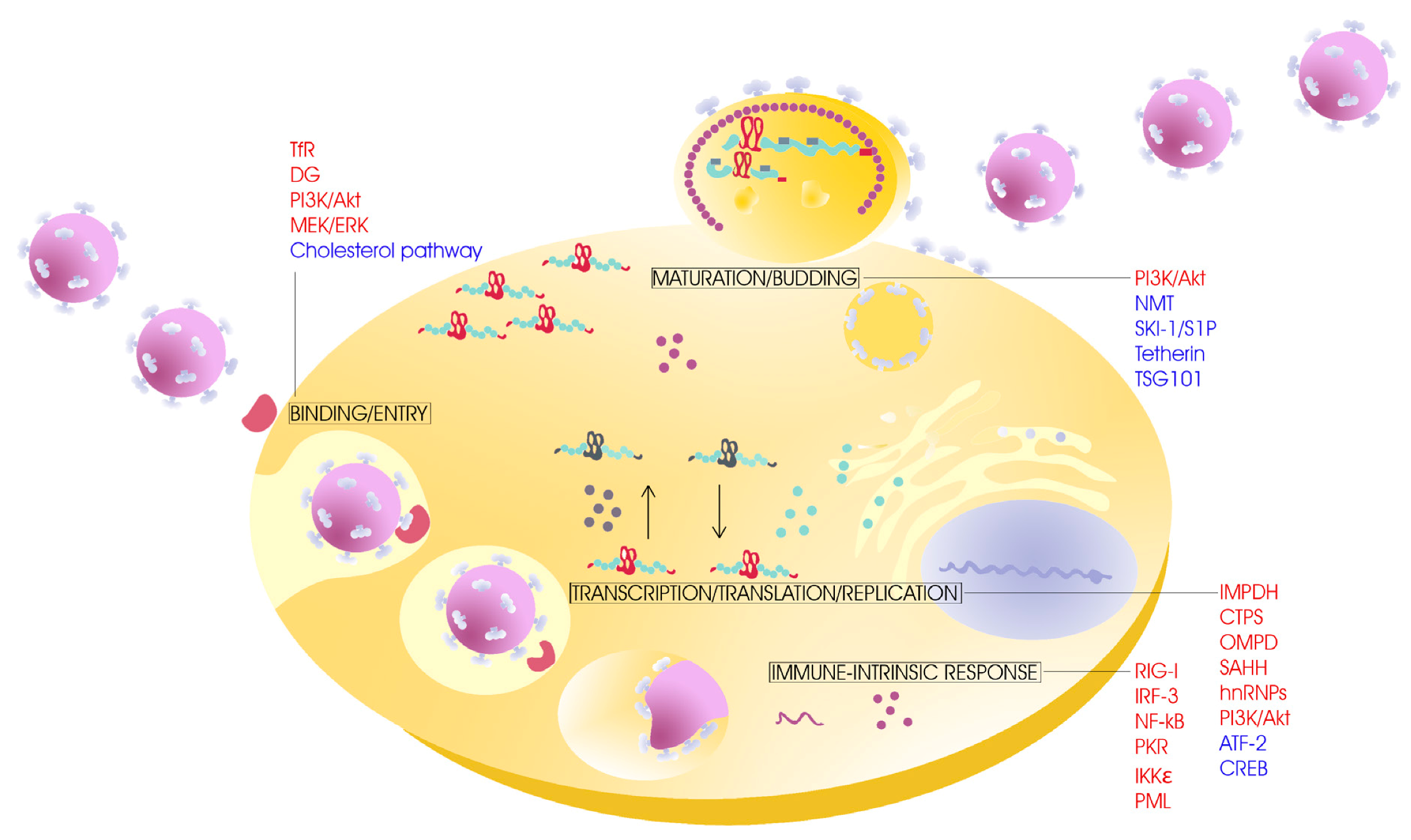{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Targeting Host Cell Factors: Advantages and Disadvantages

3. Cellular Proteins Involved in RNA Synthesis or Processing
3.1. Metabolism of Nucleosides
3.2. Heterogeneous Nuclear Ribonucleoproteins
4. Host Kinases
4.1. Kinases and Signaling Pathways
4.2. Kinases and Interferon
5. Intrinsic Antiviral Resistance

6. Conclusions and Future Perspectives
Acknowledgments
Conflict of Interest
References
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immunol. 2002, 262, 75–109. [Google Scholar]
- Enria, D.A.; Briggiler, A.M.; Sánchez, Z. Treatment of Argentine hemorrhagic fever. Antivir. Res. 2008, 78, 132–139. [Google Scholar] [CrossRef]
- Macher, A.M.; Wolfe, M.S. Historical Lassa fever reports and 30-year clinical update. Emerg. Infect. Dis. 2006, 12, 835–836. [Google Scholar] [CrossRef]
- Jamieson, D.J.; Kourtis, A.P.; Bell, M.; Rasmussen, S.A. Lymphocytic choriomeningitis virus: An emerging obstetric pathogen? Am. J. Obstet. Gynecol. 2006, 194, 1532–1536. [Google Scholar] [CrossRef]
- Fisher, S.A.; Graham, M.B.; Kuehnert, M.J.; Kotton, C.N.; Srinivasan, A.; Marty, F.M.; Comer, J.A.; Guarner, J.; Paddock, C.D.; DeMeo, D.L.; et al. LCMV in Transplant recipients investigation team. Transmission of lymphocytic choriomeningiris virus by organ transplantation. N. Engl. J. Med. 2006, 354, 2235–2249. [Google Scholar] [CrossRef]
- Charrel, R.N.; de Lamballerie, X. Zoonotic aspects of arenavirus infections. Vet. Microbiol. 2010, 140, 213–220. [Google Scholar] [CrossRef]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliot, L.H.; Belmont-Williams, R. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar]
- Peters, C.J. Human infection with arenaviruses in the Americas. Curr. Top. Microbiol. Immunol. 2002, 262, 65–74. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Ghorie, S.; Parker, L.; Huggins, J. Unexpected adverse reactions during a clinical trial in rural West Africa. Antivir. Res. 1992, 19, 139–147. [Google Scholar]
- Enria, D.A.; Maiztegui, J.I. Antiviral treatment of Argentine hemorrhagic fever. Antivir. Res. 1994, 23, 23–31. [Google Scholar]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 257–276. [Google Scholar]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Coley, W.; Kehn-Hall, K.; van Duyne, R.; Kashanchi, F. Novel HIV-1 therapeutics through targeting altered host cell pathways. Expert Opin. Biol. Ther. 2009, 9, 1369–1382. [Google Scholar] [CrossRef]
- Khattab, M.A. Targeting host factors: A novel rationale for the management of hepatitis C virus. World J. Gastroenterol. 2009, 15, 3472–3479. [Google Scholar] [CrossRef]
- Krumm, S.A.; Ndungu, J.M.; Yoon, J.J.; Dochow, M.; Sun, A.; Natchus, M.; Snyder, J.P.; Plemper, R.K. Potent host-directed small-molecule inhibitors of myxovirus RNA-dependent RNA-polymerases. PLoS One 2011, 6, e20069. [Google Scholar]
- Pastorino, B.; Nougairede, A.; Wurtz, N.; Gould, E.; de Lamballerie, X. Role of host cell factors in flavivirus infection: Implications for pathogenesis and development of antiviral drugs. Antivir. Res. 2010, 87, 281–294. [Google Scholar]
- Albariño, C.G.; Bergeron, E.; Erickson, B.R.; Khristova, M.L.; Rollin, P.E.; Nichol, S.T. Efficient reverse genetics generation of infectious junin viruses differing in glycoprotein processing. J. Virol. 2009, 83, 5606–5614. [Google Scholar] [CrossRef]
- Emonet, S.F.; Seregin, A.V.; Yun, N.E.; Poussard, A.L.; Walker, A.G.; de la Torre, J.C.; Paessler, S. Rescue from cloned cDNAs and in vivo characterization of recombinant pathogenic Romero and live-attenuated Candid #1 strains of Junin virus, the causative agent of Argentine hemorrhagic fever disease. J. Virol. 2011, 85, 1473–1483. [Google Scholar] [CrossRef]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J. Virol. 2011, 85, 12093–12097. [Google Scholar]
- Larson, R.A.; Dai, D.; Hosack, V.T.; Tan, Y.; Bolken, T.C.; Hruby, D.E.; Amberg, S.M. Identification of a broad-spectrum arenavirus entry inhibitor. J. Virol. 2008, 82, 10768–10775. [Google Scholar]
- Lee, A.M.; Rojek, J.M.; Spiropoulou, C.F.; Gundersen, A.T.; Jin, W.; Shaginian, A.; York, J.; Nunberg, J.H.; Boger, D.L.; Oldstone, M.B.; et al. Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J. Biol. Chem. 2008, 283, 18734–18742. [Google Scholar]
- Capul, A.A.; de la Torre, J.C. A cell-based luciferase assay amenable to high-throughput screening of arenavirus budding. Virology 2008, 382, 107–114. [Google Scholar] [CrossRef]
- Friedel, C.C.; Jurgen, H. Virus-Host interactomes and global models of virus-infected cells. Trends Microbiol. 2011, 19, 501–508. [Google Scholar] [CrossRef]
- Shaw, M.L. The host interactome of influenza virus presents new potential targets for antiviral drugs. Res. Med. Virol. 2011, 21, 358–369. [Google Scholar] [CrossRef]
- Tafforeau, L.; Rabourdin-Combe, C.; Lotteau, V. Virus-Human cell interactomes. Methods Mol. Biol. 2012, 812, 103–120. [Google Scholar] [CrossRef]
- Djavani, M.; Crasta, O.R.; Zhang, Y.; Zapata, J.C.; Sobral, B.; Lechner, M.G.; Bryant, J.; Davis, H.; Salvato, M.S. Gene expression in primate liver during viral hemorrhagic fever. Virol. J. 2009, 6, 20:1–20:18. [Google Scholar]
- Müller, S.; Geffers, R.; Günther, S. Analysis of gene expression in Lassa virus-infected HuH-7 cells. J. Gen. Virol. 2007, 88, 1568–1575. [Google Scholar] [CrossRef]
- Bowick, G.C.; Fennewald, S.M.; Elsom, B.L.; Aronson, J.F.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Differential signaling networks induced by mild and lethal hemorrhagic fever virus infections. J. Virol. 2006, 80, 10248–10252. [Google Scholar] [CrossRef]
- Bowick, G.C.; Fennewald, S.M.; Scott, E.P.; Zhang, L.; Elsom, B.L.; Aronson, J.F.; Spratt, H.M.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Identification of differentially activated cell-signaling networks associated with Pichinde virus pathogenesis by using systems kinomics. J. Virol. 2007, 81, 1923–1933. [Google Scholar]
- García, C.C.; Sepúlveda, C.S.; Damonte, E.B. Novel therapeutic targets for arenavirus hemorrhagic fevers. Future Virol. 2011, 6, 27–44. [Google Scholar] [CrossRef]
- Emonet, S.E.; Urata, S.; de la Torre, J.C. Arenavirus reverse genetics: New approaches for the investigation of arenavirus biology and development of antiviral strategies. Virology 2011, 411, 416–425. [Google Scholar] [CrossRef]
- Lee, A.M.; Pasquato, A.; Kunz, S. Novel approaches in anti-arenaviral drug development. Virology 2011, 411, 163–169. [Google Scholar] [CrossRef]
- Rojek, J.M.; Kunz, S. Cell entry by human pathogenic arenaviruses. Cell. Microbiol. 2008, 10, 828–835. [Google Scholar] [CrossRef]
- Streeter, D.G.; Witkowski, J.T.; Khare, G.P.; Sidwell, R.W.; Bauer, R.J.; Robins, R.K.; Simon, L.N. Mechanism of action of 1-ß-D-ribofuranosyl-1,2,4-triazole-3- carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc. Natl. Acad. Sci. USA 1973, 70, 1174–1178. [Google Scholar] [CrossRef]
- Leyssen, P.; Balzarini, J.; de Clercq, E.; Neyts, J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 2005, 79, 1943–1947. [Google Scholar] [CrossRef]
- Graci, J.D.; Cameron, C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006, 16, 37–48. [Google Scholar] [CrossRef]
- Leyssen, P.; de Clercq, E.; Neyts, J. Molecular strategies to inhibit the replication of RNA viruses. Antivir. Res. 2008, 78, 9–25. [Google Scholar]
- Ölschläger, S.; Neyts, J.; Günther, S. Depletion of GTP pool is not the predominant mechanism by which ribavirin exerts its antiviral effect on Lassa virus. Antivir. Res. 2011, 91, 89–93. [Google Scholar]
- Sepúlveda, C.S.; García, C.C.; Fascio, M.L.; D’Accorso, N.B.; Docampo Palacios, M.L.; Pellón R, F.; Damonte, E.B. Inhibition of Junín virus RNA synthesis by an antiviral acridone derivative. Antivir. Res. 2012, 96, 16–22. [Google Scholar]
- Ruiz-Jarabo, C.M.; Ly, C.; Domingo, E.; de la Torre, J.C. Lethal mutagenesis of the prototype arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 2003, 308, 37–47. [Google Scholar] [CrossRef]
- Moreno, H.; Gallego, I.; Sevilla, N.; de la Torre, J.C.; Domingo, E.; Martín, V. Ribavirin can be mutagenic for arenaviruses. J. Virol. 2011, 85, 7246–7255. [Google Scholar] [CrossRef]
- Andrei, G.; de Clercq, E. Molecular approaches for the treatment of hemorrhagic fever virus infections. Antivir. Res. 1993, 22, 45–75. [Google Scholar] [CrossRef]
- Minakawa, N.; Takeda, T.; Sasaki, T.; Matsuda, A.; Ueda, T. Nucleotides and nucleosides. 96. Synthesis and antitumor activity of 5-ethynyl-1-β-D-ribofuranosylimidazole-4-carboxamide (EICAR) and its derivatives. J. Med. Chem. 1991, 34, 778–786. [Google Scholar] [CrossRef]
- Sintchak, M.D.; Nimmesgern, E. The structure of inosine 5X-monophosphate dehydrogenase and the design of novel inhibitors. Inmunopharmacology 2000, 47, 163–184. [Google Scholar] [CrossRef]
- Goodell, J.R.; Madhok, A.A.; Hiasa, H.; Ferguson, D.M. Synthesis and evaluation of acridone- and acridone-based anti-herpes agents with topoisomerase activity. Bioorg. Med. Chem. 2006, 14, 5467–5480. [Google Scholar]
- Lowden, C.T.; Bastow, K.F. Cell culture replication of herpes simplex virus and, or human cytomegalovirus is inhibited by 3,7-dialkoxylated, 1-hydroxyacridone derivatives. Antivir. Res. 2003, 59, 143–154. [Google Scholar]
- Itoigawa, M.; Ito, C.; Wu, T.S.; Enjo, F.; Tokuda, H.; Nishino, H.; Furukawa, H. Cancer chemopreventive activity of acridone alkaloids on Epstein-Barr virus activation and two-stage mouse skin carcinogenesis. Cancer Lett. 2003, 193, 133–138. [Google Scholar] [CrossRef]
- Zarubaev, V.V.; Slita, A.V.; Krivitskaya, V.Z.; Sirotkin, A.K.; Kovalenko, A.L.; Chatterjee, N.K. Direct antiviral effect of cycloferon (10-carboxymethyl-9-acridanone) against adenovirus type 6 in vitro. Antivir. Res. 2003, 58, 131–137. [Google Scholar]
- Fujiwara, M.; Okamoto, M.; Okamoto, M.; Watanabe, M.; Machida, H.; Shigeta, S.; Konno, K.; Yokota, T.; Baba, M. Acridone derivatives are selective inhibitors of HIV-1 replication in chronically infected cells. Antivir. Res. 1999, 43, 179–189. [Google Scholar]
- Turpin, J.A.; Buckheit, R.W., Jr.; Derse, D.; Hollingshead, M.; Williamson, K.; Palamone, C.; Osterling, M.C.; Hill, S.A.; Graham, L.; Schaeffer, C.A.; et al. Inhibition of acute-, latent-, and chronic-phase human immunodeficiency virus type 1 (HIV-1) replication by a bistriazoloacridone analog that selectively inhibits HIV-1 transcription. Antimicrob. Agents Chemother. 1998, 42, 487–494. [Google Scholar]
- Tabarrini, O.; Manfroni, G.; Fravolini, A.; Cecchetti, V.; Sabatini, S.; de Clercq, E.; Rozenski, J.; Canard, B.; Dutartre, H.; Paeshuyse, J.; et al. Synthesis and anti-BVDV activity of acridones as new potential antiviral agents. J. Med. Chem. 2006, 49, 2621–2627. [Google Scholar]
- Manfroni, G.; Paeshuyse, J.; Massari, S.; Zanoli, S.; Gatto, B.; Maga, G.; Tabarrini, O.; Cecchetti, V.; Fravolini, A.; Neyts, J. Inhibition of subgenomic hepatitis C virus RNA replication by acridone derivatives: Identification of an NS3 helicase inhibitor. J. Med. Chem. 2009, 52, 3354–3365. [Google Scholar]
- Stankiewicz-Drogon, A.; Dörner, B.; Erker, T.; Boguszewska-Chachulska, A.M. Synthesis of new acridone derivatives, inhibitors of NS3 helicase, which efficiently and specifically inhibit subgenomic HCV replication. J. Med. Chem. 2010, 53, 3117–3126. [Google Scholar] [CrossRef]
- Vispé, S.; Vandenberghe, I.; Robin, M.; Annereau, J.P.; Créancier, L.; Pique, V.; Galy, J.P.; Kruczynski, A.; Barret, J.M.; Bailly, C. Novel tetra-acridine derivatives as dual inhibitors of topoisomerase II and the human proteasome. Biochem. Pharmacol. 2007, 73, 1863–1872. [Google Scholar] [CrossRef]
- Watterson, S.H.; Chen, P.; Zhao, Y.; Gu, H.H.; Dhar, T.G.; Xiao, Z.; Ballentine, S.K.; Shen, Z.; Fleener, C.A.; Rouleau, K.A.; et al. Acridone-Based inhibitors of inosine 5’-monophosphate dehydrogenase: Discovery and SAR leading to the identification of N-(2-(6-(4-ethylpiperazin-1-yl)pyridin-3-yl)propan-2-yl)-2-fluoro-9-oxo-9,10-dihydroxyacridine-3-carboxamide (BMS-566419). J. Med. Chem. 2007, 50, 3730–3742. [Google Scholar] [CrossRef]
- Adams, A. Crystal structures of acridines complexed with nucleic acids. Curr. Med. Chem. 2002, 9, 1667–1675. [Google Scholar]
- Sepúlveda, C.S.; Fascio, M.L.; Mazzucco, M.B.; Palacios, M.L.; Pellón, R.F.; García, C.C.; D’Accorso, N.B.; Damonte, E.B. Synthesis and evaluation of N-substituted acridones as antiviral agents against hemorrhagic fever viruses. Antivir. Chem. Chemother. 2008, 19, 41–47. [Google Scholar]
- Gowen, B.B.; Wong, M.H.; Larson, D.; Ye, W.; Jung, K.H.; Sefing, E.J.; Skirpstunas, R.; Smee, D.F.; Morrey, J.D.; Schneller, S.W. Development of a new Tacaribe arenavirus infection model and its use to explore antiviral activity of a novel aristeromycin analog. PLoS One 2010, 16, e12760. [Google Scholar]
- Guillerm, G.; Guillerm, D.; Vandenplas-Vitkowski, C.; Glapski, C.; de Clercq, E. Inactivation of S-adenosyl-L-homocysteine hydrolase with novel 5’-thioadenosine derivatives. Antiviral effects. Bioorg. Med. Chem. Lett. 2003, 13, 1649–1652. [Google Scholar]
- Venables, J.P.; Koh, C.S.; Froehlich, U.; Lapointe, E.; Couture, S.; Inkel, L.; Bramard, A.; Paquet, E.R.; Watier, V.; Durand, M.; et al. Multiple and specific mRNA processing targets for the major human hnRNP proteins. Mol. Cell Biol. 2008, 28, 6033–6043. [Google Scholar] [CrossRef]
- He, Y.; Smith, R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell. Mol. Life Sci. 2009, 66, 1239–1256. [Google Scholar] [CrossRef]
- Han, S.P.; Tang, Y.H.; Smith, R. Functional diversity of the hnRNPs: Past, present and perspectives. Biochem. J. 2010, 430, 379–392. [Google Scholar] [CrossRef]
- Castilla, V.; Scolaro, L.A. Involvement of heterogeneous nuclear ribonucleoproteins in viral multiplication. Future Virol. 2012, 7, 575–591. [Google Scholar] [CrossRef]
- Katoh, H.; Mori, Y.; Kambara, H.; Abe, T.; Fukuhara, T.; Morita, E.; Moriishi, K.; Kamitani, W.; Matsuura, Y. Heterogeneous nuclear ribonucleoprotein A2 participates in the replication of Japanese encephalitis virus through an interaction with viral proteins and RNA. J. Virol. 2011, 85, 10976–10988. [Google Scholar] [CrossRef]
- Shih, S.R.; Stollar, V.; Li, M.L. Host factors in enterovirus 71 replication. J. Virol. 2011, 85, 9658–9666. [Google Scholar]
- Monette, A.; Ajamian, L.; López-Lastra, M.; Mouland, A.J. Human immunodeficiency virus type 1 (HIV-1) induces the cytoplasmic retention of heterogeneous nuclear ribonucleoprotein A1 by disrupting nuclear import: Implications for HIV-1 gene expression. J. Biol. Chem. 2009, 284, 31350–31362. [Google Scholar]
- Shabman, R.S.; Gulcicek, E.E.; Stone, K.L.; Basler, C.F. The Ebola virus VP24 protein prevents hnRNP C1/C2 binding to karyopherin α1 and partially alters its nuclear import. J. Infect. Dis. 2011, 204, S904–S910. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Hesse, R.A.; Rhoderick, J.B.; Elwell, M.A.; Moe, J.B. Pathogenesis of a Pichinde virus strain adapted to produce lethal infections in guinea pigs. Infect. Immun. 1981, 32, 872–880. [Google Scholar]
- Bowick, G.C.; Spratt, H.M.; Hogg, A.E.; Endsley, J.J.; Wiktorowicz, J.E.; Kurosky, A.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Analysis of the differential host cell nuclear proteome induced by attenuated and virulent hemorrhagic arenavirus infection. J. Virol. 2009, 83, 687–700. [Google Scholar]
- Lukashevich, I.S.; Djavani, M.; Rodas, J.D.; Zapata, J.C.; Usborne, A.; Emerson, C.; Mitchen, J.; Jahrling, P.B.; Salvato, M.S. Hemorrhagic fever occurs after intravenous, but not after intragastric, inoculation of rhesus macaques with lymphocytic choriomeningitis virus. J. Med. Virol. 2002, 67, 171–186. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Rodas, J.D.; Tikhonov, I.I.; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. LCMV-mediated hepatitis in rhesus macaques: WE but not ARM strain activates hepatocytes and induces liver regeneration. Arch. Virol. 2004, 149, 2319–2336. [Google Scholar] [CrossRef]
- Djavani, M.M.; Crasta, O.R.; Zapata, J.C.; Fei, Z.; Folkerts, O.; Sobral, B.; Swindells, M.; Bryant, J.; et al. Early blood profiles of virus infection in a monkey model for Lassa fever. J. Virol. 2007, 81, 7960–7973. [Google Scholar] [CrossRef]
- Maeto, C.A.; Knott, M.E.; Linero, F.N.; Ellenberg, P.C.; Scolaro, L.A.; Castilla, V. Differential effect of acute and persistent Junin virus infections on the nucleo-cytoplasmic trafficking and expression of heterogeneous nuclear ribonucleoproteins type A and B. J. Gen. Virol. 2011, 92, 2181–2190. [Google Scholar] [CrossRef]
- Ellenberg, P.; Edreira, M.; Lozano, M.; Scolaro, L. Synthesis and expression of viral antigens in Vero cells persistently infected with Junin virus. Arch. Virol. 2002, 147, 1543–1557. [Google Scholar] [CrossRef]
- Ellenberg, P.; Edreira, M.; Scolaro, L. Resistance to superinfection of Vero cells persistently infected with Junin virus. Arch. Virol. 2004, 149, 507–522. [Google Scholar] [CrossRef]
- Keating, J.A.; Striker, R. Phosphorylation events during viral infections provide potential therapeutic targets. Rev. Med. Virol. 2012, 22, 166–181. [Google Scholar] [CrossRef]
- Dissmeyer, N.; Schnittger, A. The age of protein kinases. Methods Mol. Biol. 2011, 779, 7–52. [Google Scholar] [CrossRef]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef]
- Cooray, S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 2004, 85, 1065–1076. [Google Scholar] [CrossRef]
- Ji, W.T.; Liu, H.J. PI3K-Akt signaling and viral infection. Recent. Pat. Biotechnol. 2008, 2, 218–226. [Google Scholar] [CrossRef]
- McCormick, F. Cancer: Survival pathways meet their end. Nature 2004, 428, 267–269. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 181, 31978–31986. [Google Scholar]
- Linero, F.N.; Scolaro, L.A. Participation of the phosphatidylinositol 3-kinase/Akt pathway in Junín virus replication in vitro. Virus Res. 2009, 145, 166–170. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World hemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef]
- Ruggiero, T.; Trabucchi, M.; Ponassi, M.; Corte, G.; Chen, C.Y.; al-Haj, L.; Khabar, K.S.; Briata, P.; Gherzi, R. Identification of a set of KSRP target transcripts upregulated by PI3K-AKT signaling. BMC Mol. Biol. 2007, 8, 28:1–28:15. [Google Scholar]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.Y.; Kunz, S. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog. 2011, 7, e1002232. [Google Scholar] [CrossRef]
- Urata, S.; Ngo, N.; de la Torre, J.C. The PI3K/Akt pathway contributes to arenavirus budding. J. Virol. 2012, 86, 4578–4585. [Google Scholar] [CrossRef]
- Sullivan, J.A.; Kim, E.H.; Plisch, E.H.; Peng, S.L.; Suresh, M. FOXO3 regulates CD8 T cell memory by T cell-intrinsic mechanisms. PLoS Pathog. 2012, 8, e1002533. [Google Scholar] [CrossRef]
- Pleschka, S. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 2008, 389, 1273–1282. [Google Scholar] [CrossRef]
- Vela, E.M.; Bowick, G.C.; Herzog, N.K.; Aronson, J.F. Genistein treatment of cells inhibits arenavirus infection. Antivir. Res. 2008, 77, 153–156. [Google Scholar]
- Rojek, J.M.; Moraz, M.L.; Pythoud, C.; Rothenberger, S.; van der Goot, F.G.; Campbell, K.P.; Kunz, S. Binding of Lassa virus perturbs extracellular matrix-induced signal transduction via dystroglycan. Cell. Microbiol. 2012, 14, 1122–1134. [Google Scholar] [CrossRef]
- Vela, E.M.; Bowick, G.C.; Herzog, N.K.; Aronson, J.F. Exploring kinase inhibitors as therapies for human arenavirus Infections. Future Virol. 2008, 3, 243–251. [Google Scholar] [CrossRef]
- Castilla, V.; Merisch, S. Low pH-induced fusion of Vero cells infected with Junin virus. Arch. Virol. 1996, 141, 1307–1317. [Google Scholar] [CrossRef]
- Di Simone, C.; Zandonatti, M.; Buchmeier, M. Acidic pH triggers LCMV membrane fusion activity and conformational change in the glycoprotein spike. Virology 1994, 198, 455–465. [Google Scholar] [CrossRef]
- Di Simone, C.; Buchmeier, M. Kinetics and pH dependence of acid-induced structural changes in the Lymphoytic choriomeningitis virus glycoprotein complex. Virology 1995, 209, 3–9. [Google Scholar] [CrossRef]
- Borrow, P.; Oldstone, M.B. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology 1994, 198, 1–9. [Google Scholar] [CrossRef]
- Shah, W.A.; Peng, H.; Carbonetto, S. Role of non-raft cholesterol in lymphocytic choriomeningitis virus infection via {alpha}-dystroglycan. J. Gen. Virol. 2006, 87, 673–678. [Google Scholar] [CrossRef]
- Quirin, K.; Eschli, B.; Scheu, I.; Poort, L.; Kartenbeck, J.; Helenius, A. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology 2008, 378, 21–33. [Google Scholar] [CrossRef]
- Rojek, J.M.; Perez, M.; Kunz, S. Cellular entry of lymphocytic choriomeningitis virus. J. Virol. 2008, 82, 1505–1517. [Google Scholar] [CrossRef]
- Rojek, J.M.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.C.; Kunz, S. Different mechanisms of cell entry by human-pathogenic Old World and New World arenaviruses. J. Virol. 2008, 82, 7677–7687. [Google Scholar] [CrossRef]
- Martinez, M.G.; Cordo, S.M.; Candurra, N.A. Characterization of JUNV arenavirus cell entry. J. Gen. Virol. 2007, 88, 1776–1784. [Google Scholar] [CrossRef]
- Vela, E.M.; Colpitts, T.M.; Zhang, L.; Davey, R.A.; Aronson, J.F. Pichinde virus is trafficked through a dynamin 2 endocytic pathway that is dependent on cellular Rab5- and Rab7-mediated endosomes. Arch. Virol. 2008, 153, 1391–1396. [Google Scholar] [CrossRef]
- Martínez, M.G.; Forlenza, M.B.; Candurra, N.A. Involvement of cellular proteins in Junin arenavirus entry. Martinez MG, Forlenza MB, Candurra NA. Biotechnol. J. 2009, 4, 866–870. [Google Scholar] [CrossRef]
- Lee, A.M.; Pasquato, A.; Kunz, S. Novel approaches in anti-arenaviral drug development. Virology 2011, 411, 163–169. [Google Scholar] [CrossRef]
- Kolokoltsov, A.A.; Adhikary, S.; Garver, J.; Johnson, L.; Davey, R.A.; Vela, E.M. Inhibition of Lassa virus and Ebola virus infection in host cells treated with the kinase inhibitors genistein and tyrphostin. Arch. Virol. 2012, 157, 121–712. [Google Scholar] [CrossRef]
- Thomsen, A.R.; Nansen, A.; Andreasen, S.O.; Wodarz, D.; Christensen, J.P. Host factors influencing viral persistence. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2000, 355, 1031–1041. [Google Scholar] [CrossRef]
- Zuniga, E.I.; Hahm, B.; Oldstone, M.B. Type I interferon during viral infections: Multiple triggers for a multifunctional mediator. Curr. Top. Microbiol. Immunol. 2007, 316, 337–357. [Google Scholar]
- Borrow, P.; Martínez-Sobrido, L.; de la Torre, J.C. Inhibition of the type I interferon antiviral response during arenavirus infection. Viruses 2010, 2, 2443–2480. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Lavanya, M.; Wang, E.; Ross, S.R. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef]
- Baize, S.; Pannetier, D.; Faure, C.; Marianneau, P.; Marendat, I.; Georges-Courbot, M.C.; Deubel, V. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microbes Infect. 2006, 8, 1194–1202. [Google Scholar] [CrossRef]
- Martínez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; García-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Martínez-Sobrido, L.; Giannakas, P.; Cubitt, B.; García-Sastre, A.; de la Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKK{varepsilon}. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef]
- Rodrigo, W.W.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J. Virol. 2012, 86, 8185–8197. [Google Scholar]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef]
- Pindel, A.; Sadler, A. The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 2011, 31, 59–70. [Google Scholar] [CrossRef]
- Montero, H.; Trujillo-Alonso, V. Stress granules in the viral replication cycle. Viruses 2011, 3, 2328–2338. [Google Scholar] [CrossRef]
- Linero, F.N.; Thomas, M.G.; Boccaccio, G.L.; Scolaro, L.A. Junin virus infection impairs stress-granule formation in Vero cells treated with arsenite via inhibition of eIF2 alpha phosphorylation. J. Gen. Virol. 2011, 92, 2889–2899. [Google Scholar] [CrossRef]
- Brostrom, C.O.; Prostko, C.R.; Kaufman, R.J.; Brostrom, M.A. Inhibition of translational initiation by activators of the glucose-regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor alpha kinase. J. Biol. Chem. 1996, 271, 24995–25002. [Google Scholar]
- Cuevas, C.D.; Lavanya, M.; Wang, E.; Ross, S.R. Junin virus infects mouse cells and induces innate immune responses. J Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar] [CrossRef]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef]
- Zhou, S.; Cerny, A.M.; Bowen, G.; Chan, M.; Knipe, D.M.; Kurt-Jones, E.A.; Finberg, R.W. Discovery of a novel TLR2 signaling inhibitor with anti-viral activity. Antivir. Res. 2010, 87, 295–306. [Google Scholar]
- Hayes, M.W.; Carrion, R., Jr.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic Old World arenaviruses inhibit TLR2/Mal-dependent proinflammatory cytokines in vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef]
- Bieniasz, P.D. Intrinsic immunity: A front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef]
- Tavalai, N.; Stamminger, T. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta. 2008, 1783, 2207–2221. [Google Scholar] [CrossRef]
- Wolf, D.; Goff, S.P. Host restriction factors blocking retroviral replication. Annu. Rev. Genet. 2008, 42, 143–163. [Google Scholar] [CrossRef]
- Hinson, E.R.; Joshi, N.S.; Chen, J.H.; Rahner, C.; Jung, Y.W.; Wang, X.; Kaech, S.M.; Cresswell, P. Viperin is highly induced in neutrophils and macrophages during acute and chronic lymphocytic choriomeningitis virus infection. J. Immunol. 2010, 184, 5723–5731. [Google Scholar] [CrossRef]
- Sakuma, T.; Sakurai, A.; Yasuda, J. Dimerization of tetherin is not essential for its antiviral activity against Lassa and Marburg viruses. PLoS One 2009, 4, e6934. [Google Scholar]
- Radoshitzky, S.R.; Dong, L.; Chi, X.; Clester, J.C.; Retterer, C.; Spurgers, K.; Kuhn, J.H.; Sandwick, S.; Ruthel, G.; Kota, K.; et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J. Virol. 2010, 84, 10569–10580. [Google Scholar]
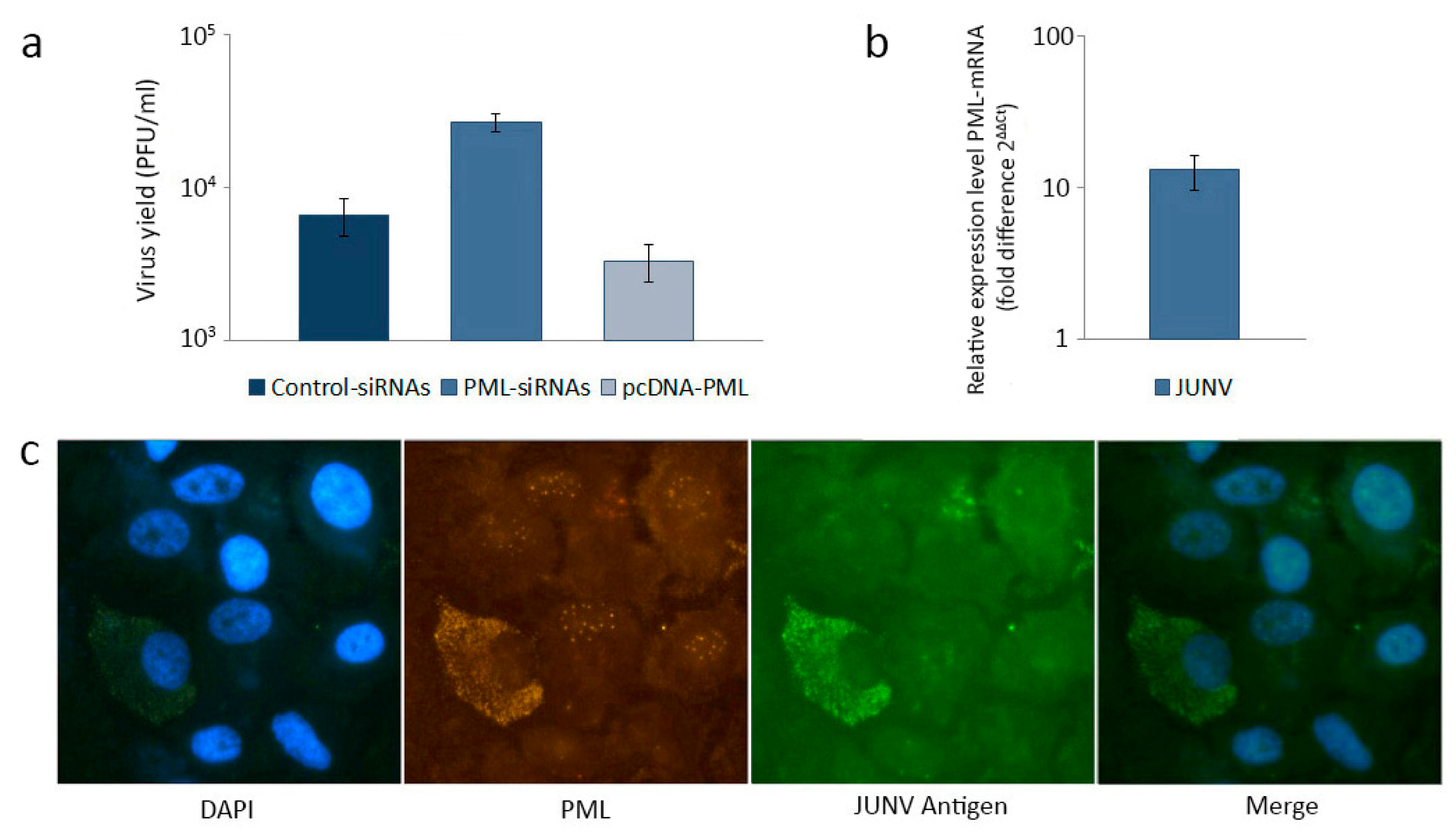
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef]
- Borden, K.L.; Culjkovic, B. Perspectives in PML: A unifying framework for PML function. Front. Biosci. 2009, 14, 497–509. [Google Scholar]
- Lang, M.; Jegou, T.; Chung, I.; Richter, K.; Münch, S.; Udvarhelyi, A.; Cremer, C.; Hemmerich, P.; Engelhardt, J.; Hell, S.W.; et al. Three-Dimensional organization of promyelocytic leukemia nuclear bodies. J. Cell Sci. 2010, 123, 392–400. [Google Scholar] [CrossRef]
- Dyck, J.A.; Maul, G.G.; Miller, W.H., Jr.; Chen, J.D.; Kakizuka, A.; Evans, R.M. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell 1994, 76, 333–343. [Google Scholar]
- Lavau, C.; Marchio, A.; Fagioli, M.; Jansen, J.; Falini, B.; Lebon, P.; Grosveld, F.; Pandolfi, P.P.; Pelicci, P.G.; Dejean, A. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene 1995, 11, 871–876. [Google Scholar]
- Chee, A.V.; Lopez, P.; Pandolfi, P.P.; Roizman, B. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J. Virol 2003, 77, 7101–7105. [Google Scholar] [CrossRef]
- Regad, T.; Chelbi-Alix, M.K. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 2001, 20, 7274–7286. [Google Scholar] [CrossRef]
- Borden, K.L.; Campbell Dwyer, E.J.; Salvato, M.S. An arenavirus RING (zinc-binding) protein binds the oncoprotein promyelocyte leukemia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J. Virol. 1998, 72, 758–766. [Google Scholar]
- García, C.C.; Topisirovic, I.; Djavani, M.; Borden, K.L.; Damonte, E.B.; Salvato, M.S. An antiviral disulfide compound blocks interaction between arenavirus Z protein and cellular promyelocytic leukemia protein. Biochem. Biophys. Res. Commun. 2010, 393, 625–630. [Google Scholar] [CrossRef]
- Borden, K.L.; Campbell Dwyer, E.J.; Salvato, M.S. The promyelocytic leukemia protein PML has a pro-apoptotic activity mediated through its RING domain. FEBS Lett. 1997, 418, 30–34. [Google Scholar] [CrossRef]
- Djavani, M.; Rodas, J.; Lukashevich, I.S.; Horejsh, D.; Pandolfi, P.P.; Borden, K.L.; Salvato, M.S. Role of the promyelocytic leukemia protein PML in the interferon sensitivity of lymphocytic choriomeningitis virus. J. Virol. 2001, 75, 6204–6208. [Google Scholar]
- Bonilla, W.V.; Pinschewer, D.D.; Klenerman, P.; Rousson, V.; Gaboli, M.; Pandolfi, P.P.; Zinkernagel, R.M.; Salvato, M.S.; Hengartner, H. Effects of promyelocytic leukemia protein on virus-host balance. J. Virol. 2002, 76, 3810–3818. [Google Scholar] [CrossRef]
- El McHichi, B.; Regad, T.; Maroui, M.A.; Rodriguez, M.S.; Aminev, A.; Gerbaud, S.; Escriou, N.; Dianoux, L.; Chelbi-Alix, M.K. SUMOylation promotes PML degradation during encephalomyocarditis virus infection. J. Virol. 2010, 84, 11634–11645. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Linero, F.N.; Sepúlveda, C.S.; Giovannoni, F.; Castilla, V.; García, C.C.; Scolaro, L.A.; Damonte, E.B. Host Cell Factors as Antiviral Targets in Arenavirus Infection. Viruses 2012, 4, 1569-1591. https://doi.org/10.3390/v4091569
Linero FN, Sepúlveda CS, Giovannoni F, Castilla V, García CC, Scolaro LA, Damonte EB. Host Cell Factors as Antiviral Targets in Arenavirus Infection. Viruses. 2012; 4(9):1569-1591. https://doi.org/10.3390/v4091569
Chicago/Turabian StyleLinero, Florencia N., Claudia S. Sepúlveda, Federico Giovannoni, Viviana Castilla, Cybele C. García, Luis A. Scolaro, and Elsa B. Damonte. 2012. "Host Cell Factors as Antiviral Targets in Arenavirus Infection" Viruses 4, no. 9: 1569-1591. https://doi.org/10.3390/v4091569
APA StyleLinero, F. N., Sepúlveda, C. S., Giovannoni, F., Castilla, V., García, C. C., Scolaro, L. A., & Damonte, E. B. (2012). Host Cell Factors as Antiviral Targets in Arenavirus Infection. Viruses, 4(9), 1569-1591. https://doi.org/10.3390/v4091569