Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway


Abstract
:1. Introduction
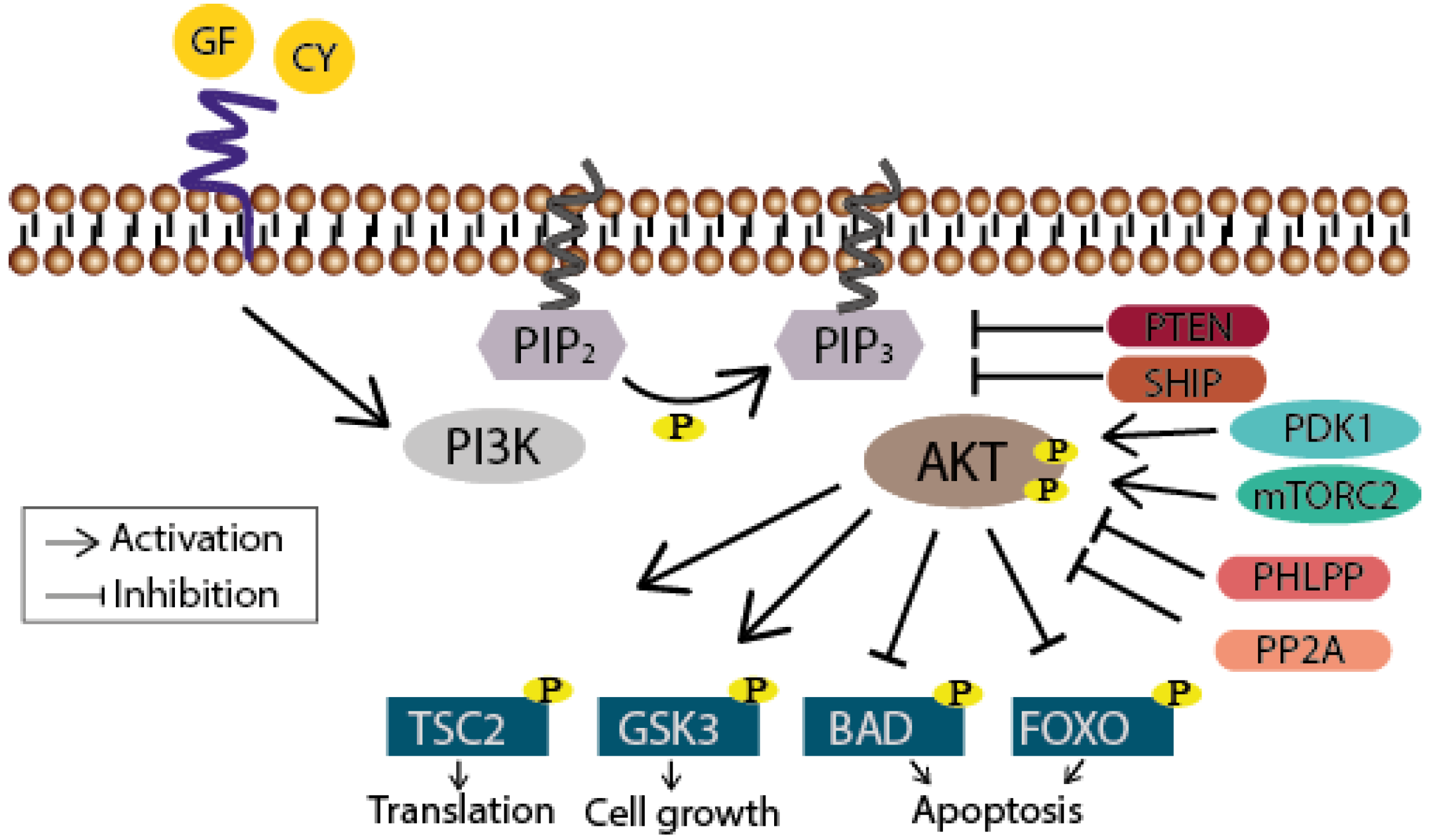
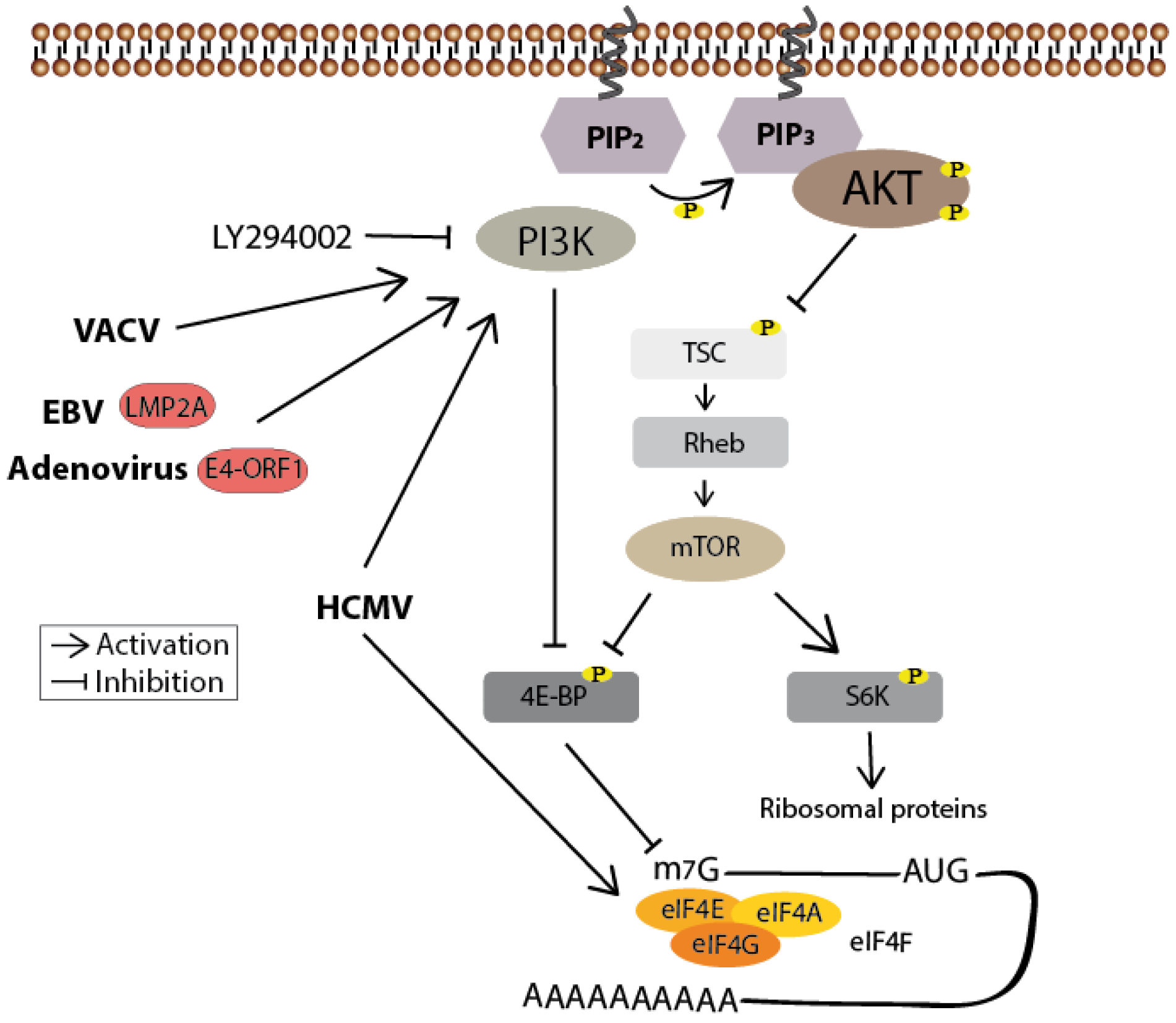
PI3K/Akt Signaling
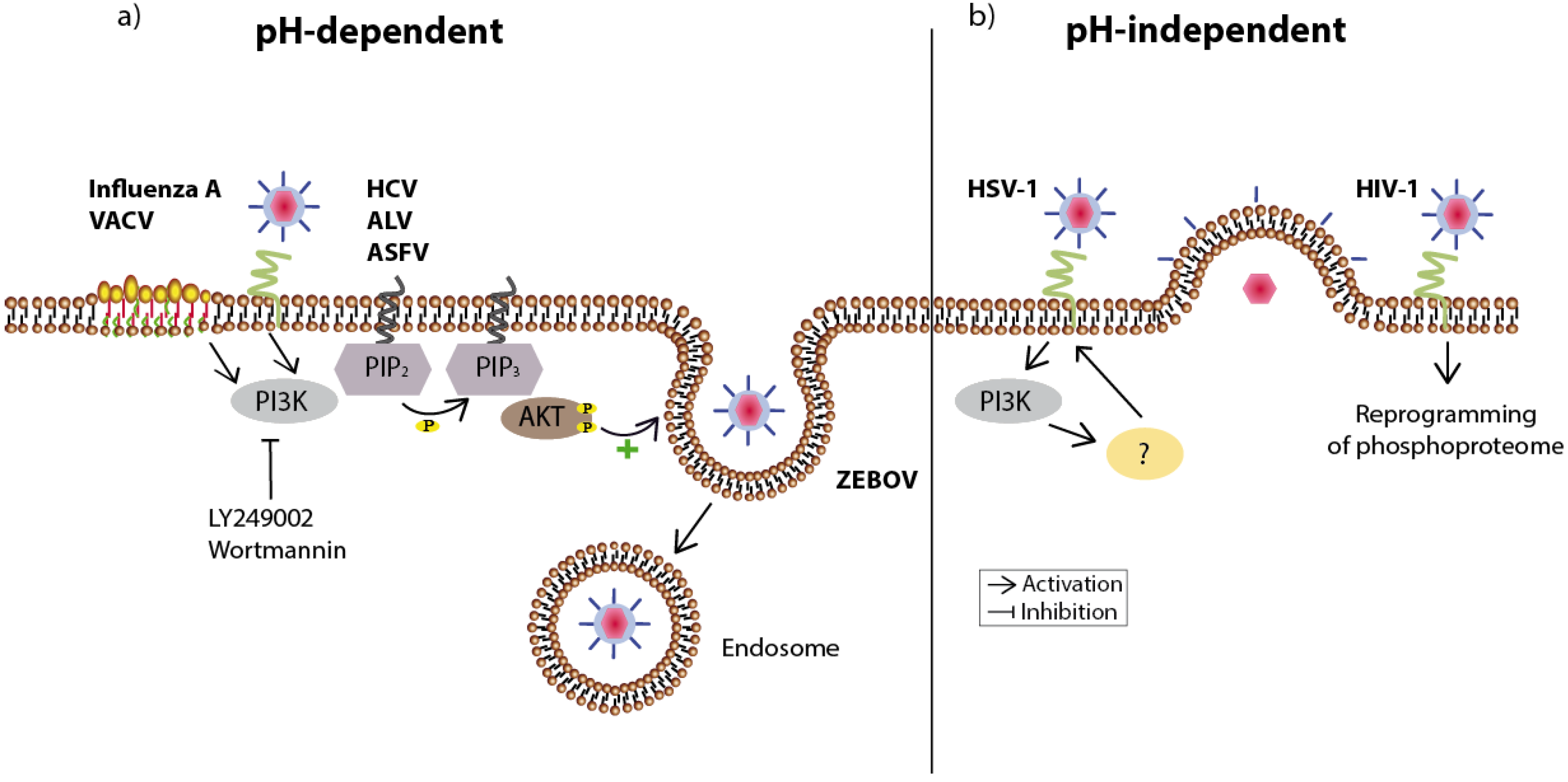
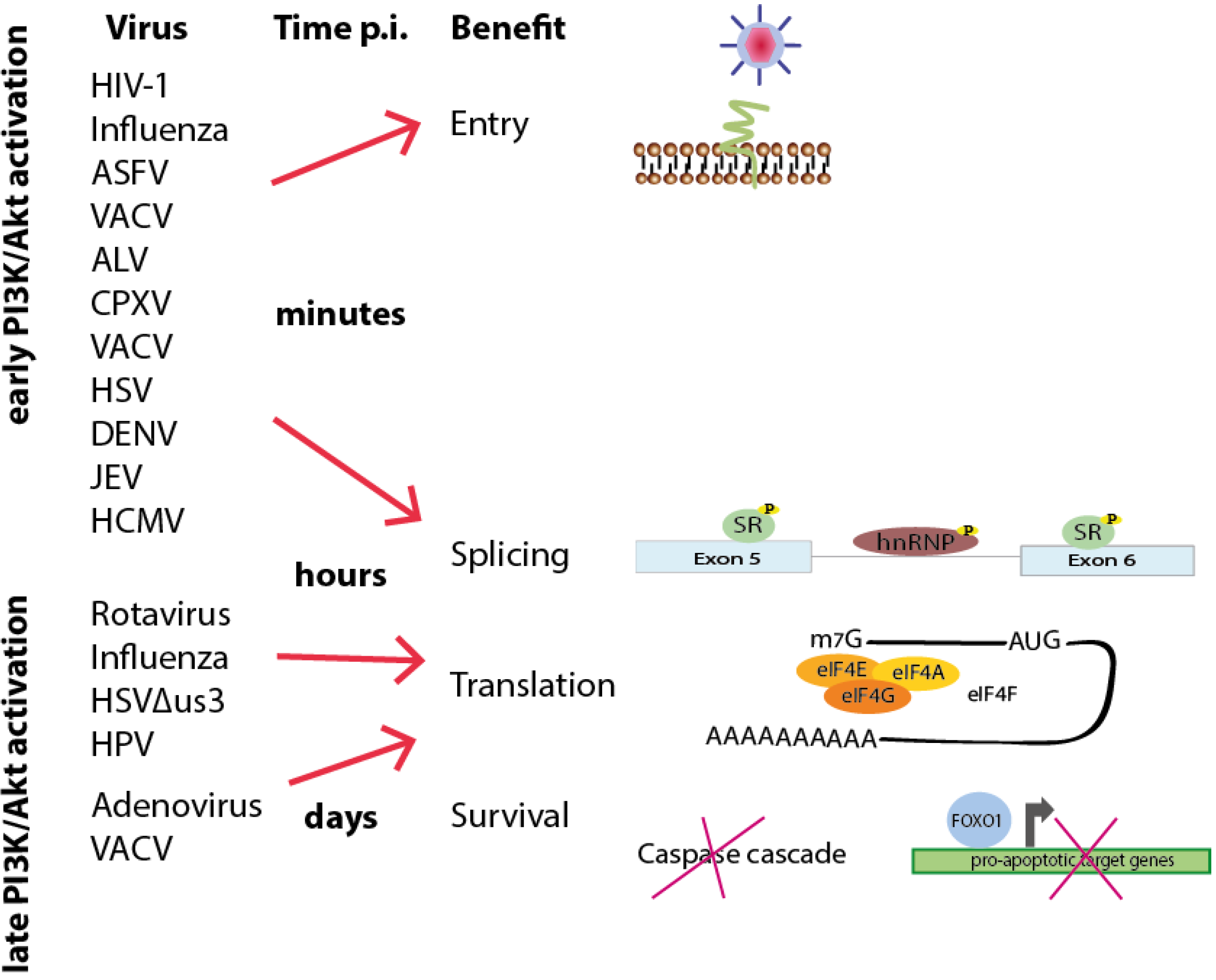
2. Viral Entry


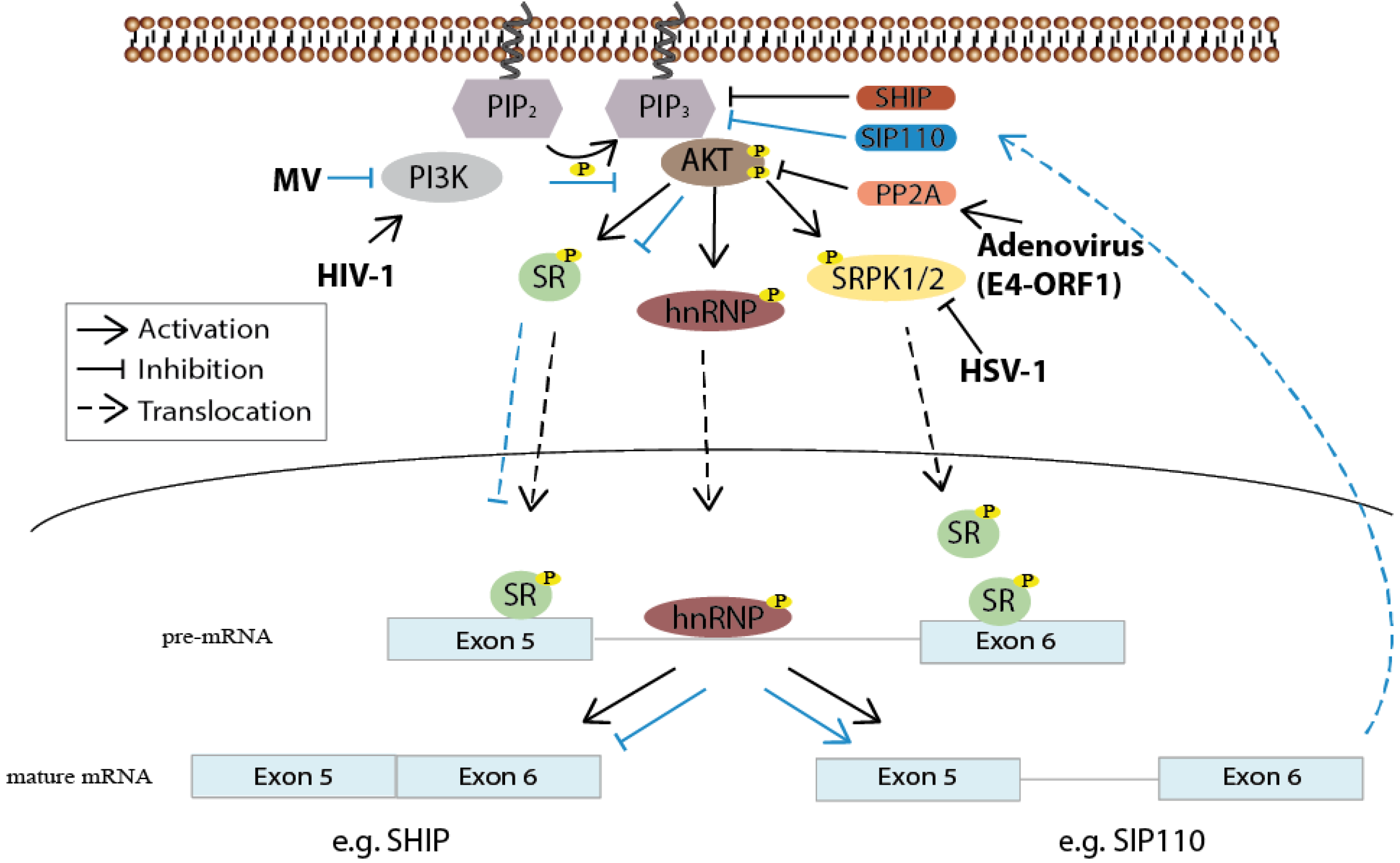
3. Pre-mRNA Splicing

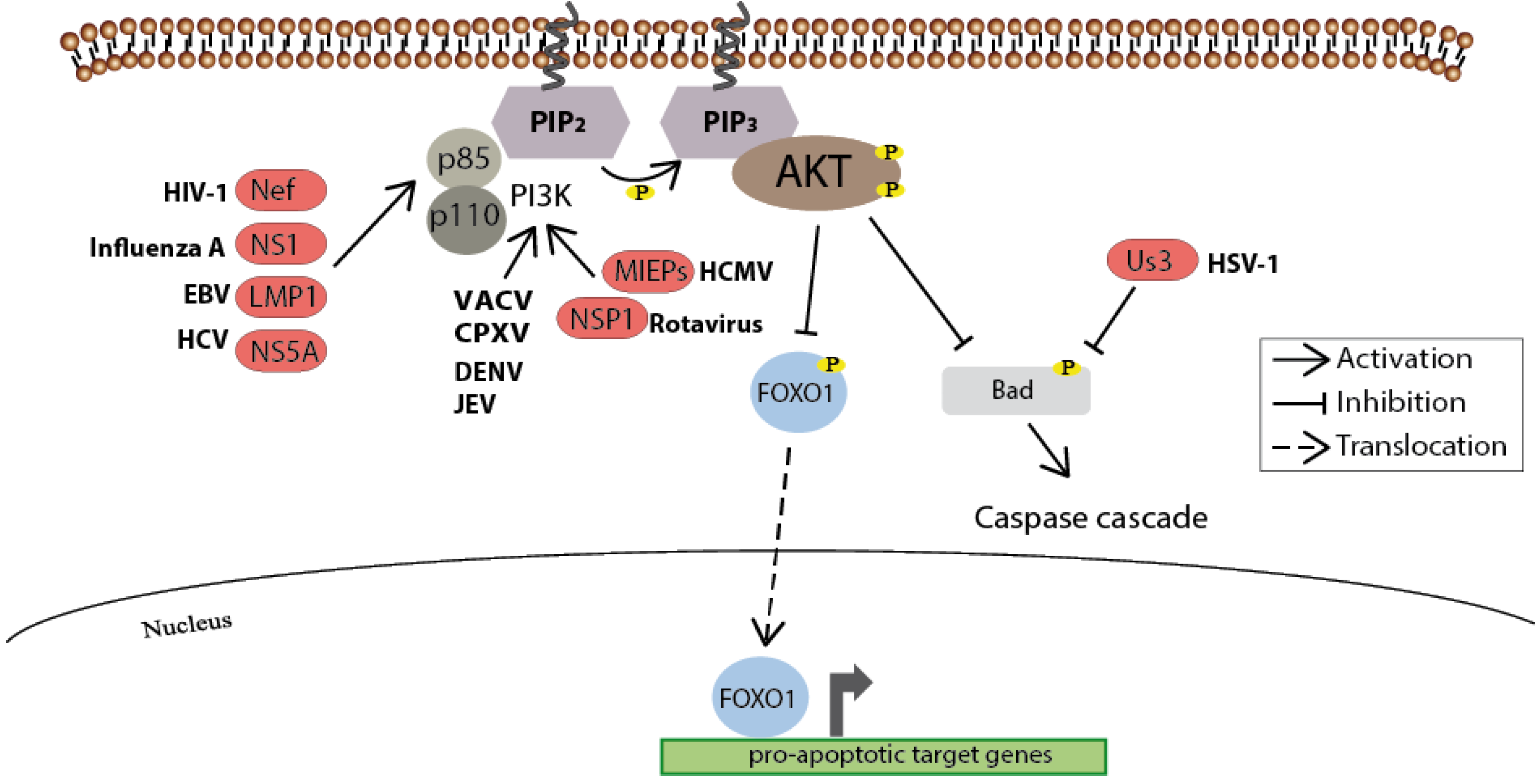
4. Cell Survival

5. Viral Translation

6. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Life Cycle | Virus | Protein | Function | Reference |
|---|---|---|---|---|
| Entry | Influenza | - | Virus internalization;Endosomal acidification | [20,22] |
| VACV | - | Integrin β1-dependent virus entry | [23] | |
| HCV | E2 | Facilitation of virus entry | [26] | |
| ALV | - | Facilitation of virus entry | [27] | |
| HSV-1 | - | Filopodia formation; Fusion | [29] | |
| ASFV | - | Macropinocytosis | [30] | |
| pre-mRNA Splicing | HIV-1 | Alternative splicing of viral mRNAs | [49] | |
| Adenovirus | E4-ORF4 | Dephosphorylation of SF2/ASF and SRp30c; splicing of viral mRNAs | [46] | |
| MV | T-cell proliferation | [44] | ||
| Cell Survival | HIV-1 | Nef | Anti-apoptotic effects | [54,55] |
| Influenza | NS1 | Anti-apoptotic effects | [57,58,59] | |
| VACV | - | Anti-apoptotic effects | [70] | |
| HCV | NS5A | Anti-apoptotic effects; viral persistence | [75,76] | |
| CPXV | - | Anti-apoptotic effects | [70] | |
| DENV | - | Anti-apoptotic effects | [74] | |
| JEV | - | Anti-apoptotic effects | [74] | |
| HCMV | MIEPs | Anti-apoptotic effects | [65] | |
| Rotavirus | NSP1 | Anti-apoptotic effects | [71] | |
| EBV | LMP1 | Persistence | [66] | |
| RSV | - | Anti-apoptotic effects | [78] | |
| CVB3 | - | Anti-apoptotic effects | [79] | |
| ARV | - | Anti-apoptotic effects | [80] | |
| Translation | VACV | - | Translation initiation via activity of 4E-BP | [91] |
| HCMV | - | Translation initiation via activity of 4E-BP | [87] | |
| Adenovirus | E4-ORF1 | mTOR activation | [95] | |
| EBV | LMP2A | mTOR activation | [90] | |
| HPV | E6, E7 | mTOR activation | [93] |

Acknowledgments
Conflicts of Interest
References and Notes
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008, 4, e1000018. [Google Scholar] [CrossRef]
- Kaur, S.; Katsoulidis, E.; Platanias, L.C. Akt and mRNA translation by interferons. Cell Cycle 2008, 7, 2112–2116. [Google Scholar] [CrossRef]
- Kaur, S.; Sassano, A.; Dolniak, B.; Joshi, S.; Majchrzak-Kita, B.; Baker, D.P.; Hay, N.; Fish, E.N.; Platanias, L.C. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 2008, 105, 4808–4813. [Google Scholar] [CrossRef]
- Kaur, S.; Sassano, A.; Joseph, A.M.; Majchrzak-Kita, B.; Eklund, E.A.; Verma, A.; Brachmann, S.M.; Fish, E.N.; Platanias, L.C. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. J. Immunol. 2008, 181, 7316–7323. [Google Scholar]
- Freudenburg, W.; Moran, J.M.; Lents, N.H.; Baldassare, J.J.; Buller, R.M.; Corbett, J.A. Phosphatidylinositol 3-kinase regulates macrophage responses to double-stranded RNA and encephalomyocarditis virus. J. Innate Immun. 2010, 2, 77–86. [Google Scholar] [CrossRef]
- Chang, T.H.; Liao, C.L.; Lin, Y.L. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 2006, 8, 157–171. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar]
- Cantrell, D.A. Phosphoinositide 3-kinase signalling pathways. J. Cell Sci. 2001, 114, 1439–1445. [Google Scholar]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Surucu, B.; Bozulic, L.; Hynx, D.; Parcellier, A.; Hemmings, B.A. In vivo analysis of protein kinase B (PKB)/Akt regulation in DNA-PKcs-null mice reveals a role for PKB/Akt in DNA damage response and tumorigenesis. J. Biol. Chem. 2008, 283, 30025–30033. [Google Scholar] [CrossRef]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [CrossRef]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef]
- Wojcechowskyj, J.A.; Didigu, C.A.; Lee, J.Y.; Parrish, N.F.; Sinha, R.; Hahn, B.H.; Bushman, F.D.; Jensen, S.T.; Seeholzer, S.H.; Doms, R.W. Quantitative phosphoproteomics reveals extensive cellular reprogramming during HIV-1 entry. Cell Host Microbe 2013, 13, 613–623. [Google Scholar] [CrossRef]
- Thorley, J.A.; McKeating, J.A.; Rappoport, J.Z. Mechanisms of viral entry: Sneaking in the front door. Protoplasma 2010, 244, 15–24. [Google Scholar] [CrossRef]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef]
- Lakadamyali, M.; Rust, M.J.; Zhuang, X. Endocytosis of influenza viruses. Microbes Infect. 2004, 6, 929–936. [Google Scholar] [CrossRef]
- Waheed, A.A.; Freed, E.O. The role of lipids in retrovirus replication. Viruses 2010, 2, 1146–1180. [Google Scholar] [CrossRef]
- Pike, L.J. Lipid rafts: Bringing order to chaos. J. Lipid Res. 2003, 44, 655–667. [Google Scholar] [CrossRef]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar] [CrossRef]
- Fujioka, Y.; Tsuda, M.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. The Ras-PI3K signaling pathway is involved in clathrin-independent endocytosis and the internalization of influenza viruses. PLoS One 2011, 6, e16324. [Google Scholar]
- Marjuki, H.; Gornitzky, A.; Marathe, B.M.; Ilyushina, N.A.; Aldridge, J.R.; Desai, G.; Webby, R.J.; Webster, R.G. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell. Microbiol. 2011, 13, 587–601. [Google Scholar] [CrossRef]
- Izmailyan, R.; Hsao, J.C.; Chung, C.S.; Chen, C.H.; Hsu, P.W.; Liao, C.L.; Chang, W. Integrin beta1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 2012, 86, 6677–6687. [Google Scholar] [CrossRef]
- Tian, B.; Lessan, K.; Kahm, J.; Kleidon, J.; Henke, C. Beta 1 integrin regulates fibroblast viability during collagen matrix contraction through a phosphatidylinositol 3-kinase/Akt/protein kinase B signaling pathway. J. Biol. Chem. 2002, 277, 24667–24675. [Google Scholar] [CrossRef]
- Saeed, M.F.; Kolokoltsov, A.A.; Freiberg, A.N.; Holbrook, M.R.; Davey, R.A. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 2008, 4, e1000141. [Google Scholar] [CrossRef]
- Liu, Z.; Tian, Y.; Machida, K.; Lai, M.M.; Luo, G.; Foung, S.K.; Ou, J.H. Transient activation of the PI3K-AKT pathway by hepatitis C virus to enhance viral entry. J. Biol. Chem. 2012, 287, 41922–41930. [Google Scholar] [CrossRef]
- Feng, S.Z.; Cao, W.S.; Liao, M. The PI3K/Akt pathway is involved in early infection of some exogenous avian leukosis viruses. J. Gen. Virol. 2011, 92, 1688–1697. [Google Scholar] [CrossRef]
- Oh, M.J.; Akhtar, J.; Desai, P.; Shukla, D. A role for heparan sulfate in viral surfing. Biochem. Biophys. Res. Commun. 2010, 391, 176–181. [Google Scholar] [CrossRef]
- Tiwari, V.; Shukla, D. Phosphoinositide 3 kinase signalling may affect multiple steps during herpes simplex virus type-1 entry. J. Gen. Virol. 2010, 91, 3002–3009. [Google Scholar] [CrossRef]
- Sanchez, E.G.; Quintas, A.; Perez-Nunez, D.; Nogal, M.; Barroso, S.; Carrascosa, A.L.; Revilla, Y. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog. 2012, 8, e1002754. [Google Scholar] [CrossRef]
- Roca, X.; Krainer, A.R.; Eperon, I.C. Pick one, but be quick: 5' splice sites and the problems of too many choices. Genes Dev. 2013, 27, 129–144. [Google Scholar] [CrossRef]
- Hertel, K.J. Combinatorial control of exon recognition. J. Biol. Chem. 2008, 283, 1211–1215. [Google Scholar] [CrossRef]
- Erkelenz, S.; Mueller, W.F.; Evans, M.S.; Busch, A.; Schoneweis, K.; Hertel, K.J.; Schaal, H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 2013, 19, 96–102. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Fu, H.; Rees, H.; Yepes, M.; Levey, A.; Ye, K. Interaction of Akt-phosphorylated SRPK2 with 14-3-3 mediates cell cycle and cell death in neurons. J. Biol. Chem. 2009, 284, 24512–24525. [Google Scholar] [CrossRef]
- Zhou, Z.; Qiu, J.; Liu, W.; Zhou, Y.; Plocinik, R.M.; Li, H.; Hu, Q.; Ghosh, G.; Adams, J.A.; Rosenfeld, M.G.; et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol. Cell 2012, 47, 422–433. [Google Scholar] [CrossRef]
- Blaustein, M.; Pelisch, F.; Tanos, T.; Munoz, M.J.; Wengier, D.; Quadrana, L.; Sanford, J.R.; Muschietti, J.P.; Kornblihtt, A.R.; Caceres, J.F.; et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat. Struct. Mol. Biol. 2005, 12, 1037–1044. [Google Scholar] [CrossRef]
- Jiang, K.; Patel, N.A.; Watson, J.E.; Apostolatos, H.; Kleiman, E.; Hanson, O.; Hagiwara, M.; Cooper, D.R. Akt2 regulation of Cdc2-like kinases (Clk/Sty), serine/arginine-rich (SR) protein phosphorylation, and insulin-induced alternative splicing of PKCbetaII messenger ribonucleic acid. Endocrinology 2009, 150, 2087–2097. [Google Scholar]
- Shultz, J.C.; Goehe, R.W.; Wijesinghe, D.S.; Murudkar, C.; Hawkins, A.J.; Shay, J.W.; Minna, J.D.; Chalfant, C.E. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 2010, 70, 9185–9196. [Google Scholar] [CrossRef]
- Vu, N.T.; Park, M.A.; Shultz, J.C.; Goehe, R.W.; Hoeferlin, L.A.; Shultz, M.D.; Smith, S.A.; Lynch, K.W.; Chalfant, C.E. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J. Biol. Chem. 2013, 288, 8575–8584. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Ahmad, M.; Guo, Y.; Zhan, Y.; Lazebnik, Y.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of an endogenous dominant-negative short isoform of caspase-9 that can regulate apoptosis. Cancer Res. 1999, 59, 999–1002. [Google Scholar]
- Schwerk, C.; Prasad, J.; Degenhardt, K.; Erdjument-Bromage, H.; White, E.; Tempst, P.; Kidd, V.J.; Manley, J.L.; Lahti, J.M.; Reinberg, D. ASAP, a novel protein complex involved in RNA processing and apoptosis. Mol. Cell. Biol. 2003, 23, 2981–2990. [Google Scholar] [CrossRef]
- Singh, K.K.; Erkelenz, S.; Rattay, S.; Dehof, A.K.; Hildebrandt, A.; Schulze-Osthoff, K.; Schaal, H.; Schwerk, C. Human SAP18 mediates assembly of a splicing regulatory multiprotein complex via its ubiquitin-like fold. RNA 2010, 16, 2442–2454. [Google Scholar] [CrossRef]
- Hu, Y.; Yao, J.; Liu, Z.; Liu, X.; Fu, H.; Ye, K. Akt phosphorylates acinus and inhibits its proteolytic cleavage, preventing chromatin condensation. EMBO J. 2005, 24, 3543–3554. [Google Scholar] [CrossRef]
- Avota, E.; Harms, H.; Schneider-Schaulies, S. Measles virus induces expression of SIP110, a constitutively membrane clustered lipid phosphatase, which inhibits T cell proliferation. Cell. Microbiol. 2006, 8, 1826–1839. [Google Scholar] [CrossRef]
- Riedel, A.; Mofolo, B.; Avota, E.; Schneider-Schaulies, S.; Meintjes, A.; Mulder, N.; Kneitz, S. Accumulation of splice variants and transcripts in response to PI3K inhibition in T cells. PLoS One 2013, 8, e50695. [Google Scholar]
- Estmer Nilsson, C.; Petersen-Mahrt, S.; Durot, C.; Shtrichman, R.; Krainer, A.R.; Kleinberger, T.; Akusjarvi, G. The adenovirus E4-ORF4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J. 2001, 20, 864–871. [Google Scholar] [CrossRef]
- Sciabica, K.S.; Dai, Q.J.; Sandri-Goldin, R.M. ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBOJ. 2003, 22, 1608–1619. [Google Scholar] [CrossRef]
- Dowling, D.; Nasr-Esfahani, S.; Tan, C.H.; O’Brien, K.; Howard, J.L.; Jans, D.A.; Purcell, D.F.; Stoltzfus, C.M.; Sonza, S. HIV-1 infection induces changes in expression of cellular splicing factors that regulate alternative viral splicing and virus production in macrophages. Retrovirology 2008, 5. [Google Scholar] [CrossRef]
- Hillebrand, F. Der Einfluss des PI3-Kinase Signalwegs auf die Regulation des alternativen HIV-1 prä-mRNA Spleißens. Ph.D. Thesis, Bayerische Julius-Maximilians-Universität Würzburg, Würzburg, Germany, 2013. [Google Scholar]
- Duronio, V. The life of a cell: Apoptosis regulation by the PI3K/PKB pathway. Biochem. J. 2008, 415, 333–344. [Google Scholar] [CrossRef]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Biggs, W.H., 3rd; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar] [CrossRef]
- Graziani, A.; Galimi, F.; Medico, E.; Cottone, E.; Gramaglia, D.; Boccaccio, C.; Comoglio, P.M. The HIV-1 nef protein interferes with phosphatidylinositol 3-kinase activation 1. J. Biol. Chem. 1996, 271, 6590–6593. [Google Scholar] [CrossRef]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 2007, 88, 13–18. [Google Scholar] [CrossRef]
- Zhirnov, O.P.; Klenk, H.D. Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis 2007, 12, 1419–1432. [Google Scholar] [CrossRef]
- Jackson, D.; Killip, M.J.; Galloway, C.S.; Russell, R.J.; Randall, R.E. Loss of function of the influenza A virus NS1 protein promotes apoptosis but this is not due to a failure to activate phosphatidylinositol 3-kinase (PI3K). Virology 2010, 396, 94–105. [Google Scholar] [CrossRef]
- Ayllon, J.; Garcia-Sastre, A.; Hale, B.G. Influenza A viruses and PI3K: Are there time, place and manner restrictions? Virulence 2012, 3, 411–414. [Google Scholar] [CrossRef]
- Ayllon, J.; Hale, B.G.; Garcia-Sastre, A. Strain-specific contribution of NS1-activated phosphoinositide 3-kinase signaling to influenza A virus replication and virulence. J. Virol. 2012, 86, 5366–5370. [Google Scholar] [CrossRef]
- Benetti, L.; Roizman, B. Protein kinase B/Akt is present in activated form throughout the entire replicative cycle of deltaU(S)3 mutant virus but only at early times after infection with wild-type herpes simplex virus 1. J. Virol. 2006, 80, 3341–3348. [Google Scholar] [CrossRef]
- Yu, Y.; Alwine, J.C. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3'-OH kinase pathway and the cellular kinase Akt. J. Virol. 2002, 76, 3731–3738. [Google Scholar] [CrossRef]
- Dawson, C.W.; Tramountanis, G.; Eliopoulos, A.G.; Young, L.S. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J. Biol. Chem. 2003, 278, 3694–3704. [Google Scholar] [CrossRef]
- Darr, C.D.; Mauser, A.; Kenney, S. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. J. Virol. 2001, 75, 6135–6142. [Google Scholar] [CrossRef]
- Peng, L.; Wu, T.T.; Tchieu, J.H.; Feng, J.; Brown, H.J.; Feng, J.; Li, X.; Qi, J.; Deng, H.; Vivanco, I.; et al. Inhibition of the phosphatidylinositol 3-kinase-Akt pathway enhances gamma-2 herpesvirus lytic replication and facilitates reactivation from latency. J. Gen. Virol. 2010, 91, 463–469. [Google Scholar] [CrossRef]
- Cooray, S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 2004, 85, 1065–1076. [Google Scholar] [CrossRef]
- Soares, J.A.; Leite, F.G.; Andrade, L.G.; Torres, A.A.; de Sousa, L.P.; Barcelos, L.S.; Teixeira, M.M.; Ferreira, P.C.; Kroon, E.G.; Souto-Padron, T.; et al. Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J. Virol. 2009, 83, 6883–6899. [Google Scholar] [CrossRef]
- Bagchi, P.; Dutta, D.; Chattopadhyay, S.; Mukherjee, A.; Halder, U.C.; Sarkar, S.; Kobayashi, N.; Komoto, S.; Taniguchi, K.; Chawla-Sarkar, M. Rotavirus nonstructural protein 1 suppresses virus-induced cellular apoptosis to facilitate viral growth by activating the cell survival pathways during early stages of infection. J. Virol. 2010, 84, 6834–6845. [Google Scholar] [CrossRef]
- Halasz, P.; Holloway, G.; Turner, S.J.; Coulson, B.S. Rotavirus replication in intestinal cells differentially regulates integrin expression by a phosphatidylinositol 3-kinase-dependent pathway, resulting in increased cell adhesion and virus yield. J. Virol. 2008, 82, 148–160. [Google Scholar] [CrossRef]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef]
- Lee, C.J.; Liao, C.L.; Lin, Y.L. Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J. Virol. 2005, 79, 8388–8399. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar]
- Mannova, P.; Beretta, L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: Control of cell survival and viral replication. J. Virol. 2005, 79, 8742–8749. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Thomas, K.W.; Monick, M.M.; Staber, J.M.; Yarovinsky, T.; Carter, A.B.; Hunninghake, G.W. Respiratory syncytial virus inhibits apoptosis and induces NF-kappa B activity through a phosphatidylinositol 3-kinase-dependent pathway. J. Biol. Chem. 2002, 277, 492–501. [Google Scholar]
- Esfandiarei, M.; Boroomand, S.; Suarez, A.; Si, X.; Rahmani, M.; McManus, B. Coxsackievirus B3 activates nuclear factor kappa B transcription factor via a phosphatidylinositol-3 kinase/protein kinase B-dependent pathway to improve host cell viability. Cell Microbiol. 2007, 9, 2358–2371. [Google Scholar] [CrossRef]
- Lin, P.Y.; Liu, H.J.; Liao, M.H.; Chang, C.D.; Chang, C.I.; Cheng, H.L.; Lee, J.W.; Shih, W.L. Activation of PI 3-kinase/Akt/NF-kappaB and Stat3 signaling by avian reovirus S1133 in the early stages of infection results in an inflammatory response and delayed apoptosis. Virology 2010, 400, 104–114. [Google Scholar] [CrossRef]
- Cooray, S.; Jin, L.; Best, J.M. The involvement of survival signaling pathways in rubella-virus induced apoptosis. Virol. J. 2005, 2. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. Importance of Akt signaling pathway for apoptosis in SARS-CoV-infected Vero E6 cells. Virology 2004, 327, 169–174. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar]
- Johnson, R.A.; Wang, X.; Ma, X.L.; Huong, S.M.; Huang, E.S. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: Inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J. Virol. 2001, 75, 6022–6032. [Google Scholar]
- Kudchodkar, S.B.; Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J. Virol. 2004, 78, 11030–11039. [Google Scholar]
- Walsh, D.; Perez, C.; Notary, J.; Mohr, I. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 2005, 79, 8057–8064. [Google Scholar]
- Kudchodkar, S.B.; Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc. Natl. Acad. Sci. USA 2006, 103, 14182–14187. [Google Scholar]
- Moody, C.A.; Scott, R.S.; Amirghahari, N.; Nathan, C.O.; Young, L.S.; Dawson, C.W.; Sixbey, J.W. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J. Virol. 2005, 79, 5499–5506. [Google Scholar]
- Zaborowska, I.; Walsh, D. PI3K signaling regulates rapamycin-insensitive translation initiation complex formation in vaccinia virus-infected cells. J. Virol. 2009, 83, 3988–3992. [Google Scholar]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar]
- O’Shea, C.; Klupsch, K.; Choi, S.; Bagus, B.; Soria, C.; Shen, J.; McCornmick, F.; Stokoe, D. Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J. 2005, 24, 1211–1221. [Google Scholar]
- Zuurbier, L.; Petricoin, E.F., 3rd; Vuerhard, M.J.; Calvert, V.; Kooi, C.; Buijs-Gladdines, J.G.; Smits, W.K.; Sonneveld, E.; Veerman, A.J.; Kamps, W.A.; et al. The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica 2012, 97, 1405–1413. [Google Scholar]
- Calzavara, E.; Chiaramonte, R.; Cesana, D.; Basile, A.; Sherbet, G.V.; Comi, P. Reciprocal regulation of Notch and PI3K/Akt signalling in T-ALL cells in vitro. J. Cell. Biochem. 2008, 103, 1405–1412. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Diehl, N.; Schaal, H. Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway. Viruses 2013, 5, 3192-3212. https://doi.org/10.3390/v5123192
Diehl N, Schaal H. Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway. Viruses. 2013; 5(12):3192-3212. https://doi.org/10.3390/v5123192
Chicago/Turabian StyleDiehl, Nora, and Heiner Schaal. 2013. "Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway" Viruses 5, no. 12: 3192-3212. https://doi.org/10.3390/v5123192
APA StyleDiehl, N., & Schaal, H. (2013). Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway. Viruses, 5(12), 3192-3212. https://doi.org/10.3390/v5123192