Abstract
Gene therapy using integrating retroviral vectors has proven its effectiveness in several clinical trials for the treatment of inherited diseases and cancer. However, vector-mediated adverse events related to insertional mutagenesis were also observed, emphasizing the need for safer therapeutic vectors. Paradoxically, alpharetroviruses, originally discovered as cancer-causing agents, have a more random and potentially safer integration pattern compared to gammaretro- and lentiviruses. In this review, we provide a short overview of the history of alpharetroviruses and explain how they can be converted into state-of-the-art gene delivery tools with improved safety features. We discuss development of alpharetroviral vectors in compliance with regulatory requirements for clinical translation, and provide an outlook on possible future gene therapy applications. Taken together, this review is a broad overview of alpharetroviral vectors spanning the bridge from their parental virus discovery to their potential applicability in clinical settings.
1. Introduction
Human gene therapy incorporating genome engineering to enhance cell functions has the potential to cure numerous life-threatening diseases, including severe combined immunodeficiencies and cancer. The evolutionary optimized ability to stably integrate DNA into cellular genomes makes retroviruses very attractive for permanent therapeutic cell modifications. During the last decades, retroviruses have been developed into valuable tools for human gene therapy with the focus on vectors derived from gammaretroviruses and lentiviruses. However, retroviral vector integration into the target cell genome can cause insertional activation of cellular proto-oncogenes, potentially leading to serious adverse events such as leukemia. This adverse effect of retroviral vectors on cellular integrity is termed genotoxicity and can be influenced by several factors, such as vector design and integration target site selection. In contrast to conventional gammaretroviral and lentiviral vectors, alpharetroviral vectors have comparatively neutral integration target site preferences, hence alpharetroviral vectors might be less genotoxic and therefore of therapeutic value. In the present review, we highlight previous alpharetroviral vector discoveries as well as current developments and the potential of alpharetroviral applications for future human gene therapy strategies.
2. History of (Alpha-) Retroviruses
More than 100 years ago, Vilhelm Ellermann and Oluf Bang demonstrated that cell-free filtrates were able to transmit leukemia in chickens [1]. A few years later, Francis Peyton Rous described the cell-free transmission of chicken sarcoma [2,3]. These groundbreaking discoveries indicated that cancer could be caused by viruses, a novel paradigm which was not widely accepted at that time. In fact, researchers supporting the concept of viruses causing cancer were said to either have “holes in their heads or holes in their filters” [4]. Nevertheless, especially the Rous sarcoma virus (RSV), a paradigmatic species of the alpharetroviral genus, played an important role in the history of retrovirology and in cancer research (Figure 1).
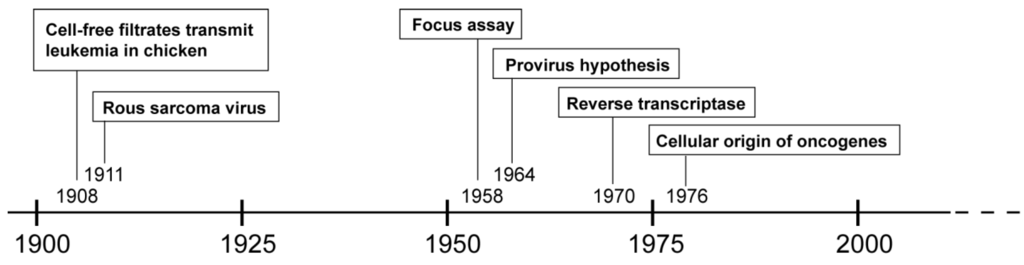
Figure 1.
Timeline of hallmark events in the history of retrovirology.
Figure 1.
Timeline of hallmark events in the history of retrovirology.
One important development made possible by the discovery of RSV was the focus assay, which provided the technical basis for studying viral infection on single viral particle and single cell levels and thus enabled researchers to investigate the molecular details of retroviral replication [5]. It was discovered that the RSV genome consisted of RNA [6], but that RSV infection required cellular DNA for replication [7]. This apparent contradiction was resolved with the provirus hypothesis published by Howard Temin in 1964: “The results presented here with RSV can be most simply explained by the following model: virus enters a cell and directs formation of a DNA containing the genetic information of the virus. This new DNA, the provirus […], then acts as a template for formation of new nucleic acid, RNA, for the virion” [8]. This hypothesis, independently proposed by Jan Svoboda and coworkers [9], was initially met with skepticism, but eventually became widely accepted upon identification of reverse transcriptase [10,11]. Many additional discoveries concerning the retroviral life cycle soon followed. However, the mechanism of RSV-induced cancer remained an unsettled issue until 1976, when molecular hybridization studies by Harold E. Varmus, J. Michael Bishop and coworkers revealed that, “[…] part or all of the transforming gene(s) […] was derived from the chicken genome or a species closely related to chicken […]” [12]. The transforming gene transferred by RSV was then identified as the SRC proto-oncogene, and soon followed by the discovery of additional oncogenes transferred by other retroviral species. Discovery of the cellular origin of viral oncogenes drastically changed the perspective of cancer research and immensely contributed to the understanding of cancer development. However, the transfer of proto-oncogenes was not the only mechanism through which retroviruses could cause cancer. In a process called insertional transformation, the retroviral promoter elements were shown to increase expression of cellular proto-oncogenes, such as MYC, thereby causing neoplasms, albeit as rare events and with much longer latencies than the acute transforming oncogene-transferring counterparts [13,14]. The genotoxic effect of retroviruses, first described in 1981, led to a severe setback in human gene therapy more than a decade later. Consequently, the mechanisms of insertional genotoxicity have been intensely studied and retroviral vector designs have been markedly improved, thus increasing the safety of human gene therapy.
3. From the Virus to the Vector
3.1. Taking a Different Perspective: Retroviruses for Human Gene Therapy
The discovery of viral transfer of genes other than those required for viral replication [12] served as a paradigm for the development of human gene therapy vectors. With the increasing knowledge of the genetic origin of many diseases in the genomics era, researchers envisioned that by inserting a corrected version of a defective gene into a patient’s genome, a permanent cure could be achieved on a molecular basis. Gene therapy aims at curing life-threatening diseases, such as severe combined immunodeficiencies and cancer by exploiting the retroviral life cycle for the transfer of such therapeutic transgenes.
While aforementioned historical discoveries were made with avian-infectious alpharetroviral vectors, researchers working on gene therapy initially exploited gammaretroviral vectors derived from MLV (murine leukemia virus), which naturally replicates in mice. From a safety perspective, the availability of cell lines for the production of replication-defective retroviruses in 1983 marked an important step towards the development of retroviral vectors for human gene therapy [15]. Soon thereafter, pioneering studies in mice proved the concept of transferring genes into hematopoietic stem cells, albeit with low levels of gene transfer and gene expression [16,17]. During the next two decades, retroviral vectors were markedly improved with greater focus on vectors derived from gammaretroviruses (MLV) and lentiviruses (human immunodeficiency virus-1, HIV-1). Clinical benefit to previously terminally ill patients was demonstrated in several preclinical studies and clinical trials have employed these vectors to treat primary immunodeficiencies, such as severe combined immunodeficiency (SCID) and chronic granulomatous disease (CGD). However, despite therapeutic benefits, some of these patients suffered from severe adverse events due to clonal expansion of transduced cells, eventually leading to myelodysplastic syndromes and/or leukemias [18]. Furthermore, the clinical success in the CGD trial was hampered by the occurrence of vector silencing, which resulted in only a transient benefit [19,20]. These adverse events led to the temporary postponement of many gene therapy clinical trials. Intensive investigations revealed that they resulted from genotoxic integrations of retroviral vectors in the genome, leading to upregulation of cellular proto-oncogenes, such as LMO2 and MDS1-EVI1 [20,21,22,23,24]. First described for replicating retroviruses more than a decade earlier, the risk of insertional proto-oncogene activation had initially been anticipated to be low for replication-defective retroviral vectors. However, insertional activation still occurred in clinical trials, thus raising the urgent question of how to improve the safety of retroviral gene therapy.
3.2. Genotoxicity of Retroviral Vectors
Identification of the mechanisms and reduction of underlying genotoxicity have become major objectives of the field of human gene therapy. In general, retroviral vector genotoxicity is due to upregulation of cellular proto-oncogene expression and can theoretically be caused by several mechanisms, such as (i) promoter insertion; (ii) promoter activation and (iii) gene transcript truncation (Figure 2).
(i) The genotoxic mechanism of promoter insertion describes the insertion of promoter sequences directly upstream of cellular transcription units, thereby adversely influencing their expression (Figure 2i). This can either be mediated by read-through transcription from the inserted promoter into the adjacent gene or by a combination of read-through transcription and splicing events involving vector and cellular splice sites. In 1981, promoter insertion became the first described mechanism of insertional transformation for replicating alpharetroviruses and caused neoplastic transformation in birds. In the reported cases, the viral promoter, which resides in the 5' and 3' long terminal repeats (LTRs) of the retrovirus, caused read-through transcription into the cellular MYC proto-oncogene and was detected by the presence of virus-MYC fusion transcripts [13,14]. In addition to the overexpression of adjacent genes, promoter insertion can also lead to oncogene capture by replication-competent retroviral vectors and thus to the development of acute transforming viruses. These viruses have an enormous genotoxic potential as oncogene expression occurs irrespective of their integration sites. However, while promoter insertion can be induced by any retroviral vector harboring promoter elements, oncogene capture is a very rare event and is restricted to replication-competent vectors. Additionally, in contrast to RSV, most acute transforming viruses become replication-defective, as they lose part of their viral coding sequences and thus require helper viruses for infectious viral particle formation.
(ii) In most clinical trials, which used replication-defective gammaretroviral vectors, promoter activation was identified as the primary cause of transformation (Figure 2ii). Here, the powerful enhancer in the gammaretroviral LTR most likely caused upregulated expression of cellular proto-oncogenes, such as LMO2 or MDS1-EVI1 [20,21,22,23,24]. While the mechanism of promoter insertion is restricted to upstream and in sense-oriented integrations of the retroviral vector adjacent to the affected cellular transcription unit, enhancer-mediated promoter activation can occur at different orientations and loci up to several hundred kilobases from the insertion site [25,26].
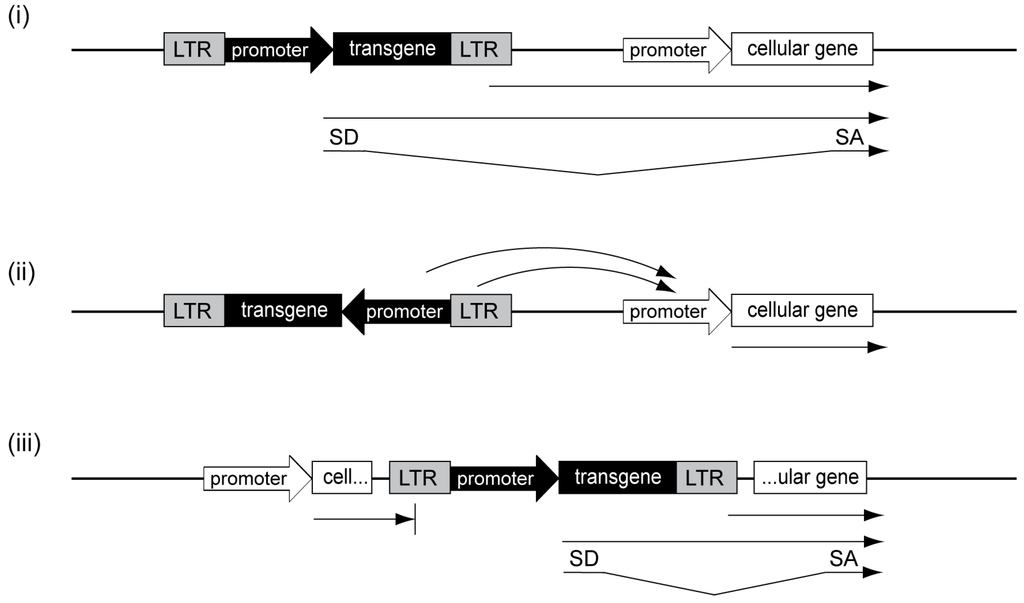
Figure 2.
Genotoxicity mechanisms. (i) Promoter insertion upstream and in sense to cellular transcription units can lead to read-through transcription into adjacent cellular genes, either from the internal promoter or from the long terminal repeat (LTR) as indicated by the arrows. If splice acceptor (SA) and donor (SD) sites are present, promoter insertion can be accompanied by splice events; (ii) promoter activation is mediated by enhancer-interactions by respective elements in the internal promoter or in the LTR with cellular promoters; (iii) gene transcript truncation can lead to shortened cellular transcripts, either lacking 3' (left example) or 5' sequences (right example).
Figure 2.
Genotoxicity mechanisms. (i) Promoter insertion upstream and in sense to cellular transcription units can lead to read-through transcription into adjacent cellular genes, either from the internal promoter or from the long terminal repeat (LTR) as indicated by the arrows. If splice acceptor (SA) and donor (SD) sites are present, promoter insertion can be accompanied by splice events; (ii) promoter activation is mediated by enhancer-interactions by respective elements in the internal promoter or in the LTR with cellular promoters; (iii) gene transcript truncation can lead to shortened cellular transcripts, either lacking 3' (left example) or 5' sequences (right example).
(iii) There is increasing evidence that gene transcript truncation is also a safety concern (Figure 2iii). In contrast to promoter insertion, gene transcript truncation is caused by intragenic retroviral vector integrations. These intragenic integrations frequently occur in intronic regions, involve aberrant splicing events and result in a loss of either 5' or 3' sequences of cellular genes. When transcription initiates from an inserted promoter, 5' sequences of the affected cellular gene are lost (Figure 2iii; right example). In contrast, when transcription is initiated from a cellular promoter followed by read-through into the retroviral vector, loss of 3' sequences can be caused by premature polyadenylation at polyadenylation sites introduced within the vector (Figure 2iii; left example). In several murine studies using lentiviral vectors, gene transcript truncations of 5' or 3' sequences led to leukemia development by either removal of regulatory regions of proto-oncogenes [27] or by downregulation of full-length transcripts of supposedly haploinsufficient tumor suppressor genes [28]. Functional consequences of gene transcript truncation were also demonstrated in a clinical trial involving one patient suffering from β-thalassemia [29]. After lentiviral β-globin gene transfer, this patient showed therapeutic benefit largely attributed to a dominant clone, in which intragenic vector integration in HMGA2 had occurred. This insertion led to a 3' gene transcript truncation, rendering the shortened mRNA insensitive to microRNA-mediated downregulation, thus upregulating HMGA2, which led to clonal expansion. While this benign clonal expansion has not resulted in a serious adverse event to date, and is actually associated with clinical benefit, it clearly demonstrates the potency of gene transcript truncation to contribute to clonal imbalance in a clinical setting.
Promoter insertion, promoter activation and gene transcript truncation are the three most prevalent mechanisms of retroviral vector genotoxicity described to date. While these mechanisms have been described individually for the sake of clarity, insertional mutagenic events show a higher layer of complexity. For example, a single integration may affect more than one gene and induce transcriptional deregulation by more than one mechanism simultaneously [30]. Importantly, our current understanding of genotoxicity mechanisms allows several prevention strategies to be envisioned. Since insertional deregulation of cellular transcription is dependent on the presence of strong promoter/enhancer sequences and splice sites, optimized vector design should omit these elements. In addition, integration target sites influence genotoxicity, with potentially detrimental integrations occurring near or within genes, especially proto-oncogenes and tumor suppressor genes. In this regard, genome-wide studies of retroviral DNA integration have elucidated distinct integration target site preferences for different retroviral vectors. While gammaretroviral vectors preferentially integrate in the proximity of transcription start sites, CpG islands, and genes with implications in cancer, lentiviral vectors tend to integrate within transcription units of actively transcribed genes [27,31,32,33,34,35,36,37,38,39,40,41]. These integration target site preferences are influenced by retrovirus-specific interactions of the retroviral integrase with cellular tethering factors, such as lens-epithelium-derived growth factor/p75 (LEDGF) in the lentiviral [42,43,44,45,46] and bromodomain and extraterminal domain (BET) proteins in the gammaretroviral context [47,48,49]. These tethering factors direct retroviral integrations to specific regions in the genome and thus largely contribute to integration target site preferences. There have been attempts to modify retroviral vectors and/or their tethering factors with the aim to obtain potentially safer integration characteristics [50,51,52,53,54,55,56]. However, some of these modifications suffered from reduced gene transfer efficacy or incomplete redirection of integration site preference and clinical applicability remains to be shown. Importantly, in contrast to gammaretroviral and lentiviral vectors, alpharetroviral vectors have a relatively neutral integration spectrum [57,58,59,60,61]. It is currently unknown whether alpharetroviral integration follows a pattern yet to be identified or if it occurs independently from tethering factors. Nevertheless, the comparatively neutral integration pattern of alpharetroviral vectors renders them less genotoxic [59,60,61] and thus increases their therapeutic value for future human gene therapy.
3.4. Alpharetroviral Vector Developments
Compliance with the aforementioned retroviral safety criteria is necessary in order to take advantage of the comparatively neutral alpharetroviral integration pattern for clinical applications. The most widely used alpharetroviral vector system is the RCAS (replication-competent avian leukosis virus LTR with a splice acceptor) system [74,75]. RCAS is based on RSV, in which the captured SRC oncogene has been exchanged by a unique restriction site, via which a transgene of interest can be inserted. The original RCAS vector is replication-competent in avian cells (its natural host) and expresses the transgene from a spliced message. Due to its replication-competence, high viral titers can be achieved in avian cells. In order to transduce mammalian cells, the alpharetroviral envelope glycoprotein was replaced by envelope glycoproteins derived from other retroviruses in a process called pseudotyping. While this allows transduction of mammalian cells, it generally precludes alpharetroviral replication in mammalian cells due to several blocks in the life cycle (as reviewed by [76]). Nevertheless, for clinical applications, the potential for viral replication/mobilization should be entirely eliminated, and thus pseudotyped RCAS vectors are unlikely to fulfill current clinical safety criteria. However, they are extremely useful for nonclinical applications and have been successfully used to transduce rhesus macaque hematopoietic stem and progenitor cells. After transplantation, long-term polyclonal engraftment with gene marking in myeloid and lymphoid lineages was observed, thus clearly underlining the clinical relevance of alpharetroviral vectors [77]. RCAS-derived vectors lacking the envelope gene have been designed to facilitate the generation of variably pseudotyped alpharetroviral vectors. Pseudotyped alpharetroviral vectors can be produced to allow the transduction of mammalian cells by providing the genetic information for the envelope in trans in packaging cells [78,79]. Since the vectors lacking the envelope gene do not contain all the information needed for viral replication, these vectors can be considered replication-defective even in avian cells. However, they lack the advanced split-packaging design and the SIN deletion, which are present in state-of-the-art gammaretroviral and lentiviral vectors and which are required for clinical applications [80,81,82].
Aiming for a clinically applicable alpharetroviral vector system, we designed an advanced split-packaging design completely avoiding sequence overlaps between the constructs expressing retroviral proteins in trans and the actual genomic message to be packaged [83] (Figure 3), thus minimizing the risk of RCR formation. The resulting transfer vector is also devoid of splice sites as the alpharetroviral splice donor is within the gag reading frame, further preventing potentially genotoxic splicing events. This is in contrast to gammaretroviral and lentiviral vectors, which still contain retroviral splice sites. Using the alpharetroviral split-packaging system, we successfully generated alpharetroviral particles from human 293T cells via transient plasmid transfection, albeit with low titers. However, codon-optimization of the alpharetroviral gag-pro/pol sequence for human tRNA codon-usage preferences spared ~300 bp in the gag-pro/pol transition region required for ribosomal frameshifting (Figure 3), and drastically increased titers by several orders of magnitude (approx. 107 transducing units (TU)/mL) [83].
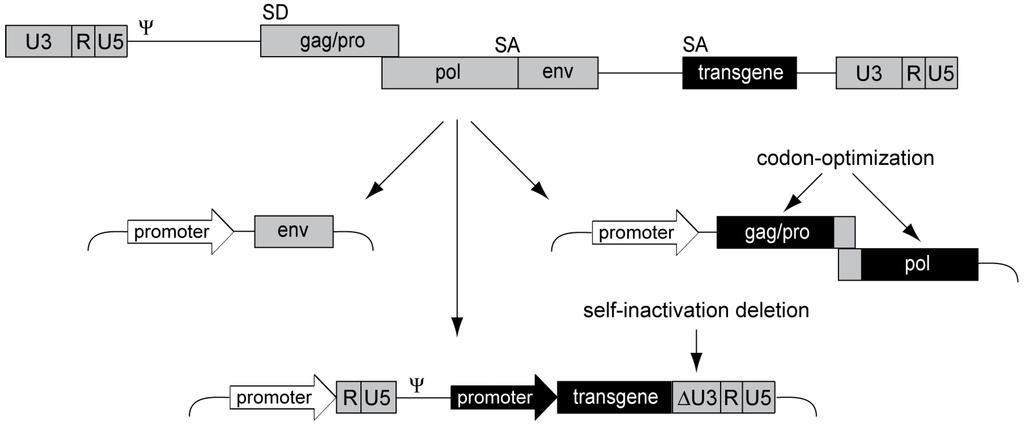
Figure 3.
Alpharetroviral SIN split-packaging system. Schematic depiction of the alpharetroviral split-packaging system, with a replication-competent alpharetrovirus shown on top and the three respective split-packaging system components shown below. The viral coding sequence components are split onto two plasmids, one encoding the viral envelope (env) and one encoding the gag-pro/pol polyprotein. The latter was codon-optimized, sparing approximately 300 bp in the gag-pro/pol transition region to ensure proper frameshifting. The vector constitutes the third component of the split-packaging system. An external promoter drives the expression of the RNA, which contains an internal transgene expression cassette, a packaging signal (Ψ), and R, U5 and U3 regions, allowing for packaging, reverse transcription and integration. An alpharetroviral SIN vector was designed by removing transcriptional control elements from the U3 region (ΔU3) of the LTR.
Figure 3.
Alpharetroviral SIN split-packaging system. Schematic depiction of the alpharetroviral split-packaging system, with a replication-competent alpharetrovirus shown on top and the three respective split-packaging system components shown below. The viral coding sequence components are split onto two plasmids, one encoding the viral envelope (env) and one encoding the gag-pro/pol polyprotein. The latter was codon-optimized, sparing approximately 300 bp in the gag-pro/pol transition region to ensure proper frameshifting. The vector constitutes the third component of the split-packaging system. An external promoter drives the expression of the RNA, which contains an internal transgene expression cassette, a packaging signal (Ψ), and R, U5 and U3 regions, allowing for packaging, reverse transcription and integration. An alpharetroviral SIN vector was designed by removing transcriptional control elements from the U3 region (ΔU3) of the LTR.
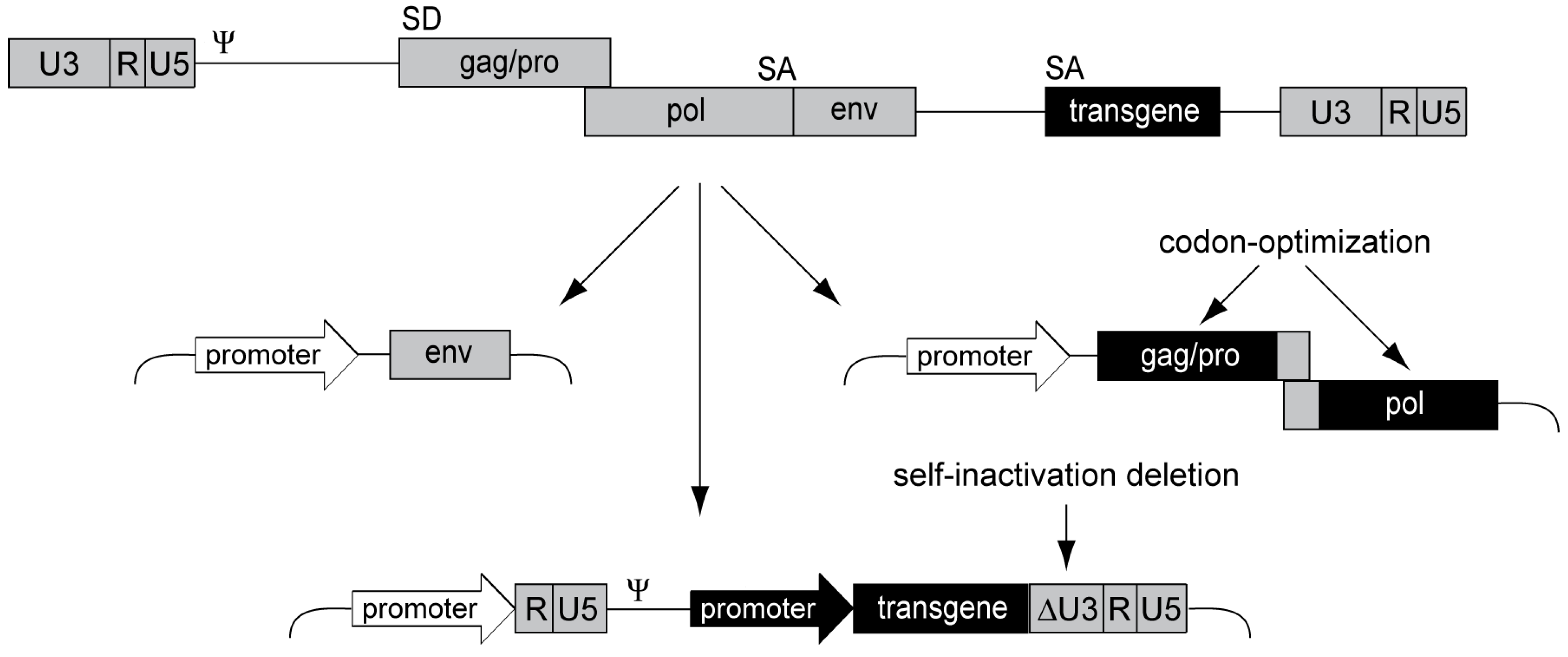
In a next step, we removed transcriptional control elements from the LTRs to generate alpharetroviral SIN vectors with reduced genotoxic potential and the ability to mediate physiologic transgene expression levels via internal promoters (Figure 3). Initially, we deleted 159 nucleotides of the wild-type U3 region, preserving the terminal 3' and 5' U3 sequences to maintain integrase attachment and polyadenylation sites [83]. While we showed that this initial SIN design reduced U3 transcriptional activity to background levels, this vector still contained the TATA box, a core promoter element of retroviruses. To exclude potential transcription due to this promoter element, we generated a second generation SIN vector by deleting the TATA box [60]. Importantly, both SIN designs eliminated several enhancer elements from the U3 region, such as CAAT enhancers, Y box motifs, and CArG boxes [84], thus increasing retroviral vector genosafety [69,85,86] and avoiding interference with the regulation of the internal promoter.
Using this latest generation of alpharetroviral SIN vectors, we and others demonstrated a comparatively neutral integration pattern in murine hematopoietic stem and progenitor cells, reduced genotoxicity in sensitive in vitro immortalization assays [60], and lack of aberrant splicing [87] compared to clinically used gammaretroviral and/or lentiviral SIN vectors. In more sophisticated integration site analyses in human hematopoietic stem and progenitor cells, alpharetroviral vectors exhibited a comparatively neutral integration pattern with regard to several annotated genomic features and potentially dangerous genomic loci, as well as with regard to epigenetically-defined functional genomic regions, such asenhancers (H3K4me1+) and promoters (H3K4me3+) when compared to gammaretroviral or transcribed gene bodies (H3K36me3) when compared to lentiviral vectors [61]. Finally, proof-of-principle was provided for the alpharetroviral genetic modification of hematopoietic stem and progenitor cells in mice [60], the modification of human T lymphocytes and NK cells (unpublished data), and phenotypic correction of X-linked CGD in a humanized mouse model [87].
In general, retroviral gene expression can be hampered by silencing, one of the cellular defense mechanisms against foreign DNA [88], leading to the downregulation of transgene expression. Silencing is mediated by cellular factors, which can recognize repressive elements on the vector, such as the retroviral primer binding site [89] or parts of the LTRs [90]. Silencing results in the formation of repressive epigenetic marks, including DNA methylation and histone deacetylation. In addition to gammaretro- and lentiviral vectors (reviewed in [91]), silencing has also been described in the context of alpharetroviral vectors [92,93,94,95,96,97,98]. It can generally be influenced by the target cell species [96], the differentiation status of the cell [99], the cell type [100] and the position of the integration site [101,102,103]. As alpharetroviral silencing has been reported to be more pronounced in mammalian than in avian cells [96], future modifications of alpharetroviral vectors should contain “antisilencing” modifications to ensure sustained therapeutic transgene expression. Such modifications include the removal of repressive elements from the vector and/or the insertion of supporting transcriptional regulatory elements. To this effect, the removal of viral coding sequences (split-packaging design) and the removal of transcriptional elements (SIN design) might already have eliminated repressive elements from the vector. In addition, transgene expression in SIN vectors is independent from LTR-mediated transcription and has been reported to be less susceptible to silencing in the context of gammaretro- and lentiviral SIN vectors (reviewed in [91]). Nevertheless, future incorporation of supporting transcriptional regulatory elements, such as scaffold/matrix attachment regions, insulators, locus control regions, ubiquitous chromatin opening elements [104] or chimeric versions thereof [105], is likely to enhance the therapeutic potential of alpharetroviral SIN vectors [76,106,107].
5. From Bench to Bedside: Regulatory Requirements for Clinical Translation
Gene therapy using retroviral vectors has proven to be very effective in several clinical trials targeting hematopoietic stem cells and T-lymphocytes. Previous trials primarily employed gammaretroviral and lentiviral vectors, but the knowledge gained from experiences with these retroviral family members can be applied to advance new generation alpharetroviral SIN vectors to the clinical arena.
Generally, regulatory agencies, e.g. the U.S. Food and Drug Administration (www.fda.gov) and the European Medicines Agency (www.ema.europa.eu), are responsible for reviewing and approving planned gene therapy clinical trials. In light of previous severe adverse events, it is imperative to prove that the intended alpharetroviral gene therapy strategy is safe. To meet this goal, several recommendations for advanced therapy medicinal products (ATMP, a therapeutic product based on genes, cells or tissues) have been established (see recommendations on aforementioned webpages). For the regulatory review process of ATMPs, careful risk-benefit assessments need to be performed in the context of the particular clinical indication under study. This includes characterization of the alpharetroviral vector, such as content (including the transgene), delivery mode, and the somatic cell therapy product’s behavior in different contexts (e.g., isolated tissues and living organisms). Kaufmann et al. [87] provided a good example of validation of alpharetroviral gene therapy for a specific disease in a preclinical mouse model employing transduced mouse hematopoietic stem cells and, as a second step, in a humanized mouse model using alpharetrovirally corrected patient cells.
In general, design of alpharetroviral vectors for clinical applications should comply with replication-incompetence, low genotoxicity, low immunogenicity, and the exclusion of germ-line integrations. Also the following points should be taken into account for the transgene and its product (see FDA guidelines): (i) preferably localized or lineage-specific expression rather than systemic/ubiquitous expression; (ii) low level and duration of expression (to avoid phenotoxicity); and (iii) anticipation of acute vs. long-term effects. With respect to transgenes, especially growth factors, growth factor receptors [128] and immune modulators should be handled with care.
There are basically two different ways to deliver therapeutic transgenes into patient cells—in vivo or ex vivo. While in vivo transgene delivery is relatively simple since it only requires the injection of purified virus into the target organ or into the bloodstream, it is associated with several concerns including toxicity, immunogenicity, lack of specificity and dose-control. In contrast, ex vivo transgene delivery is more complex. It requires isolation of the target cells, in vitro culture, and re-infusion after cell modification. However, ex vivo transgene delivery enables a tight control of specificity, vector dose, and circumvents human complement inactivation of retroviral particles. As for alpharetroviral gene therapy, the ex vivo method is thus preferred.
Preclinical investigation cannot be applied directly to patients because of regulatory requirements (except for the “hospital exemption clause/compassionate use”) and ethical concerns. Therefore, appropriate animal models have to be chosen, which should ideally be biologically relevant and should model the disease phenotype. In the case of a specific monogenetic disease, the corresponding knock out models should be used. Ideally, also the transplantation of patients’ cells in so called “humanized” mouse models, which accept human grafts, e.g., the NOD/Prkdcscid/Il2rg (NSG) knock out model, should be performed.
Candidate target cells for clinical application include human CD34+ hematopoietic precursor cells and T cells. Both cell types can be efficiently transduced with alpharetroviral vectors, as demonstrated by our group [60] (and unpublished data). Moreover, Kaufmann et al. [87] showed efficient transduction of X-linked chronic granulomatous disease (CGD) patient derived CD34+ cells with an alpharetroviral vector expressing a functional version of the defective gene. Importantly, upon transplantation of the gene-modified cells into the NSG mouse model, they demonstrated functional disease correction and long-term expression of the transgene without obvious side effects. Interestingly, no aberrant splicing, caused by interference of viral and cellular splice sites, was observed for alpharetroviral in contrast to lentiviral vectors, implying that the safety profile of alpharetroviral vectors may even extend beyond their more neutral integration pattern in clinically relevant human CD34+ cells [61,87].
In addition, some overall safety considerations of alpharetrovirally genetically modified cells as somatic cell therapy medicinal products need to be addressed in vivo. This includes the analysis of (i) biodistribution and toxicology (to identify potential sites of toxicity), (ii) immuno-toxicity/immunogenicity (vector/transgene products affecting the immune system, pre-exisiting immunity and cross-reactivity) and (iii) shedding issues of retroviral vectors [129]. Ideally, most analyses should be accomplished with the clinical vector production lot and, if possible, in a controlled environment and in GMP-like compliance.
7. Conclusions and Outlook
Alpharetroviruses were discovered more than 100 years ago. However they have come into play as potential retroviral vectors for human gene therapy only very recently, when serious adverse events in clinical trials underscored the need for safer vectors. Intensive research efforts revealed that retroviral vector genotoxicity is largely influenced by the vector architecture and the integration pattern. Thus, alpharetroviral vectors, which have a relatively neutral integration pattern compared to clinically used gammaretroviral and lentiviral vectors, represent attractive and potentially safer tools for human gene therapy. We applied the latest concepts of retroviral vector design to develop state-of-the-art alpharetroviral SIN vectors with a split-packaging design. Proof-of-principle for these vectors was provided by the genetic modification of clinically relevant target cells, such as hematopoietic stem and progenitor cells and T-lymphocytes. Moreover, future incorporation of supporting transcriptional regulatory elements protecting from cellular silencing mechanisms is likely to further enhance the therapeutic potential of alpharetroviral vectors. Since upscaling retroviral vector production represents one of the major challenges on the road to clinical translation, it is especially promising that alpharetroviral SIN vectors can be produced transiently as well as from stable human packaging cell lines. Compliance with several regulatory requirements, including performance of preclinical biodistribution and toxicology studies of the gene therapy product and careful risk-benefit assessments are still needed to further advance alpharetroviral vectors to the clinical arena. Altogether, the reduced genotoxicity in combination with the perspective of economic, stable vector production underlines the potential of alpharetroviral SIN vectors as useful tools for future human gene therapy strategies. Thus, as paradoxical as it seems, a viral genus, first noted for its transforming potential, might contribute to safer human gene therapy in the near future.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB738, Cluster of Excellence REBIRTH (EXC 62/1), SPP1230), the Bundesministerium für Bildung und Forschung (BMBF, Joint Research Project IFB-Tx, PidNet), the DAAD (Modern Applications in Biotechnology) and the European Union (FP7 project PERSIST). We would like to thank Christopher Baum for his contribution and support for developing alpharetroviral SIN vectors and Michael Morgan for proofreading the final version of the manuscript.
This review article is dedicated to Axel Rethwilm, who initiated this Special Issue on Retroviral Vectors together with Dirk Lindemann.
Conflicts of Interest
The authors declare that they have no conflict of interest, except that Axel Schambach and Julia D. Suerth are inventors on a patent application describing alpharetroviral SIN vectors.
References and Notes
- Ellermann, V.; Bang, O. Experimentelle Leukämie bei Hühnern. Zentralbl. Bakteriol. Parasitenkd. Infektionskr. Hyg. Abt. I. 1908, 46, 595–609. [Google Scholar]
- Rous, P. A transmissible avian neoplasm. (sarcoma of the common fowl.). J. Exp. Med. 1910, 12, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Diamond, L.; Wolman, S.R. Charlotte friend, ph.D. 1921–1987. A scientist’s life. Ann. N. Y. Acad. Sci. 1989, 567, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Rubin, H. Characteristics of an assay for Rous sarcoma virus and Rous sarcoma cells in tissue culture. Virology 1958, 6, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Bather, R. The nucleic acid of partially purified Rous No. I sarcoma virus. Br. J. Cancer 1957, 11, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Rubin, H.; Temin, H.M. A radiological study of cell-virus interaction in the Rous sarcoma. Virology 1959, 7, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M. The Participation of DNA in Rous Sarcoma Virus Production. Virology 1964, 23, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, J.; Chyle, P.; Simkovic, D.; Hilgert, I. Demonstration of the absence of infectious Rous virus in rat tumour XC, whose structurally intact cells produce Rous sarcoma when transferred to chicks. Folia Biol. 1963, 9, 77–81. [Google Scholar]
- Baltimore, D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Mizutani, S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 1970, 226, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Hayward, W.S.; Neel, B.G.; Astrin, S.M. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 1981, 290, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Hayward, W.S.; Robinson, H.L.; Fang, J.; Astrin, S.M. Avian leukosis virus-induced tumors have common proviral integration sites and synthesize discrete new RNAs: Oncogenesis by promoter insertion. Cell 1981, 23, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.; Mulligan, R.C.; Baltimore, D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 1983, 33, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.A.; Lemischka, I.R.; Nathan, D.G.; Mulligan, R.C. Introduction of new genetic material into pluripotent haematopoietic stem cells of the mouse. Nature 1984, 310, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E.; Magli, M.C.; Huszar, D.; Phillips, R.A.; Bernstein, A. Introduction of a selectable gene into primitive stem cells capable of long-term reconstitution of the hemopoietic system of W/Wv mice. Cell 1985, 42, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B. Update on gene therapy for immunodeficiencies. Clin. Immunol. 2010, 135, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kuhlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Gohring, G.; Steinemann, D.; et al. Gene therapy for Wiskott-Aldrich syndrome—Long-term efficacy and genotoxicity. Sci. Transl. Med. 2014, 6, 227ra233. [Google Scholar] [CrossRef]
- Lazo, P.A.; Lee, J.S.; Tsichlis, P.N. Long-distance activation of the Myc protooncogene by provirus insertion in Mlvi-1 or Mlvi-4 in rat T-cell lymphomas. Proc. Natl. Acad. Sci. USA 1990, 87, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, C.; Ihle, J.N. Retroviral insertions 90 kilobases proximal to the Evi-1 myeloid transforming gene activate transcription from the normal promoter. Mol. Cell Biol. 1991, 11, 1820–1828. [Google Scholar] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Invest. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Heckl, D.; Schwarzer, A.; Haemmerle, R.; Steinemann, D.; Rudolph, C.; Skawran, B.; Knoess, S.; Krause, J.; Li, Z.; Schlegelberger, B.; et al. Lentiviral vector induced insertional haploinsufficiency of Ebf1 causes murine leukemia. Mol. Ther. 2012, 20, 1187–1195. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Sokol, M.; Wabl, M.; Ruiz, I.R.; Pedersen, F.S. Novel principles of gamma-retroviral insertional transcription activation in murine leukemia virus-induced end-stage tumors. Retrovirology 2014, 11, 36. [Google Scholar] [CrossRef]
- Mooslehner, K.; Karls, U.; Harbers, K. Retroviral integration sites in transgenic Mov mice frequently map in the vicinity of transcribed DNA regions. J. Virol. 1990, 64, 3056–3058. [Google Scholar] [PubMed]
- Scherdin, U.; Rhodes, K.; Breindl, M. Transcriptionally active genome regions are preferred targets for retrovirus integration. J. Virol. 1990, 64, 907–912. [Google Scholar] [PubMed]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Hematti, P.; Hong, B.K.; Ferguson, C.; Adler, R.; Hanawa, H.; Sellers, S.; Holt, I.E.; Eckfeldt, C.E.; Sharma, Y.; Schmidt, M.; et al. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004, 2, e423. [Google Scholar] [CrossRef]
- Barr, S.D.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. Integration targeting by avian sarcoma-leukosis virus and human immunodeficiency virus in the chicken genome. J. Virol. 2005, 79, 12035–12044. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Laufs, S.; Blake, J.; Schwager, C.; Wu, X.; Zeller, J.W.; Ho, A.D.; Fruehauf, S. Retroviral integration sites correlate with expressed genes in hematopoietic stem cells. Stem Cells 2005, 23, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Beard, B.C.; Dickerson, D.; Beebe, K.; Gooch, C.; Fletcher, J.; Okbinoglu, T.; Miller, D.G.; Jacobs, M.A.; Kaul, R.; Kiem, H.P.; et al. Comparison of HIV-derived lentiviral and MLV-based gammaretroviral vector integration sites in primate repopulating cells. Mol. Ther. 2007, 15, 1356–1365. [Google Scholar] [CrossRef]
- Beard, B.C.; Keyser, K.A.; Trobridge, G.D.; Peterson, L.J.; Miller, D.G.; Jacobs, M.; Kaul, R.; Kiem, H.P. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum. Gene Ther. 2007, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Brady, T.; Agosto, L.M.; Malani, N.; Berry, C.C.; O’Doherty, U.; Bushman, F. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. Aids 2009, 23, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One 2007, 2, e1340. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; de Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Vanegas, M.; Fregoso, O.; Saenz, D.; Chung, S.; Peretz, M.; Poeschla, E.M. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 2004, 78, 9524–9537. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J.; de Kogel, C.; Demeulemeester, J.; Vets, S.; El Ashkar, S.; Malani, N.; Bushman, F.D.; Landuyt, B.; Husson, S.J.; Busschots, K.; et al. The bet family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell. Rep. 2013, 5, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, R.; Ronen, K.; Vets, S.; Malani, N.; de Rijck, J.; McNeely, M.; Bushman, F.D.; Debyser, Z. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 2010, 18, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Vets, S.; De Rijck, J.; Brendel, C.; Grez, M.; Bushman, F.; Debyser, Z.; Gijsbers, R. Transient Expression of an LEDGF/p75 Chimera Retargets Lentivector Integration and Functionally Rescues in a Model for X-CGD. Mol. Ther. Nucleic Acids 2013, 2, e77. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Shun, M.C.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 2009, 5, e1000259. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Diamond, T.L.; Hwang, Y.; Marshall, H.M.; Bushman, F.D. Modulating target site selection during human immunodeficiency virus DNA integration in vitro with an engineered tethering factor. Hum. Gene Ther. 2006, 17, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Ferris, A.L.; Wu, X.; Hughes, C.M.; Stewart, C.; Smith, S.J.; Milne, T.A.; Wang, G.G.; Shun, M.C.; Allis, C.D.; Engelman, A.; et al. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. USA 2010, 107, 3135–3140. [Google Scholar] [CrossRef] [PubMed]
- Aiyer, S.; Swapna, G.V.; Malani, N.; Aramini, J.M.; Schneider, W.M.; Plumb, M.R.; Ghanem, M.; Larue, R.C.; Sharma, A.; Studamire, B.; et al. Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res. 2014, 42, 5917–5928. [Google Scholar] [CrossRef] [PubMed]
- El Ashkar, S.; de Rijck, J.; Demeulemeester, J.; Vets, S.; Madlala, P.; Cermakova, K.; Debyser, Z.; Gijsbers, R. BET-independent MLV-based vectors target away from promoters and regulatory elements. Mol. Ther. Nucleic Acids 2014, 3, e179. [Google Scholar]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef] [PubMed]
- Narezkina, A.; Taganov, K.D.; Litwin, S.; Stoyanova, R.; Hayashi, J.; Seeger, C.; Skalka, A.M.; Katz, R.A. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 2004, 78, 11656–11663. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Renaud, G.; Gomes, T.J.; Ferris, A.; Hendrie, P.C.; Donahue, R.E.; Hughes, S.H.; Wolfsberg, T.G.; Russell, D.W.; Dunbar, C.E. Reduced genotoxicity of avian sarcoma leukosis virus vectors in rhesus long-term repopulating cells compared to standard murine retrovirus vectors. Mol. Ther. 2008, 16, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.D.; Maetzig, T.; Brugman, M.H.; Heinz, N.; Appelt, J.U.; Kaufmann, K.B.; Schmidt, M.; Grez, M.; Modlich, U.; Baum, C.; et al. Alpharetroviral self-inactivating vectors: Long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol. Ther. 2012, 20, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Moiani, A.; Suerth, J.D.; Gandolfi, F.; Rizzi, E.; Severgnini, M.; de Bellis, G.; Schambach, A.; Mavilio, F. Genome-wide analysis of alpharetroviral integration in human hematopoietic stem/progenitor cells. Genes 2014, 5, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.E.; Kessler, S.W.; Bodine, D.; McDonagh, K.; Dunbar, C.; Goodman, S.; Agricola, B.; Byrne, E.; Raffeld, M.; Moen, R. Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J. Exp. Med. 1992, 176, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Ranzani, M.; Volpin, M.; Bartholomae, C.; Duros, C.; Artus, A.; Merella, S.; Benedicenti, F.; Sergi Sergi, L.; Sanvito, F.; et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol. Ther. 2014, 22, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Temin, H.M. Construction of a helper cell line for avian reticuloendotheliosis virus cloning vectors. Mol. Cell Biol. 1983, 3, 2241–2249. [Google Scholar] [PubMed]
- Miller, A.D.; Rosman, G.J. Improved retroviral vectors for gene transfer and expression. Biotechniques 1989, 7, 980–990. [Google Scholar] [PubMed]
- Soneoka, Y.; Cannon, P.M.; Ramsdale, E.E.; Griffiths, J.C.; Romano, G.; Kingsman, S.M.; Kingsman, A.J. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 1995, 23, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; von Ruden, T.; Kantoff, P.W.; Garber, C.; Seiberg, M.; Ruther, U.; Anderson, W.F.; Wagner, E.F.; Gilboa, E. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc. Natl. Acad. Sci. USA 1986, 83, 3194–3198. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [PubMed]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Baum, C.; Dullmann, J.; Li, Z.; Fehse, B.; Meyer, J.; Williams, D.A.; von Kalle, C. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood 2003, 101, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Willemsen, R.; van Elzakker, P.; van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Kondo, E.; Akatsuka, Y.; Nawa, A.; Kuzushima, K.; Tsujimura, K.; Tanimoto, M.; Kodera, Y.; Morishima, Y.; Kuzuya, K.; Takahashi, T. Retroviral vector backbone immunogenicity: Identification of cytotoxic t-cell epitopes in retroviral vector-packaging sequences. Gene Ther. 2005, 12, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Federspiel, M.J.; Hughes, S.H. Retroviral gene delivery. Methods Cell Biol. 1997, 52, 179–214. [Google Scholar] [PubMed]
- Hughes, S.H. The RCAS vector system. Folia Biol. 2004, 50, 107–119. [Google Scholar]
- Svoboda, J. Molecular biology of cell nonpermissiveness to retroviruses. Has the time come? Gene 1998, 206, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ferris, A.; Larochelle, A.; Krouse, A.E.; Metzger, M.E.; Donahue, R.E.; Hughes, S.H.; Dunbar, C.E. Transduction of rhesus macaque hematopoietic stem and progenitor cells with avian sarcoma and leukosis virus vectors. Hum. Gene Ther. 2007, 18, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Roth, M.G.; Hunter, E. A chimeric avian retrovirus containing the influenza virus hemagglutinin gene has an expanded host range. J. Virol. 1992, 66, 7374–7382. [Google Scholar] [PubMed]
- Barsov, E.V.; Hughes, S.H. Gene transfer into mammalian cells by a Rous sarcoma virus-based retroviral vector with the host range of the amphotropic murine leukemia virus. J. Virol. 1996, 70, 3922–3929. [Google Scholar] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [PubMed]
- Kraunus, J.; Schaumann, D.H.S.; Meyer, J.; Modlich, U.; Fehse, B.; Brandenburg, G.; von Laer, D.; Klump, H.; Schambach, A.; Bohne, J.; et al. Self-inactivating retroviral vectors with improved RNA processing. Gene Therapy 2004, 11, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Schambach, A.; Mueller, D.; Galla, M.; Verstegen, M.M.; Wagemaker, G.; Loew, R.; Baum, C.; Bohne, J. Overcoming promoter competition in packaging cells improves production of self-inactivating retroviral vectors. Gene Ther. 2006, 13, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.D.; Maetzig, T.; Galla, M.; Baum, C.; Schambach, A. Self-inactivating alpharetroviral vectors with a split-packaging design. J. Virol. 2010, 84, 6626–6635. [Google Scholar] [CrossRef] [PubMed]
- Ruddell, A. Transcription regulatory elements of the avian retroviral long terminal repeat. Virology 1995, 206, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Russ, J.L.; Eiden, M.V. Evaluation of residual promoter activity in gamma-retroviral self-inactivating (SIN) vectors. Mol. Ther. 2012, 20, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.B.; Brendel, C.; Suerth, J.D.; Mueller-Kuller, U.; Chen-Wichmann, L.; Schwable, J.; Pahujani, S.; Kunkel, H.; Schambach, A.; Baum, C.; et al. Alpharetroviral vector-mediated gene therapy for X-CGD: Functional correction and lack of aberrant splicing. Mol. Ther. 2012, 21, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Barklis, E.; Mulligan, R.C.; Jaenisch, R. Chromosomal position or virus mutation permits retrovirus expression in embryonal carcinoma cells. Cell 1986, 47, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.R.; Krieg, A.M.; Max, E.E.; Khan, A.S. Negative control region at the 5' end of murine leukemia virus long terminal repeats. Mol. Cell Biol. 1989, 9, 739–746. [Google Scholar] [PubMed]
- Ellis, J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005, 16, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Katz, R.A.; Ishov, A.M.; Maul, G.G.; Skalka, A.M. The cellular protein daxx interacts with avian sarcoma virus integrase and viral DNA to repress viral transcription. J. Virol. 2005, 79, 4610–4618. [Google Scholar] [CrossRef] [PubMed]
- Poleshko, A.; Palagin, I.; Zhang, R.; Boimel, P.; Castagna, C.; Adams, P.D.; Skalka, A.M.; Katz, R.A. Identification of cellular proteins that maintain retroviral epigenetic silencing: Evidence for an antiviral response. J. Virol. 2008, 82, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Katz, R.A.; Jack-Scott, E.; Narezkina, A.; Palagin, I.; Boimel, P.; Kulkosky, J.; Nicolas, E.; Greger, J.G.; Skalka, A.M. High-frequency epigenetic repression and silencing of retroviruses can be antagonized by histone deacetylase inhibitors and transcriptional activators, but uniform reactivation in cell clones is restricted by additional mechanisms. J. Virol. 2007, 81, 2592–2604. [Google Scholar] [CrossRef] [PubMed]
- Shalginskikh, N.; Poleshko, A.; Skalka, A.M.; Katz, R.A. Retroviral DNA methylation and epigenetic repression are mediated by the antiviral host protein Daxx. J. Virol. 2013, 87, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Hejnar, J.; Plachy, J.; Geryk, J.; Machon, O.; Trejbalova, K.; Guntaka, R.V.; Svoboda, J. Inhibition of the rous sarcoma virus long terminal repeat-driven transcription by in vitro methylation: Different sensitivity in permissive chicken cells versus mammalian cells. Virology 1999, 255, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Hejnar, J.; Svoboda, J.; Geryk, J.; Fincham, V.J.; Hak, R. High rate of morphological reversion in tumor cell line H-19 associated with permanent transcriptional suppression of the LTR, v-src, LTR provirus. Cell Growth Differ. 1994, 5, 277–285. [Google Scholar] [PubMed]
- Searle, S.; Gillespie, D.A.; Chiswell, D.J.; Wyke, J.A. Analysis of the variations in proviral cytosine methylation that accompany transformation and morphological reversion in a line of Rous sarcoma virus-infected Rat-1 cells. Nucleic Acids Res. 1984, 12, 5193–5210. [Google Scholar] [CrossRef] [PubMed]
- Jahner, D.; Stuhlmann, H.; Stewart, C.L.; Harbers, K.; Lohler, J.; Simon, I.; Jaenisch, R. De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature 1982, 298, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Klug, C.A.; Cheshier, S.; Weissman, I.L. Inactivation of a GFP retrovirus occurs at multiple levels in long-term repopulating stem cells and their differentiated progeny. Blood 2000, 96, 894–901. [Google Scholar] [PubMed]
- Hoeben, R.C.; Migchielsen, A.A.; van der Jagt, R.C.; van Ormondt, H.; van der Eb, A.J. Inactivation of the Moloney murine leukemia virus long terminal repeat in murine fibroblast cell lines is associated with methylation and dependent on its chromosomal position. J. Virol. 1991, 65, 904–912. [Google Scholar] [PubMed]
- Plachy, J.; Kotab, J.; Divina, P.; Reinisova, M.; Senigl, F.; Hejnar, J. Proviruses selected for high and stable expression of transduced genes accumulate in broadly transcribed genome areas. J. Virol. 2010, 84, 4204–4211. [Google Scholar] [CrossRef] [PubMed]
- Senigl, F.; Auxt, M.; Hejnar, J. Transcriptional provirus silencing as a crosstalk of de novo DNA methylation and epigenomic features at the integration site. Nucleic Acids Res. 2012, 40, 5298–5312. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, M.N.; Skipper, K.A.; Anakok, O. Optimizing retroviral gene expression for effective therapies. Hum. Gene Ther. 2013, 24, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Benabdellah, K.; Gutierrez-Guerrero, A.; Cobo, M.; Munoz, P.; Martin, F. A chimeric HS4-SAR insulator (IS2) that prevents silencing and enhances expression of lentiviral vectors in pluripotent stem cells. PLoS One 2014, 9, e84268. [Google Scholar] [CrossRef] [PubMed]
- Senigl, F.; Plachy, J.; Hejnar, J. The core element of a CpG island protects avian sarcoma and leukosis virus-derived vectors from transcriptional silencing. J. Virol. 2008, 82, 7818–7827. [Google Scholar] [CrossRef] [PubMed]
- Hejnar, J.; Hajkova, P.; Plachy, J.; Elleder, D.; Stepanets, V.; Svoboda, J. CpG island protects Rous sarcoma virus-derived vectors integrated into nonpermissive cells from DNA methylation and transcriptional suppression. Proc. Natl. Acad. Sci. USA 2001, 98, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Merten, O.W. State-of-the-art of the production of retroviral vectors. J. Gene Med. 2004, 6, S105–S124. [Google Scholar] [CrossRef] [PubMed]
- Coroadinha, A.S.; Gama-Norton, L.; Amaral, A.I.; Hauser, H.; Alves, P.M.; Cruz, P.E. Production of retroviral vectors: Review. Curr. Gene Ther. 2010, 10, 456–473. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Starkey, W.; Vile, R.G. A replication-competent retrovirus arising from a split-function packaging cell line was generated by recombination events between the vector, one of the packaging constructs, and endogenous retroviral sequences. J. Virol. 1998, 72, 2663–2670. [Google Scholar] [PubMed]
- U.S. Food and Drug Administration. Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy. Available online: http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm072987.htm (accessed on 1 December 2014).
- Ni, Y.; Sun, S.; Oparaocha, I.; Humeau, L.; Davis, B.; Cohen, R.; Binder, G.; Chang, Y.N.; Slepushkin, V.; Dropulic, B. Generation of a packaging cell line for prolonged large-scale production of high-titer HIV-1-based lentiviral vector. J. Gene Med. 2005, 7, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Otto, E.; Jones-Trower, A.; Vanin, E.F.; Stambaugh, K.; Mueller, S.N.; Anderson, W.F.; McGarrity, G.J. Characterization of a replication-competent retrovirus resulting from recombination of packaging and vector sequences. Hum. Gene Ther. 1994, 5, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Von Werder, A.; Seidler, B.; Schmid, R.M.; Schneider, G.; Saur, D. Production of avian retroviruses and tissue-specific somatic retroviral gene transfer in vivo using the RCAS/TVA system. Nat. Protoc. 2012, 7, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Morgan, R.A.; Cornetta, K.; June, C.H.; Binder-Scholl, G.; Dudley, M.E.; Feldman, S.A.; Rosenberg, S.A.; Shurtleff, S.A.; Rooney, C.M.; et al. Replication-competent retroviruses in gene-modified T cells used in clinical trials: Is it time to revise the testing requirements? Mol. Ther. 2012, 20, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Takeuchi, Y.; Martin, F.; Cosset, F.L.; Mitrophanous, K.; Collins, M. Continuous high-titer HIV-1 vector production. Nat. Biotechnol. 2003, 21, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Cosset, F.L.; Legras, C.; Chebloune, Y.; Savatier, P.; Thoraval, P.; Thomas, J.L.; Samarut, J.; Nigon, V.M.; Verdier, G. A new avian leukosis virus-based packaging cell line that uses two separate transcomplementing helper genomes. J. Virol. 1990, 64, 1070–1078. [Google Scholar] [PubMed]
- Schaefer-Klein, J.; Givol, I.; Barsov, E.V.; Whitcomb, J.M.; VanBrocklin, M.; Foster, D.N.; Federspiel, M.J.; Hughes, S.H. The EV-O-derived cell line DF-1 supports the efficient replication of avian leukosis-sarcoma viruses and vectors. Virology 1998, 248, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Himly, M.; Foster, D.N.; Bottoli, I.; Iacovoni, J.S.; Vogt, P.K. The DF-1 chicken fibroblast cell line: Transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 1998, 248, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Loftus, S.K.; Larson, D.M.; Watkins-Chow, D.; Church, D.M.; Pavan, W.J. Generation of RCAS vectors useful for functional genomic analyses. DNA Res. 2001, 8, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.D.; Schambach, A.; Baum, C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr. Opin. Immunol. 2012, 24, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Hotta, A.; Saito, Y.; Kyogoku, K.; Kawabe, Y.; Nishijima, K.; Kamihira, M.; Iijima, S. Characterization of transient expression system for retroviral vector production. J. Biosci. Bioeng. 2006, 101, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Sandrin, V.; Boson, B.; Salmon, P.; Gay, W.; Negre, D.; Le Grand, R.; Trono, D.; Cosset, F.L. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 2002, 100, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Ghani, K.; Cottin, S.; Kamen, A.; Caruso, M. Generation of a high-titer packaging cell line for the production of retroviral vectors in suspension and serum-free media. Gene Ther. 2007, 14, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Le Doux, J.M.; Morgan, J.R.; Snow, R.G.; Yarmush, M.L. Proteoglycans secreted by packaging cell lines inhibit retrovirus infection. J. Virol. 1996, 70, 6468–6473. [Google Scholar] [PubMed]
- Suomalainen, M.; Garoff, H. Incorporation of homologous and heterologous proteins into the envelope of Moloney murine leukemia virus. J. Virol. 1994, 68, 4879–4889. [Google Scholar] [PubMed]
- Cosset, F.L.; Takeuchi, Y.; Battini, J.L.; Weiss, R.A.; Collins, M.K. High-titer packaging cells producing recombinant retroviruses resistant to human serum. J. Virol. 1995, 69, 7430–7436. [Google Scholar] [PubMed]
- Wicke, D.C.; Meyer, J.; Buesche, G.; Heckl, D.; Kreipe, H.; Li, Z.; Welte, K.H.; Ballmaier, M.; Baum, C.; Modlich, U. Gene therapy of MPL deficiency: Challenging balance between leukemia and pancytopenia. Mol. Ther. 2010, 18, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, C.; Galla, M.; Dannhauser, P.N.; Maetzig, T.; Sodeik, B.; Schambach, A.; Baum, C. Pseudotype-independent nonspecific uptake of gammaretroviral and lentiviral particles in human cells. Hum. Gene Ther. 2012, 23, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Hauer, J.; Lim, A.; Picard, C.; Wang, G.P.; Berry, C.C.; Martinache, C.; Rieux-Laucat, F.; Latour, S.; Belohradsky, B.H.; et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010, 363, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Cooray, S.; Gilmour, K.C.; Parsley, K.L.; Adams, S.; Howe, S.J.; Al Ghonaium, A.; Bayford, J.; Brown, L.; Davies, E.G.; et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2011, 3, 97ra79. [Google Scholar] [PubMed]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Aebersold, P.; Cornetta, K.; Kasid, A.; Morgan, R.A.; Moen, R.; Karson, E.M.; Lotze, M.T.; Yang, J.C.; Topalian, S.L.; et al. Gene transfer into humans—Immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J. Med. 1990, 323, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.P.; Xue, S.A.; Thomas, S.; Cesco-Gaspere, M.; Tranter, A.; Willcox, B.; Lee, S.P.; Steven, N.; Morris, E.C.; Stauss, H.J. Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther. 2008, 15, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Riviere, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Bonini, C.; Ferrari, G.; Verzeletti, S.; Servida, P.; Zappone, E.; Ruggieri, L.; Ponzoni, M.; Rossini, S.; Mavilio, F.; Traversari, C.; et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 1997, 276, 1719–1724. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 132ra153. [Google Scholar] [CrossRef]
- Newrzela, S.; Al-Ghaili, N.; Heinrich, T.; Petkova, M.; Hartmann, S.; Rengstl, B.; Kumar, A.; Jack, H.M.; Gerdes, S.; Roeder, I.; et al. T-cell receptor diversity prevents T-cell lymphoma development. Leukemia 2012, 26, 2499–2507. [Google Scholar] [CrossRef] [PubMed]
- Newrzela, S.; Cornils, K.; Li, Z.; Baum, C.; Brugman, M.H.; Hartmann, M.; Meyer, J.; Hartmann, S.; Hansmann, M.L.; Fehse, B.; et al. Resistance of mature T cells to oncogene transformation. Blood 2008, 112, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Rengstl, B.; Muik, A.; Petkova, M.; Schmid, F.; Wistinghausen, R.; Warner, K.; Crispatzu, G.; Hansmann, M.L.; Herling, M.; et al. Mature T-cell lymphomagenesis induced by retroviral insertional activation of janus kinase 1. Mol. Ther. 2013, 21, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).