Matrix and Backstage: Cellular Substrates for Viral Vaccines


Abstract
:1. Introduction
2. Finite Cell Cultures
2.1. Primary Monkey Kidney Cells

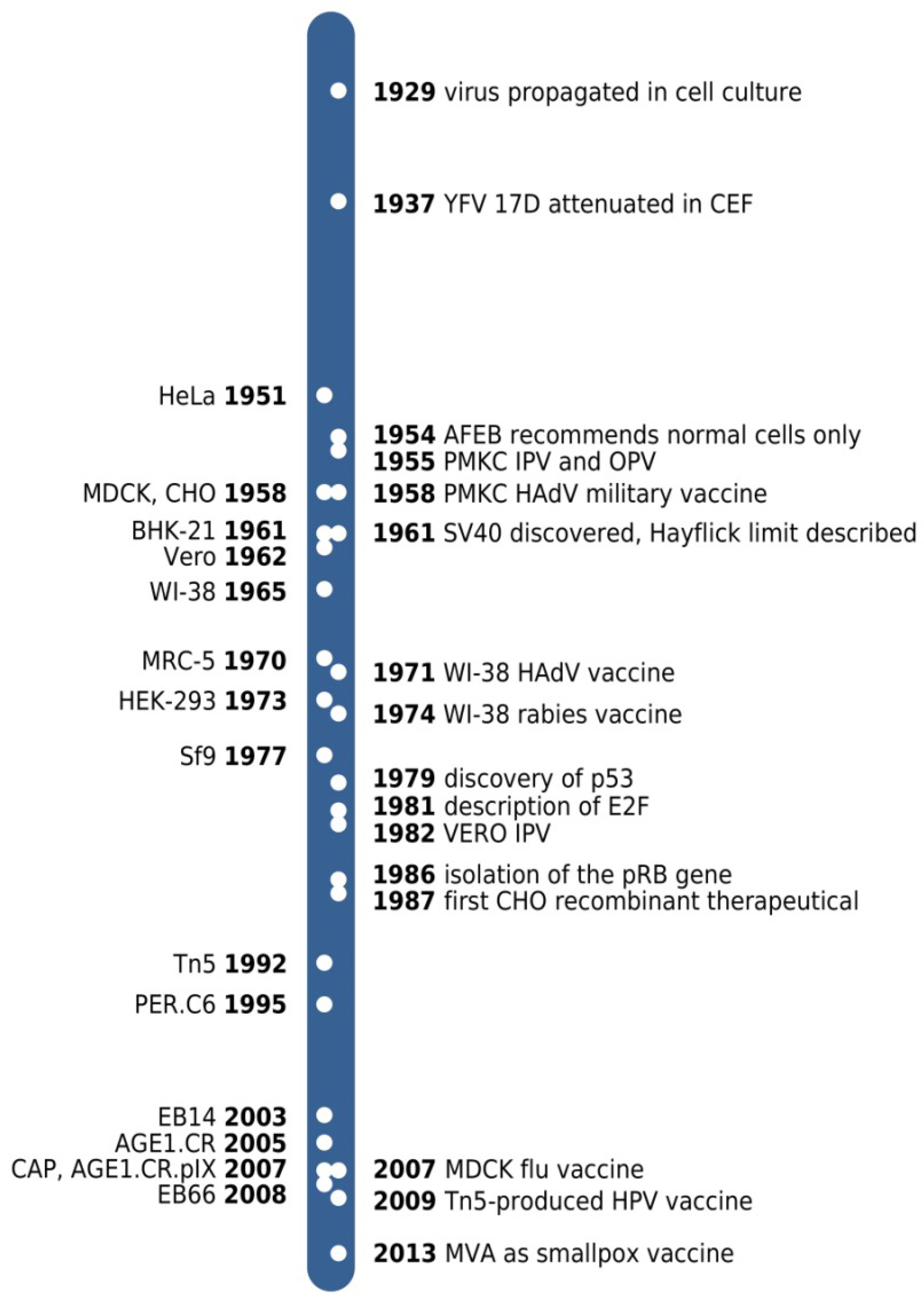{kind=link}
| killed | live | |||
|---|---|---|---|---|
| finite cultures | primary | monkey kidney | PV, HAdV | PV |
| mouse brain | HNTV, JEV | |||
| hamster kidney | JEV | |||
| embryonated eggs | IAV, IBV | LAIV, YFV | ||
| CEF | IAV, IBV, RABV, TBEV | MV, MuV MVA | ||
| passaged | FRhL-2 | RV | ||
| WI-38 | RUBV, HAdV | |||
| MRC-5 | HAV | PV, VZV | ||
| continuous | spontaneous | Vero | PV, JEV, RABV | RV,VACV |
| MDCK | IAV, IBV | |||
| Sf9, Tn5 | AcNPV | |||
| EB66 | ||||
| designed | HEK 293, PER.C6, CAP | |||
| AGE1.CR.pIX |
2.2. Chicken Embryo Fibroblasts
2.3. Diploid Cells
2.4. Disadvantages and Advantages of Finite Cell Substrates
3. Continuous Cell Lines
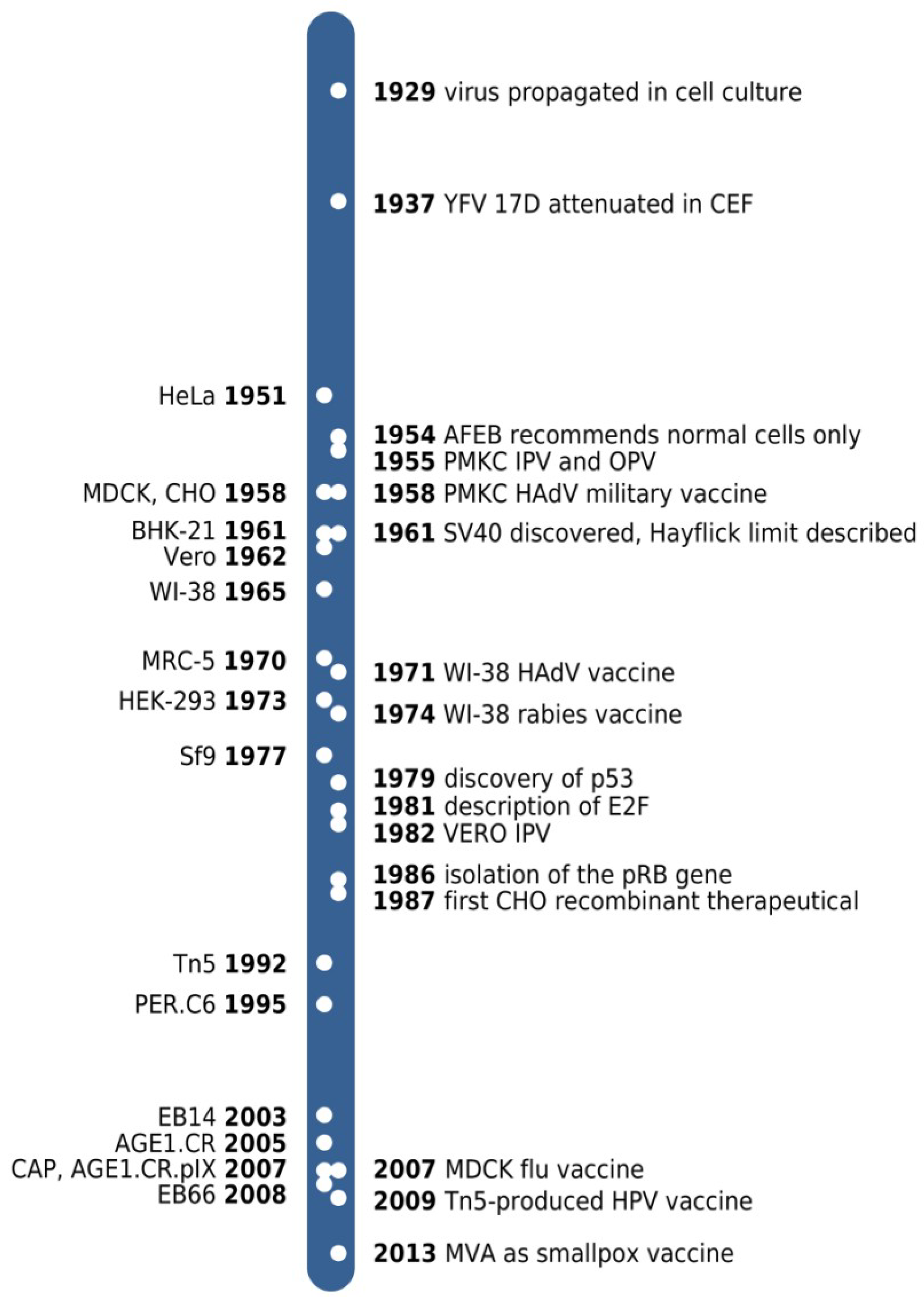
3.1. Vero
3.2. MDCK
3.3. BHK, CHO and EB66
3.4. Insect Cell Lines
3.5. Designed Cell Lines: Regulation of Cell Cycle Progression, Apoptosis and Senescence
3.6. Designed Cell Lines: HEK 293, PER.C6, CAP, CR.pIX
4. Regulatory Properties
4.1. Tumorigenicity
4.2. Oncogenicity
4.3. Adventitious Agents
4.4. Public Concerns and Misconceptions
5. Conclusions
Conflicts of Interest
References and Notes
- Rivers, T.M.; Haagen, E.; Muckenfuss, R.S. Observations concerning the persistence of living cells in maitland’s medium for the cultivation of vaccine virus. J. Exp. Med. 1929, 50, 181–187. [Google Scholar] [CrossRef]
- Theiler, M.; Smith, H.H. The use of yellow fever virus modified by in vitro cultivation for human immunization. J. Exp. Med. 1937, 65, 787–800. [Google Scholar] [CrossRef]
- Li, C.P.; Rivers, T.M. Cultivation of vaccine virus. J. Exp. Med. 1930, 52, 465–470. [Google Scholar] [CrossRef]
- Parker, F.; Nye, R.N. Studies on filterable viruses: I. Cultivation of vaccine virus. Am. J. Pathol. 1925, 1, 325–335. [Google Scholar]
- Webster, L.T.; Clow, A.D. Propagation of rabies virus in tissue culture. J. Exp. Med. 1937, 66, 125–131. [Google Scholar] [CrossRef]
- Schaeffer, W.I. Terminology associated with cell, tissue, and organ culture, molecular biology, and molecular genetics. Tissue culture association terminology committee. Vitro Cell Dev. Biol. J. Tissue Cult. Assoc. 1990, 26, 97–101. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The illusion of cell immortality. Br. J. Cancer 2000, 83, 841–846. [Google Scholar] [CrossRef]
- Hilleman, M.R. Efficacy of and indications for use of adenovirus vaccine. Am. J. Public Health Nations Health 1958, 48, 153–158. [Google Scholar] [CrossRef]
- Russell, K.L.; Hawksworth, A.W.; Ryan, M.A.K.; Strickler, J.; Irvine, M.; Hansen, C.J.; Gray, G.C.; Gaydos, J.C. Vaccine-preventable adenoviral respiratory illness in US military recruits, 1999–2004. Vaccine 2006, 24, 2835–2842. [Google Scholar]
- Salk, J.E. Studies in human subjects on active immunization against poliomyelitis. I. A preliminary report of experiments in progress. J. Am. Med. Assoc. 1953, 151, 1081–1098. [Google Scholar]
- Sabin, A.B.; Ramos-Alvarez, M.; Alvarez-Amezquita, J.; Pelon, W.; Michaels, R.H.; Spigland, I.; KOCH, M.A.; Barnes, J.M.; Rhim, J.S. Live, orally given poliovirus vaccine. Effects of rapid mass immunization on population under conditions of massive enteric infection with other viruses. JAMA J. Am. Med. Assoc. 1960, 173, 1521–1526. [Google Scholar] [CrossRef]
- Courtois, G.; Flack, A.; Jervis, G.A.; Koprowski, H.; Ninane, G. Preliminary report on mass vaccination of man with live attenuated poliomyelitis virus in the Belgian Congo and Ruanda-Urundi. Br. Med. J. 1958, 2, 187–190. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Lebrun, A.; Courtois, G.; Koprowski, H. Vaccination with the CHAT strain of type 1 attenuated poliomyelitis virus in Leopoldville, Congo. 3. Safety and efficacy during the first 21 months of study. Bull. World Health Org. 1961, 24, 785–792. [Google Scholar]
- Rhodes, A.J. Research on the development of a poliomyelitis vaccine: Toronto, 1950–1953. Can. Med. Assoc. J. 1956, 75, 48–49. [Google Scholar]
- Lambert, S.M.; Markel, H. Making history: Thomas Francis, MD, Jr., and the 1954 salk poliomyelitis vaccine field trial. Arch. Pediatr. Adolesc. Med. 2000, 154, 512–517. [Google Scholar]
- Smorodintsev, A.A.; Drobyshevskaya, A.I.; Bulychev, N.P.; Chalkina, O.M.; Groisman, G.M.; Ilyenko, V.I.; Kantorovich, R.A.; Kurnosova, L.M.; Vasilyev, K.G.; Votyakov, V.I.; et al. Immunological and epidemiological effectiveness of live poliomyelitis vaccine in the USSR. Bull. World Health Org. 1960, 23, 705–725. [Google Scholar]
- Southwick, C.H.; Siddiqi, M.F.; Oppenheimer, J.R. Twenty-year changes in rhesus monkey populations in agricultural areas of Northern India. Ecology 1983, 64, 434. [Google Scholar] [CrossRef]
- Koprowski, H. Historical aspects of the development of live virus vaccine in poliomyelitis. Br. Med. J. 1960, 2, 85–91. [Google Scholar] [CrossRef]
- WHO. Inactivated poliovirus vaccine following oral poliovirus vaccine cessation. Relevé Épidémiologique Hebd. Sect. Hygiène Secrétariat Société Nations Wkly. Epidemiol. Rec. Health Sect. Secr. Leag. Nations 2006, 81, 137–144. [Google Scholar]
- Pearce, J.M.S. Salk and Sabin: Poliomyelitis immunisation. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1552. [Google Scholar] [CrossRef]
- Langmuir, A.D.; Nathanson, N.; Hall, W.J. Surveillance of poliomyelitis in the United States in 1955. Am. J. Public Health Nations Health 1956, 46, 75–88. [Google Scholar] [CrossRef]
- Salk, J.E.; Krech, U.; Youngner, J.S.; Bennett, B.L.; Lewis, L.J.; Bazeley, P.L. Formaldehyde treatment and safety testing of experimental poliomyelitis vaccines. Am. J. Public Health Nations Health 1954, 44, 563–570. [Google Scholar] [CrossRef]
- Melnick, J.L. Problems associated with the use of live poliovirus vaccine. Am. J. Public Health Nations Health 1960, 50, 1013–1031. [Google Scholar]
- Blume, S.; Geesink, I. A brief history of polio vaccines. Science 2000, 288, 1593–1594. [Google Scholar] [CrossRef]
- Cox, H.R. Active immunization against poliomyelitis. Bull. N. Y. Acad. Med. 1953, 29, 943–960. [Google Scholar]
- Koprowski, H. Biological modification of rabies virus as a result of its adaptation to chicks and developing chick embryos. Bull. World Health Org. 1954, 10, 709–724. [Google Scholar]
- Enders, J.F.; Robbins, F.C.; Weller, T.H. Classics in infectious diseases. The cultivation of the poliomyelitis viruses in tissue culture by John F. Enders, Frederick C. Robbins, and Thomas H. Weller. Rev. Infect. Dis. 1980, 2, 493–504. [Google Scholar] [CrossRef]
- Theiler, M.; Smith, H.H. The effect of prolonged cultivation in vitro upon the pathogenicity of yellow fever virus. J. Exp. Med. 1937, 65, 767–786. [Google Scholar] [CrossRef]
- Mayr, A.; Munz, E. Changes in the vaccinia virus through continuing passages in chick embryo fibroblast cultures. Zentralblatt Für Bakteriol. Parasitenkd. Infekt. Hyg. 1 Abt Med.-Hyg. Bakteriol. Virusforsch. Parasitol. Orig. 1964, 195, 24–35. [Google Scholar]
- Mayr, A.; Hochstein-Mintzel, V.; Stickl, H. Abstammung, eigenschaften und verwendung des attenuierten vaccinia-stammes MVA. Infect. 1975, 3, 6–14. [Google Scholar] [CrossRef]
- Meyer, H.; Sutter, G.; Mayr, A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 1991, 72, 1031–1038. [Google Scholar] [CrossRef]
- Meisinger-Henschel, C.; Schmidt, M.; Lukassen, S.; Linke, B.; Krause, L.; Konietzny, S.; Goesmann, A.; Howley, P.; Chaplin, P.; Suter, M.; et al. Genomic sequence of chorioallantois vaccinia virus Ankara, the ancestor of modified vaccinia virus Ankara. J. Gen. Virol. 2007, 88, 3249–3259. [Google Scholar] [CrossRef]
- Carroll, M.W.; Moss, B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: Propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 1997, 238, 198–211. [Google Scholar] [CrossRef]
- Blanchard, T.J.; Alcami, A.; Andrea, P.; Smith, G.L. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: Implications for use as a human vaccine. J. Gen. Virol. 1998, 79, 1159–1167. [Google Scholar]
- Drexler, I.; Heller, K.; Wahren, B.; Erfle, V.; Sutter, G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J. Gen. Virol. 1998, 79, 347–352. [Google Scholar]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef]
- Sutter, G.; Wyatt, L.S.; Foley, P.L.; Bennink, J.R.; Moss, B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine 1994, 12, 1032–1040. [Google Scholar] [CrossRef]
- Drillien, R.; Spehner, D.; Hanau, D. Modified vaccinia virus Ankara induces moderate activation of human dendritic cells. J. Gen. Virol. 2004, 85, 2167–2175. [Google Scholar] [CrossRef]
- Liu, L.; Chavan, R.; Feinberg, M.B. Dendritic cells are preferentially targeted among hematolymphocytes by modified vaccinia virus Ankara and play a key role in the induction of virus-specific T cell responses in vivo. BMC Immunol. 2008, 9, 15. [Google Scholar] [CrossRef]
- Cottingham, M.G.; Carroll, M.W. Recombinant MVA vaccines: Dispelling the myths. Vaccine 2013, 31, 4247–4251. [Google Scholar] [CrossRef]
- Kremer, M.; Volz, A.; Kreijtz, J.H.C.M.; Fux, R.; Lehmann, M.H.; Sutter, G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. Methods Mol. Biol. Clifton NJ 2012, 890, 59–92. [Google Scholar] [CrossRef]
- Smith, G.L.; Moss, B. Infectious poxvirus vectors have capacity for at least 25,000 base pairs of foreign DNA. Gene 1983, 25, 21–28. [Google Scholar] [CrossRef]
- Stickl, H.; Hochstein-Mintzel, V.; Mayr, A.; Huber, H.C.; Schäfer, H.; Holzner, A. MVA vaccination against smallpox: Clinical tests with an attenuated live vaccinia virus strain (MVA) (author’s transl). Dtsch. Med. Wochenschr. 1974, 99, 2386–2392. [Google Scholar] [CrossRef]
- Mayr, A. Smallpox vaccination and bioterrorism with pox viruses. Comp. Immunol. Microbiol. Infect. Dis. 2003, 26, 423–430. [Google Scholar] [CrossRef]
- Webster, D.P.; Dunachie, S.; Vuola, J.M.; Berthoud, T.; Keating, S.; Laidlaw, S.M.; McConkey, S.J.; Poulton, I.; Andrews, L.; Andersen, R.F.; et al. Enhanced T cell-mediated protection against malaria in human challenges by using the recombinant poxviruses FP9 and modified vaccinia virus Ankara. Proc. Natl. Acad. Sci. USA 2005, 102, 4836–4841. [Google Scholar] [CrossRef]
- Cebere, I.; Dorrell, L.; McShane, H.; Simmons, A.; McCormack, S.; Schmidt, C.; Smith, C.; Brooks, M.; Roberts, J.E.; Darwin, S.C.; et al. Phase I clinical trial safety of DNA- and modified virus Ankara-vectored human immunodeficiency virus type 1 (HIV-1) vaccines administered alone and in a prime-boost regime to healthy HIV-1-uninfected volunteers. Vaccine 2006, 24, 417–425. [Google Scholar] [CrossRef]
- Gilbert, S.C.; Moorthy, V.S.; Andrews, L.; Pathan, A.A.; McConkey, S.J.; Vuola, J.M.; Keating, S.M.; Berthoud, T.; Webster, D.; McShane, H.; et al. Synergistic DNA-MVA prime-boost vaccination regimes for malaria and tuberculosis. Vaccine 2006, 24, 4554–4561. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Jacobs, J.P.; Jones, C.M.; Baille, J.P. Characteristics of a human diploid cell designated MRC-5. Nature 1970, 227, 168–170. [Google Scholar] [CrossRef]
- Top, F.H., Jr.; Dudding, B.A.; Russell, P.K.; Buescher, E.L. Control of respiratory disease in recruits with types 4 and 7 adenovirus vaccines. Am. J. Epidemiol. 1971, 94, 142–146. [Google Scholar]
- Gray, G.C.; Goswami, P.R.; Malasig, M.D.; Hawksworth, A.W.; Trump, D.H.; Ryan, M.A.; Schnurr, D.P. Adult adenovirus infections: Loss of orphaned vaccines precipitates military respiratory disease epidemics. For the Adenovirus Surveillance Group. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2000, 31, 663–670. [Google Scholar] [CrossRef]
- Barraza, E.M.; Ludwig, S.L.; Gaydos, J.C.; Brundage, J.F. Reemergence of adenovirus type 4 acute respiratory disease in military trainees: Report of an outbreak during a lapse in vaccination. J. Infect. Dis. 1999, 179, 1531–1533. [Google Scholar] [CrossRef]
- Wadman, M. Medical research: Cell division. Nature 2013, 498, 422–426. [Google Scholar] [CrossRef]
- Kleiman, M.B.; Carver, D.H. Failure of the RA 27/3 strain of rubella virus to induce intrinsic interference. J. Gen. Virol. 1977, 36, 335–340. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Farquhar, J.; Katz, M.; Ingalls, T.H. A new attenuated rubella virus grown in human fibroblasts: Evidence for reduced nasopharyngeal excretion. Am. J. Epidemiol. 1967, 86, 468–477. [Google Scholar]
- Wallace, R.E.; Vasington, P.J.; Petricciani, J.C.; Hopps, H.E.; Lorenz, D.E.; Kadanka, Z. Development of a diploid cell line from fetal rhesus monkey lung for virus vaccine production. In Vitro 1973, 8, 323–332. [Google Scholar] [CrossRef]
- Petricciani, J.C.; Hopps, H.E.; Lorenz, D.E.; Vasington, P.J.; Wallace, R.E. Sub-human primate diploid cell lines as substrates for virus vaccine production. US4040905 A, 9 August 1977. [Google Scholar]
- Simonsen, L.; Morens, D.; Elixhauser, A.; Gerber, M.; van Raden, M.; Blackwelder, W. Effect of rotavirus vaccination programme on trends in admission of infants to hospital for intussusception. Lancet 2001, 358, 1224–1229. [Google Scholar] [CrossRef]
- Trent, D.; Shin, J.; Hombach, J.; Knezevic, I.; Minor, P. WHO Working Group on technical specifications for manufacture and evaluation of dengue vaccines, Geneva, Switzerland, 11–12 May 2009. Vaccine 2010, 28, 8246–8255. [Google Scholar] [CrossRef]
- Enserink, M. Influenza. Crisis underscores fragility of vaccine production system. Science 2004, 306, 385. [Google Scholar] [CrossRef]
- Uscher-Pines, L.; Barnett, D.J.; Sapsin, J.W.; Bishai, D.M.; Balicer, R.D. A systematic analysis of influenza vaccine shortage policies. Public Health 2008, 122, 183–191. [Google Scholar]
- Nichols, W.W.; Murphy, D.G.; Cristofalo, V.J.; Toji, L.H.; Greene, A.E.; Dwight, S.A. Characterization of a new human diploid cell strain, IMR-90. Science 1977, 196, 60–63. [Google Scholar]
- Takahashi, M. Development of a live varicella vaccine–Past and future. Jpn. J. Infect. Dis. 2000, 53, 47–55. [Google Scholar]
- WHO Technical Report Series; WHO Expert Committee on Biological Standardization, Fifty-seventh report. TRS 962; World Health Organization: Geneva, Switzerland, 2011.
- Rheingans, R.D.; Antil, L.; Dreibelbis, R.; Podewils, L.J.; Bresee, J.S.; Parashar, U.D. Economic costs of rotavirus gastroenteritis and cost-effectiveness of vaccination in developing countries. J. Infect. Dis. 2009, 200, S16–S27. [Google Scholar] [CrossRef]
- Scherer, S.F. The utilization of a pure strain of mammalian cells (Earle) for the cultivation of viruses in vitro. I. Multiplication of pseudorabies and herpes simplex viruses. Am. J. Pathol. 1953, 29, 113–137. [Google Scholar]
- Scherer, W.F.; Syverton, J.T.; Gey, G.O. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 1953, 97, 695–710. [Google Scholar] [CrossRef]
- Montagnon, B.J. Polio and rabies vaccines produced in continuous cell lines: A reality for Vero cell line. Dev. Biol. Stand. 1989, 70, 27–47. [Google Scholar]
- Vincent-Falquet, J.C.; Peyron, L.; Souvras, M.; Moulin, J.C.; Tektoff, J.; Patet, J. Qualification of working cell banks for the Vero cell line to produce licensed human vaccines. Dev. Biol. Stand. 1989, 70, 153–156. [Google Scholar]
- Montagnon, B.J.; Vincent-Falquet, J.C. Experience with the Vero cell line. Dev. Biol. Stand. 1998, 93, 119–123. [Google Scholar]
- Knezevic, I.; Stacey, G.; Petricciani, J.; Sheets, R. Evaluation of cell substrates for the production of biologicals: Revision of WHO recommendations. Report of the WHO Study Group on Cell Substrates for the Production of Biologicals, 22–23 April 2009, Bethesda, USA. Biol. J. Int. Assoc. Biol. Stand. 2010, 38, 162–169. [Google Scholar]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar]
- Dukes, J.D.; Whitley, P.; Chalmers, A.D. The MDCK variety pack: Choosing the right strain. BMC Cell Biol. 2011, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Omeir, R.L.; Teferedegne, B.; Foseh, G.S.; Beren, J.J.; Snoy, P.J.; Brinster, L.R.; Cook, J.L.; Peden, K.; Lewis, A.M., Jr. Heterogeneity of the tumorigenic phenotype expressed by Madin-Darby canine kidney cells. Comp. Med. 2011, 61, 243–250. [Google Scholar]
- Gaush, C.R.; Smith, T.F. Replication and plaque assay of influenza virus in an established line of canine kidney cells. Appl. Microbiol. 1968, 16, 588–594. [Google Scholar]
- Doroshenko, A.; Halperin, S.A. Trivalent MDCK cell culture-derived influenza vaccine Optaflu (Novartis Vaccines). Exp. Rev. Vaccines 2009, 8, 679–688. [Google Scholar] [CrossRef]
- Gregersen, J.-P.; Schmitt, H.-J.; Trusheim, H.; Bröker, M. Safety of MDCK cell culture-based influenza vaccines. Fut. Microbiol. 2011, 6, 143–152. [Google Scholar] [CrossRef]
- Macpherson, I.; Stoker, M. Polyoma transformation of hamster cell clones–An investigation of genetic factors affecting cell competence. Virology 1962, 16, 147–151. [Google Scholar] [CrossRef]
- Stoker, M.; Macpherson, I. Syrian hamster fibroblast cell line bhk21 and its derivatives. Nature 1964, 203, 1355–1357. [Google Scholar] [CrossRef]
- Albariño, C.G.; Ghiringhelli, P.D.; Posik, D.M.; Lozano, M.E.; Ambrosio, A.M.; Sanchez, A.; Romanowski, V. Molecular characterization of attenuated Junin virus strains. J. Gen. Virol. 1997, 78, 1605–1610. [Google Scholar]
- Polo, J.M.; Belli, B.A.; Driver, D.A.; Frolov, I.; Sherrill, S.; Hariharan, M.J.; Townsend, K.; Perri, S.; Mento, S.J.; Jolly, D.J.; et al. Stable alphavirus packaging cell lines for Sindbis virus and Semliki Forest virus-derived vectors. Proc. Natl. Acad. Sci. USA 1999, 96, 4598–4603. [Google Scholar] [CrossRef]
- Oehmig, A.; Büttner, M.; Weiland, F.; Werz, W.; Bergemann, K.; Pfaff, E. Identification of a calicivirus isolate of unknown origin. J. Gen. Virol. 2003, 84, 2837–2845. [Google Scholar] [CrossRef]
- Tjio, J.H.; Puck, T.T. Genetics of somatic mammalian cells. II. Chromosomal constitution of cells in tissue culture. J. Exp. Med. 1958, 108, 259–268. [Google Scholar] [CrossRef]
- Marsh, M.; Bron, R. SFV infection in CHO cells: Cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 1997, 110, 95–103. [Google Scholar]
- Berting, A.; Farcet, M.R.; Kreil, T.R. Virus susceptibility of Chinese hamster ovary (CHO) cells and detection of viral contaminations by adventitious agent testing. Biotechnol. Bioeng. 2010, 106, 598–607. [Google Scholar] [CrossRef]
- Xu, X.; Nagarajan, H.; Lewis, N.E.; Pan, S.; Cai, Z.; Liu, X.; Chen, W.; Xie, M.; Wang, W.; Hammond, S.; et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 2011, 29, 735–741. [Google Scholar] [CrossRef]
- Anderson, K.P.; Low, M.A.; Lie, Y.S.; Keller, G.A.; Dinowitz, M. Endogenous origin of defective retroviruslike particles from a recombinant Chinese hamster ovary cell line. Virology 1991, 181, 305–311. [Google Scholar] [CrossRef]
- Lubiniecki, A.S. Historical reflections on cell culture engineering. Cytotechnology 1998, 28, 139–145. [Google Scholar] [CrossRef]
- Derouazi, M.; Martinet, D.; Besuchet Schmutz, N.; Flaction, R.; Wicht, M.; Bertschinger, M.; Hacker, D.L.; Beckmann, J.S.; Wurm, F.M. Genetic characterization of CHO production host DG44 and derivative recombinant cell lines. Biochem. Biophys. Res. Commun. 2006, 340, 1069–1077. [Google Scholar] [CrossRef]
- Adams, H.P., Jr.; del Zoppo, G.; Alberts, M.J.; Bhatt, D.L.; Brass, L.; Furlan, A.; Grubb, R.L.; Higashida, R.T.; Jauch, E.C.; Kidwell, C.; et al. Atherosclerotic peripheral vascular disease and quality of care outcomes in research interdisciplinary working groups guidelines for the early management of adults with ischemic stroke: A guideline from the American heart association/American stroke association stroke council, clinical cardiology council, cardiovascular radiology and intervention council, and the atherosclerotic peripheral vascular disease and quality of care outcomes in research interdisciplinary working groups: The American academy of neurology affirms the value of this guideline as an educational tool for neurologists. Stroke J. Cereb. Circ. 2007, 38, 1655–1711. [Google Scholar] [CrossRef]
- Collen, D.; Lijnen, H.R. New approaches to thrombolytic therapy. Arterioscler. Dallas Tex 1984, 4, 579–585. [Google Scholar] [CrossRef]
- Barnes, D.; Hughes, R.A. Guillain-Barré syndrome after treatment with streptokinase. BMJ 1992, 304, 1225. [Google Scholar] [CrossRef]
- Curran, J.W.; Morgan, W.M.; Hardy, A.M.; Jaffe, H.W.; Darrow, W.W.; Dowdle, W.R. The epidemiology of AIDS: Current status and future prospects. Science 1985, 229, 1352–1357. [Google Scholar]
- Walsh, G. Biopharmaceutical benchmarks 2010. Nat. Biotechnol. 2010, 28, 917–924. [Google Scholar] [CrossRef]
- Olivier, S.; Jacoby, M.; Brillon, C.; Bouletreau, S.; Mollet, T.; Nerriere, O.; Angel, A.; Danet, S.; Souttou, B.; Guehenneux, F.; et al. EB66 cell line, a duck embryonic stem cell-derived substrate for the industrial production of therapeutic monoclonal antibodies with enhanced ADCC activity. mAbs 2010, 2, 405–415. [Google Scholar]
- Brown, S.W.; Mehtali, M. The avian EB66(R) cell line, application to vaccines, and therapeutic protein production. PDA J. Pharm. Sci. Technol. PDA 2010, 64, 419–425. [Google Scholar]
- Guehenneux, F.; Moreau, K.; Esnault, M.; Mehtali, M. Duck Embryonic Derived Stem Cell Lines for the Production of Viral Vaccines. WO/2008/129058, 24 April 2007. [Google Scholar]
- Wickham, T.J.; Davis, T.; Granados, R.R.; Shuler, M.L.; Wood, H.A. Screening of insect cell lines for the production of recombinant proteins and infectious virus in the baculovirus expression system. Biotechnol. Progr. 1992, 8, 391–396. [Google Scholar] [CrossRef]
- Vaughn, J.L.; Goodwin, R.H.; Tompkins, G.J.; McCawley, P. The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae). In Vitro 1977, 13, 213–217. [Google Scholar] [CrossRef]
- McPherson, C.E. Development of a novel recombinant influenza vaccine in insect cells. Biol. J. Int. Assoc. Biol. Stand. 2008, 36, 350–353. [Google Scholar]
- Sáenz-Robles, M.T.; Sullivan, C.S.; Pipas, J.M. Transforming functions of simian virus 40. Oncogene 2001, 20, 7899–7907. [Google Scholar] [CrossRef]
- Frisch, S.M.; Mymryk, J.S. Adenovirus-5 E1A: Paradox and paradigm. Nat. Rev. Mol. Cell Biol. 2002, 3, 441–452. [Google Scholar] [CrossRef]
- Korzeniewski, N.; Spardy, N.; Duensing, A.; Duensing, S. Genomic instability and cancer: Lessons learned from human papillomaviruses. Cancer Lett. 2011, 305, 113–122. [Google Scholar] [CrossRef]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef]
- Kovesdi, I.; Reichel, R.; Nevins, J.R. Identification of a cellular transcription factor involved in E1A trans-activation. Cell 1986, 45, 219–228. [Google Scholar] [CrossRef]
- Rich, T.; Allen, R.L.; Wyllie, A.H. Defying death after DNA damage. Nature 2000, 407, 777–783. [Google Scholar] [CrossRef]
- Crawford, L.V.; Pim, D.C.; Gurney, E.G.; Goodfellow, P.; Taylor-Papadimitriou, J. Detection of a common feature in several human tumor cell lines–A 53,000-dalton protein. Proc. Natl. Acad. Sci. USA 1981, 78, 41–45. [Google Scholar] [CrossRef]
- Berk, A.J. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005, 24, 7673–7685. [Google Scholar] [CrossRef]
- Linzer, D.I.; Levine, A.J. Characterization of a 54 K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Bocchetta, M.; Eliasz, S.; de Marco, M.A.; Rudzinski, J.; Zhang, L.; Carbone, M. The SV40 large T antigen-p53 complexes bind and activate the insulin-like growth factor-I promoter stimulating cell growth. Cancer Res. 2008, 68, 1022–1029. [Google Scholar] [CrossRef]
- Cheng, J.; DeCaprio, J.A.; Fluck, M.M.; Schaffhausen, B.S. Cellular transformation by Simian Virus 40 and Murine Polyoma Virus T antigens. Semin. Cancer Biol. 2009, 19, 218–228. [Google Scholar] [CrossRef]
- Topalis, D.; Andrei, G.; Snoeck, R. The large tumor antigen: A “Swiss Army knife” protein possessing the functions required for the polyomavirus life cycle. Antiv. Res. 2013, 97, 122–136. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Münger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef]
- Cuconati, A.; Mukherjee, C.; Perez, D.; White, E. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 2003, 17, 2922–2932. [Google Scholar] [CrossRef]
- Hall, A.H.S.; Alexander, K.A. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J. Virol. 2003, 77, 6066–6069. [Google Scholar] [CrossRef]
- Jensen, F.; Koprowski, H.; Ponten, J.A. Rapid transformation of human fibroblast cultures by simian virus. Proc. Natl. Acad. Sci. USA 1963, 50, 343–348. [Google Scholar] [CrossRef]
- Jensen, F.C.; Girardi, A.J.; Gilden, R.V.; Koprowski, H. Infection of human and simian tissue cultures with rous sarcoma virus. Proc. Natl. Acad. Sci. USA 1964, 52, 53–59. [Google Scholar] [CrossRef]
- Aaronson, S.A.; Todaro, G.J. SV40 T antigen induction and transformation in human fibroblast cell strains. Virology 1968, 36, 254–261. [Google Scholar] [CrossRef]
- Todaro, G.J.; Aaronson, S.A. Human cell strains susceptible to focus formation by human adenovirus type 12. Proc. Natl. Acad. Sci. USA 1968, 61, 1272–1278. [Google Scholar] [CrossRef]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef]
- Adey, A.; Burton, J.N.; Kitzman, J.O.; Hiatt, J.B.; Lewis, A.P.; Martin, B.K.; Qiu, R.; Lee, C.; Shendure, J. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 2013, 500, 207–211. [Google Scholar] [CrossRef]
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 2006, 44, 1755–1762. [Google Scholar] [CrossRef]
- Ziegert, C.; Wentzensen, N.; Vinokurova, S.; Kisseljov, F.; Einenkel, J.; Hoeckel, M.; von Knebel Doeberitz, M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 2003, 22, 3977–3984. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Graham, F.L. Covalently closed circles of human adenovirus DNA are infectious. EMBO J. 1984, 3, 2917–2922. [Google Scholar]
- Louis, N.; Evelegh, C.; Graham, F.L. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology 1997, 233, 423–429. [Google Scholar] [CrossRef]
- Lochmüller, H.; Jani, A.; Huard, J.; Prescott, S.; Simoneau, M.; Massie, B.; Karpati, G.; Acsadi, G. Emergence of early region 1-containing replication-competent adenovirus in stocks of replication-defective adenovirus recombinants (delta E1 + delta E3) during multiple passages in 293 cells. Hum. Gene Ther. 1994, 5, 1485–1491. [Google Scholar] [CrossRef]
- Fallaux, F.J.; Bout, A.; van der Velde, I.; van den Wollenberg, D.J.; Hehir, K.M.; Keegan, J.; Auger, C.; Cramer, S.J.; van Ormondt, H.; van der Eb, A.J.; et al. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum. Gene Ther. 1998, 9, 1909–1917. [Google Scholar] [CrossRef]
- Fallaux, F.J.; Hoeben, R.C.; Bout, A.; Valerio, D.; van Der Eb, A.J. Packaging Systems for Human Recombinant Adenovirus to be Used in Gene Therapy. EP0833934, 14 June 1996. [Google Scholar]
- Samina, I.; Havenga, M.; Koudstaal, W.; Khinich, Y.; Koldijk, M.; Malkinson, M.; Simanov, M.; Perl, S.; Gijsbers, L.; Weverling, G.J.; et al. Safety and efficacy in geese of a PER.C6-based inactivated West Nile virus vaccine. Vaccine 2007, 25, 8338–8345. [Google Scholar] [CrossRef]
- Le Ru, A.; Jacob, D.; Transfiguracion, J.; Ansorge, S.; Henry, O.; Kamen, A.A. Scalable production of influenza virus in HEK-293 cells for efficient vaccine manufacturing. Vaccine 2010, 28, 3661–3671. [Google Scholar] [CrossRef]
- Sanders, B.P.; Edo-Matas, D.; Custers, J.H.H.V.; Koldijk, M.H.; Klaren, V.; Turk, M.; Luitjens, A.; Bakker, W.A.M.; Uytdehaag, F.; Goudsmit, J.; et al. PER.C6(®) cells as a serum-free suspension cell platform for the production of high titer poliovirus: A potential low cost of goods option for world supply of inactivated poliovirus vaccine. Vaccine 2013, 31, 850–856. [Google Scholar] [CrossRef]
- “Designer” cells as Substrate for Manufacture of Viral Vaccines. Available online: http://www.fda.gov/ohrms/dockets/ac/01/briefing/3750b1_01.pdf (accessed on 17 January 2014).
- Schiedner, G.; Gaitatzis, N.; Hertel, S.; Bialek, C.; Kewes, H.; Volpers, C.; Waschütza, G. Human Cell Lines for Production of Biopharmaceuticals. In Cells and Culture, Proceedings of the 20th ESACT Meeting, Dresden, Germany, June 17-20, 2007; Noll, T., Ed.; Springer: Amsterdam, The Netherlands, 2010; pp. 503–511. [Google Scholar]
- Schiedner, G.; Hertel, S.; Kochanek, S. Efficient transformation of primary human amniocytes by E1 functions of Ad5: Generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 2000, 11, 2105–2116. [Google Scholar] [CrossRef]
- Kochanek, S.; Schiedner, G. Permanent Amniocyte Cell Line, the Production Thereof and Its Use for Producing Gene Transfer Vectors. WO/2001/036615, 18 November 1999. [Google Scholar]
- Sandig, V.; Jordan, I. Immortalized Avian Cell Lines for Virus Production. WO/2005/042728, 3 November 2003. [Google Scholar]
- Jordan, I.; Vos, A.; Beilfuss, S.; Neubert, A.; Breul, S.; Sandig, V. An avian cell line designed for production of highly attenuated viruses. Vaccine 2009, 27, 748–756. [Google Scholar]
- Jordan, I.; Northoff, S.; Thiele, M.; Hartmann, S.; Horn, D.; Höwing, K.; Bernhardt, H.; Oehmke, S.; von Horsten, H.; Rebeski, D.; et al. A chemically defined production process for highly attenuated poxviruses. Biol. J. Int. Assoc. Biol. Stand. 2011, 39, 50–58. [Google Scholar]
- Sandig, V.; Jordan, I. Productivity augmenting protein factor, novel cell lines and uses thereof. WO/2007/054516, 8 November 2005. [Google Scholar]
- Lohr, V.; Rath, A.; Genzel, Y.; Jordan, I.; Sandig, V.; Reichl, U. New avian suspension cell lines provide production of influenza virus and MVA in serum-free media: Studies on growth, metabolism and virus propagation. Vaccine 2009, 27, 4975–4982. [Google Scholar]
- Lohr, V.; Genzel, Y.; Jordan, I.; Katinger, D.; Mahr, S.; Sandig, V.; Reichl, U. Live attenuated influenza viruses produced in a suspension process with avian AGE1.CR.pIX cells. BMC Biotechnol. 2012, 12, 79. [Google Scholar] [CrossRef]
- Ehreth, J. The global value of vaccination. Vaccine 2003, 21, 596–600. [Google Scholar] [CrossRef]
- Rosolowsky, M.; McKee, R.; Nichols, W.; Garfinkle, B. Chromosomal characterization of MRC-5 cell banks utilizing G-banding technique. Dev. Biol. Stand. 1998, 93, 109–117. [Google Scholar]
- WHO Technical Report Series; Recommendations for the Evaluation of Animal Cell Cultures as Substrates for the Manufacture of Biological Medicinal Products and for the Characterization of Cell Banks. TRS 978, Annex 3; World Health Organization: Geneva, Switzerland, 2013.
- Manohar, M.; Orrison, B.; Peden, K.; Lewis, A.M., Jr. Assessing the tumorigenic phenotype of VERO cells in adult and newborn nude mice. Biol. J. Int. Assoc. Biol. Stand. 2008, 36, 65–72. [Google Scholar]
- Hilleman, M.R. Cells, vaccines, and the pursuit of precedent. Natl. Cancer Inst. Monogr. 1968, 29, 463–469. [Google Scholar]
- Hilleman, M.R. History, precedent, and progress in the development of mammalian cell culture systems for preparing vaccines: Safety considerations revisited. J. Med. Virol. 1990, 31, 5–12. [Google Scholar] [CrossRef]
- WHO Technical Report Series; WHO Technical Report Series. Recommendations for the Evaluation of Animal Cell Cultures as Substrates for the Manufacture of Biological Medicinal Products and for the Characterization of Cell Banks. TRS 878, Annex 1; World Health Organization: Geneva, Switzerland, 2010.
- Hahn, W.C.; Weinberg, R.A. Modelling the molecular circuitry of cancer. Nat. Rev. Cancer 2002, 2, 331–341. [Google Scholar] [CrossRef]
- Grachev, V.; Magrath, D.; Griffiths, E. WHO requirements for the use of animal cells as in vitro substrates for the production of biologicals (Requirements for biological susbstances No. 50). Biol. J. Int. Assoc. Biol. Stand. 1998, 26, 175–193. [Google Scholar]
- Yang, H.; Zhang, L.; Galinski, M. A probabilistic model for risk assessment of residual host cell DNA in biological products. Vaccine 2010, 28, 3308–3311. [Google Scholar] [CrossRef]
- Gong, M.N.; Sai, Y.; Zhou, W.; Thompson, B.T.; Xu, L.-L.; Christiani, D.C. Genotyping patients with recent blood transfusions. Epidemiol. Camb. Mass 2003, 14, 744–747. [Google Scholar] [CrossRef]
- Bolhassani, A.; Safaiyan, S.; Rafati, S. Improvement of different vaccine delivery systems for cancer therapy. Mol. Cancer 2011, 10, 3. [Google Scholar] [CrossRef]
- Wierenga, D.E.; Cogan, J.; Petricciani, J.C. Administration of tumor cell chromatin to immunosuppressed and non-immunosuppressed non-human primates. Biol. J. Int. Assoc. Biol. Stand. 1995, 23, 221–224. [Google Scholar]
- Jordan, I.; Lohr, V.; Genzel, Y.; Reichl, U.; Sandig, V. Elements in the development of a production process for modified vaccinia virus Ankara. Microorganisms 2013, 1, 100–121. [Google Scholar] [CrossRef]
- Li, T.-C.; Scotti, P.D.; Miyamura, T.; Takeda, N. Latent infection of a new alphanodavirus in an insect cell line. J. Virol. 2007, 81, 10890–10896. [Google Scholar] [CrossRef]
- Frierson, J.G. The yellow fever vaccine: A history. Yale J. Biol. Med. 2010, 83, 77–85. [Google Scholar]
- Rollison, D.E.M.; Page, W.F.; Crawford, H.; Gridley, G.; Wacholder, S.; Martin, J.; Miller, R.; Engels, E.A. Case-control study of cancer among US Army veterans exposed to simian virus 40-contaminated adenovirus vaccine. Am. J. Epidemiol. 2004, 160, 317–324. [Google Scholar] [CrossRef]
- Vilchez, R.A.; Butel, J.S. Emergent human pathogen simian virus 40 and its role in cancer. Clin. Microbiol. Rev. 2004, 17, 495–508. [Google Scholar] [CrossRef]
- Poulin, D.L.; deCaprio, J.A. Is there a role for SV40 in human cancer? J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4356–4365. [Google Scholar] [CrossRef]
- Rizzo, P.; Di Resta, I.; Powers, A.; Ratner, H.; Carbone, M. Unique strains of SV40 in commercial poliovaccines from 1955 not readily identifiable with current testing for SV40 infection. Cancer Res. 1999, 59, 6103–6108. [Google Scholar]
- Peden, K.; Sheng, L.; Omeir, R.; Yacobucci, M.; Klutch, M.; Laassri, M.; Chumakov, K.; Pal, A.; Murata, H.; Lewis, A.M., Jr. Recovery of strains of the polyomavirus SV40 from rhesus monkey kidney cells dating from the 1950s to the early 1960s. Virology 2008, 370, 63–76. [Google Scholar] [CrossRef]
- Lundstig, A.; Dejmek, A.; Eklund, C.; Filinic, I.; Dillner, J. No detection of SV40 DNA in mesothelioma tissues from a high incidence area in Sweden. Anticancer Res. 2007, 27, 4159–4161. [Google Scholar]
- Qi, F.; Carbone, M.; Yang, H.; Gaudino, G. Simian virus 40 transformation, malignant mesothelioma and brain tumors. Exp. Rev. Respir. Med. 2011, 5, 683–697. [Google Scholar] [CrossRef]
- Slenczka, W.; Klenk, H.D. Forty years of marburg virus. J. Infect. Dis. 2007, 196, S131–S135. [Google Scholar] [CrossRef]
- Centers for Disease Control (CDC). Ebola virus infection in imported primates–Virginia, 1989. MMWR Morb. Mortal. Wkly. Rep. 1989, 38, 831–832, 837–838. [Google Scholar]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Johnson, K.M.; Kawaoka, Y.; Lipkin, W.I.; Negredo, A.I.; Netesov, S.V.; Nichol, S.T.; et al. Proposal for a revised taxonomy of the family Filoviridae: Classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010, 155, 2083–2103. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Ebola-Reston virus infection among quarantined nonhuman primates–Texas, 1996. MMWR Morb. Mortal. Wkly. Rep. 1996, 45, 314–316. [Google Scholar]
- Barrette, R.W.; Metwally, S.A.; Rowland, J.M.; Xu, L.; Zaki, S.R.; Nichol, S.T.; Rollin, P.E.; Towner, J.S.; Shieh, W.-J.; Batten, B.; et al. Discovery of swine as a host for the Reston ebolavirus. Science 2009, 325, 204–206. [Google Scholar] [CrossRef]
- Centers for Disease Control (CDC). Update: Filovirus infections among persons with occupational exposure to nonhuman primates. MMWR Morb. Mortal. Wkly. Rep. 1990, 39, 266–267, 273. [Google Scholar]
- Southwick, C.H.; Beg, M.A.; Siddiqi, M.R. A population survey of rhesus monkeys in villages, towns and temples of Northern India. Ecology 1961, 42, 538. [Google Scholar] [CrossRef]
- Merten, O.-W. Virus contaminations of cell cultures-A biotechnological view. Cytotechnology 2002, 39, 91–116. [Google Scholar] [CrossRef]
- Böni, J.; Stalder, J.; Reigel, F.; Schüpbach, J. Detection of reverse transcriptase activity in live attenuated virus vaccines. Clin. Diagn. Virol. 1996, 5, 43–53. [Google Scholar] [CrossRef]
- Weissmahr, R.N.; Schüpbach, J.; Böni, J. Reverse transcriptase activity in chicken embryo fibroblast culture supernatants is associated with particles containing endogenous avian retrovirus EAV-0 RNA. J. Virol. 1997, 71, 3005–3012. [Google Scholar]
- Herniou, E.; Martin, J.; Miller, K.; Cook, J.; Wilkinson, M.; Tristem, M. Retroviral diversity and distribution in vertebrates. J. Virol. 1998, 72, 5955–5966. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. International human genome sequencing consortium initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Belshaw, R.; Pereira, V.; Katzourakis, A.; Talbot, G.; Paces, J.; Burt, A.; Tristem, M. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2004, 101, 4894–4899. [Google Scholar] [CrossRef]
- Huda, A.; Polavarapu, N.; Jordan, I.K.; McDonald, J.F. Endogenous retroviruses of the chicken genome. Biol. Direct 2008, 3, 9. [Google Scholar] [CrossRef]
- Unknown Author Reverse transcriptase activity in chicken-cell derived vaccine. Relevé Épidémiologique Hebd. Sect. Hygiène Secrétariat Société Nations Wkly. Epidemiol. Rec. Health Sect. Secr. Leag. Nations 1998, 73, 209–212.
- Pain, B.; Guehenneux, F. Avian cell lines for the production of useful substances. WO/2003/076601, 8 March 2002. [Google Scholar]
- Kuehn, B.M. FDA: Benefits of rotavirus vaccination outweigh potential contamination risk. JAMA J. Am. Med. Assoc. 2010, 304, 30–31. [Google Scholar] [CrossRef]
- Victoria, J.G.; Wang, C.; Jones, M.S.; Jaing, C.; McLoughlin, K.; Gardner, S.; Delwart, E.L. Viral nucleic acids in live-attenuated vaccines: Detection of minority variants and an adventitious virus. J. Virol. 2010, 84, 6033–6040. [Google Scholar] [CrossRef]
- Hess, R.D.; Weber, F.; Watson, K.; Schmitt, S. Regulatory, biosafety and safety challenges for novel cells as substrates for human vaccines. Vaccine 2012, 30, 2715–2727. [Google Scholar] [CrossRef]
- Khan, A.S.; Ma, W.; Ma, Y.; Kumar, A.; Williams, D.K.; Muller, J.; Ma, H.; Galvin, T.A. Proposed algorithm to investigate latent and occult viruses in vaccine cell substrates by chemical induction. Biol. J. Int. Assoc. Biol. Stand. 2009, 37, 196–201. [Google Scholar]
- Luciani, F.; Bull, R.A.; Lloyd, A.R. Next generation deep sequencing and vaccine design: Today and tomorrow. Trends Biotechnol. 2012, 30, 443–452. [Google Scholar]
- Katz, S.L.; Enders, J.F.; Holloway, A. The development and evaluation of an attenuated measles virus vaccine. Am. J. Public Health Nations Health 1962, 52, 5–10. [Google Scholar] [CrossRef]
- Ni, H.; Chang, G.J.; Xie, H.; Trent, D.W.; Barrett, A.D. Molecular basis of attenuation of neurovirulence of wild-type Japanese encephalitis virus strain SA14. J. Gen. Virol. 1995, 76, 409–413. [Google Scholar]
- Ni, H.; Burns, N.J.; Chang, G.J.; Zhang, M.J.; Wills, M.R.; Trent, D.W.; Sanders, P.G.; Barrett, A.D. Comparison of nucleotide and deduced amino acid sequence of the 5’ non-coding region and structural protein genes of the wild-type Japanese encephalitis virus strain SA14 and its attenuated vaccine derivatives. J. Gen. Virol. 1994, 75, 1505–1510. [Google Scholar] [CrossRef]
- Añez, G.; Men, R.; Eckels, K.H.; Lai, C.-J. Passage of dengue virus type 4 vaccine candidates in fetal rhesus lung cells selects heparin-sensitive variants that result in loss of infectivity and immunogenicity in rhesus macaques. J. Virol. 2009, 83, 10384–10394. [Google Scholar] [CrossRef]
- Jegede, A.S. What led to the Nigerian boycott of the polio vaccination campaign? PLoS Med. 2007, 4, e73. [Google Scholar] [CrossRef]
- Henderson, L.; Millett, C.; Thorogood, N. Perceptions of childhood immunization in a minority community: Qualitative study. J. R. Soc. Med. 2008, 101, 244–251. [Google Scholar] [CrossRef]
- Hooper, E. Experimental oral polio vaccines and acquired immune deficiency syndrome. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2001, 356, 803–814. [Google Scholar]
- Gao, F.; Bailes, E.; Robertson, D.L.; Chen, Y.; Rodenburg, C.M.; Michael, S.F.; Cummins, L.B.; Arthur, L.O.; Peeters, M.; Shaw, G.M.; et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 1999, 397, 436–441. [Google Scholar] [CrossRef]
- Koprowski, H. Hypotheses and facts. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2001, 356, 831–833. [Google Scholar] [CrossRef]
- Weiss, R.A. Polio vaccines exonerated. Nature 2001, 410, 1035–1036. [Google Scholar]
- Vartanian, J.-P.; Wain-Hobson, S. Analysis of a library of macaque nuclear mitochondrial sequences confirms macaque origin of divergent sequences from old oral polio vaccine samples. Proc. Natl. Acad. Sci. USA 2002, 99, 7566–7569. [Google Scholar] [CrossRef]
- Worobey, M.; Santiago, M.L.; Keele, B.F.; Ndjango, J.-B.N.; Joy, J.B.; Labama, B.L.; Dhed’A, B.D.; Rambaut, A.; Sharp, P.M.; Shaw, G.M.; et al. Origin of AIDS: Contaminated polio vaccine theory refuted. Nature 2004, 428, 820. [Google Scholar] [CrossRef]
- Grabenstein, J.D. What the World’s religions teach, applied to vaccines and immune globulins. Vaccine 2013, 31, 2011–2023. [Google Scholar] [CrossRef]
- Leiva, R. A brief history of human diploid cell strains. Natl. Cathol. Bioeth. Q. 2006, 6, 443–451. [Google Scholar]
- Furton, E.J. Vaccines and the right of conscience. Natl. Cathol. Bioeth. Q. 2004, 4, 53–62. [Google Scholar]
- Rappuoli, R. From Pasteur to genomics: Progress and challenges in infectious diseases. Nat. Med. 2004, 10, 1177–1185. [Google Scholar] [CrossRef]
- Plotkin, S.A. Vaccines: The fourth century. Clin. Vacc. Immunol. CVI 2009, 16, 1709–1719. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jordan, I.; Sandig, V. Matrix and Backstage: Cellular Substrates for Viral Vaccines. Viruses 2014, 6, 1672-1700. https://doi.org/10.3390/v6041672
Jordan I, Sandig V. Matrix and Backstage: Cellular Substrates for Viral Vaccines. Viruses. 2014; 6(4):1672-1700. https://doi.org/10.3390/v6041672
Chicago/Turabian StyleJordan, Ingo, and Volker Sandig. 2014. "Matrix and Backstage: Cellular Substrates for Viral Vaccines" Viruses 6, no. 4: 1672-1700. https://doi.org/10.3390/v6041672
APA StyleJordan, I., & Sandig, V. (2014). Matrix and Backstage: Cellular Substrates for Viral Vaccines. Viruses, 6(4), 1672-1700. https://doi.org/10.3390/v6041672