Contribution of Viral Mimics of Cellular Genes to KSHV Infection and Disease


{kind=link}
Abstract
:1. Introduction
2. KSHV-related diseases: KS, MCD, KICS and PEL
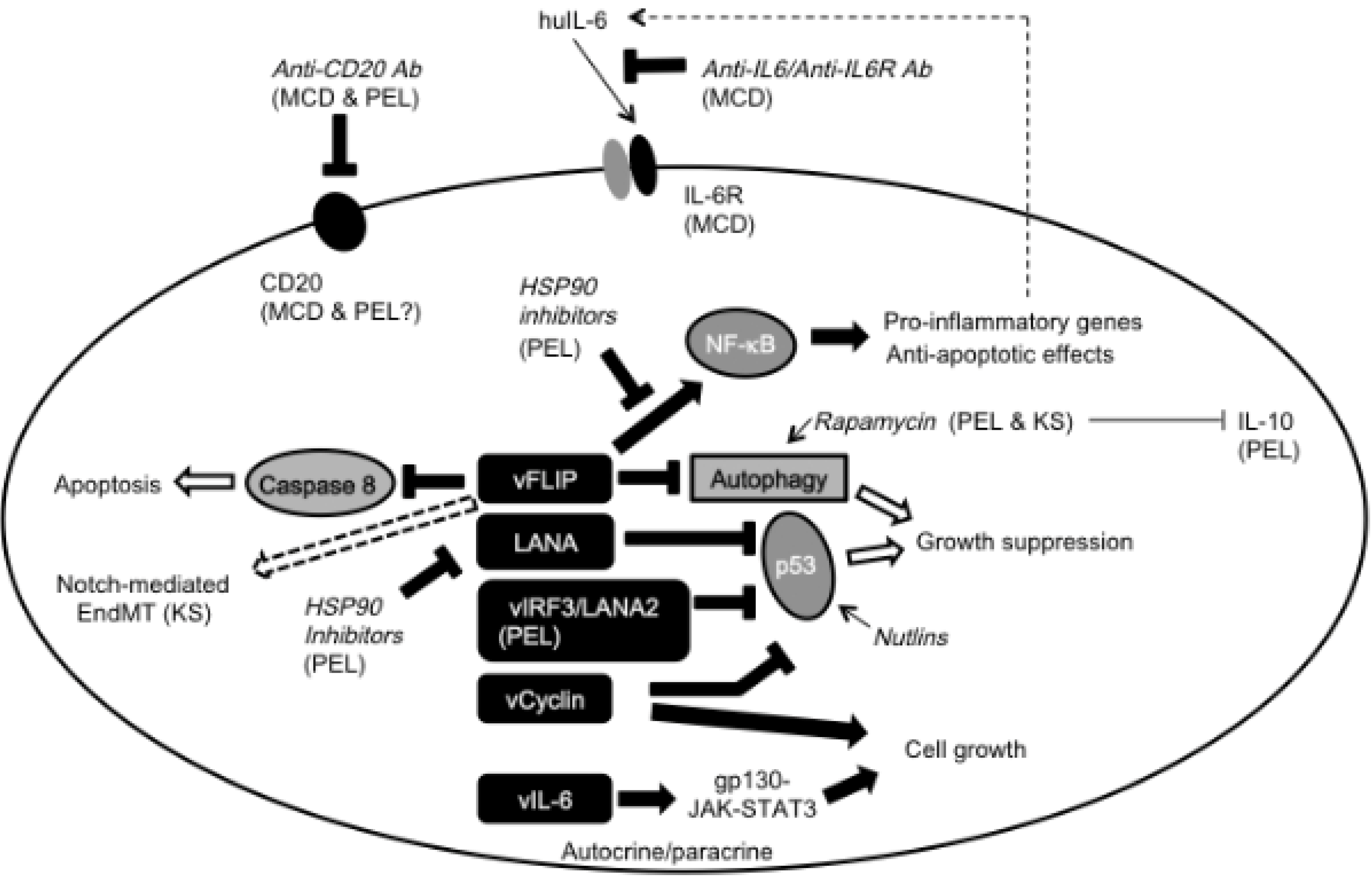
2.1. KS
2.2. MCD and KICS

2.3. PEL
3. KSHV-pirated Inflammatory Genes: vIL-6, vFLIP and vMIPs
3.1. vFLIP
3.2. vMIPs
4. The NF-κB and p53 Pathways: Common Targets of KSHV Gene Products Relevant to KSHV Malignancies
5. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Nador, R.G.; Cesarman, E.; Knowles, D.M.; Said, J.W. Herpes-like DNA sequences in a body-cavity-based lymphoma in an HIV-negative patient. N. Engl. J. Med. 1995, 333, 943. [Google Scholar] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; D’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Dupin, N.; Diss, T.L.; Kellam, P.; Tulliez, M.; Du, M.Q.; Sicard, D.; Weiss, R.A.; Isaacson, P.G.; Boshoff, C. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 2000, 95, 1406–1412. [Google Scholar] [PubMed]
- Uldrick, T.S.; Wang, V.; O’Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin. Infect. Dis. 2010, 51, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.N.; Ganem, D.E.; Osmond, D.H.; Page-Shafer, K.A.; Macrae, D.; Kedes, D.H. Sexual transmission and the natural history of human herpesvirus 8 infection. N. Engl. J. Med. 1998, 338, 948–954. [Google Scholar] [CrossRef]
- Dollard, S.C.; Butler, L.M.; Jones, A.M.; Mermin, J.H.; Chidzonga, M.; Chipato, T.; Shiboski, C.H.; Brander, C.; Mosam, A.; Kiepiela, P.; et al. Substantial regional differences in human herpesvirus 8 seroprevalence in sub-Saharan Africa: Insights on the origin of the “Kaposi’s sarcoma belt”. Int. J. Cancer 2010, 127, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Niedt, G.W.; Prioleau, P.G. Kaposi’s sarcoma occurring in a dermatome previously involved by herpes zoster. J. Am. Acad. Dermatol. 1988, 18, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, C.J.; Harrington, R.D.; Dhanireddy, S.; Crane, H.M.; Casper, C.; Kitahata, M.M. Paradoxical immune reconstitution inflammatory syndrome in HIV-infected patients treated with combination antiretroviral therapy after AIDS-defining opportunistic infection. Clin. Infect. Dis. 2012, 54, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Browning, P.J.; Sechler, J.M.; Kaplan, M.; Washington, R.H.; Gendelman, R.; Yarchoan, R.; Ensoli, B.; Gallo, R.C. Identification and culture of Kaposi’s sarcoma-like spindle cells from the peripheral blood of human immunodeficiency virus-1-infected individuals and normal controls. Blood 1994, 84, 2711–2720. [Google Scholar]
- Yao, L.; Salvucci, O.; Cardones, A.R.; Hwang, S.T.; Aoki, Y.; De La Luz Sierra, M.; Sajewicz, A.; Pittaluga, S.; Yarchoan, R.; Tosato, G. Selective expression of stromal-derived factor-1 in the capillary vascular endothelium plays a role in Kaposi sarcoma pathogenesis. Blood 2003, 102, 3900–3905. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, S.; Taddeo, A.; Calabro, M.L.; Brambilla, L.; Bellinvia, M.; Bergamo, E.; Clerici, M.; Villa, M.L. Peripheral blood endothelial progenitors as potential reservoirs of Kaposi’s sarcoma-associated herpesvirus. PLoS One 2008, 3, e1520. [Google Scholar] [CrossRef]
- Duus, K.M.; Lentchitsky, V.; Wagenaar, T.; Grose, C.; Webster-Cyriaque, J. Wild-type Kaposi’s sarcoma-associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J. Virol. 2004, 78, 4074–4084. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Brazeau, E.; Lagunoff, M. Kaposi’s sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 2004, 328, 7–18. [Google Scholar] [CrossRef]
- Grossmann, C.; Podgrabinska, S.; Skobe, M.; Ganem, D. Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J. Virol. 2006, 80, 7179–7185. [Google Scholar] [CrossRef] [PubMed]
- Matta, H.; Surabhi, R.M.; Zhao, J.; Punj, V.; Sun, Q.; Schamus, S.; Mazzacurati, L.; Chaudhary, P.M. Induction of spindle cell morphology in human vascular endothelial cells by human herpesvirus 8-encoded viral FLICE inhibitory protein K13. Oncogene 2007, 26, 1656–1660. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Pise-Masison, C.A.; Brady, J.N.; Tosato, G. Gene regulation and functional alterations induced by Kaposi’s sarcoma-associated herpesvirus-encoded ORFK13/vFLIP in endothelial cells. J. Virol. 2009, 83, 2140–2153. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, C.; Whitby, D.; Hatziioannou, T.; Fisher, C.; van der Walt, J.; Hatzakis, A.; Weiss, R.; Schulz, T. Kaposi’s-sarcoma-associated herpesvirus in HIV-negative Kaposi’s sarcoma. Lancet 1995, 345, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.A.; Punjabi, A.S.; Wells, R.C.; Wittkopp, C.J.; Vart, R.; Lagunoff, M. The KSHV viral IL-6 homolog is sufficient to induce blood to lymphatic endothelial cell differentiation. Virology 2012, 428, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Pekkonen, P.; Laurinavicius, S.; Sugiyama, N.; Henderson, S.; Gunther, T.; Rantanen, V.; Kaivanto, E.; Aavikko, M.; Sarek, G.; et al. KSHV-initiated notch activation leads to membrane-type-1 matrix metalloproteinase-dependent lymphatic endothelial-to-mesenchymal transition. Cell Host Microbe 2011, 10, 577–590. [Google Scholar] [CrossRef]
- Gasperini, P.; Espigol-Frigole, G.; McCormick, P.J.; Salvucci, O.; Maric, D.; Uldrick, T.S.; Polizzotto, M.N.; Yarchoan, R.; Tosato, G. Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through Notch-dependent signaling. Cancer Res. 2012, 72, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Emuss, V.; Lagos, D.; Pizzey, A.; Gratrix, F.; Henderson, S.R.; Boshoff, C. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog. 2009, 5, e1000616. [Google Scholar] [CrossRef] [PubMed]
- Oksenhendler, E.; Carcelain, G.; Aoki, Y.; Boulanger, E.; Maillard, A.; Clauvel, J.P.; Agbalika, F. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected patients. Blood 2000, 96, 2069–2073. [Google Scholar] [PubMed]
- Polizzotto, M.N.; Uldrick, T.S.; Hu, D.; Yarchoan, R. Clinical manifestations of kaposi sarcoma herpesvirus lytic activation: Multicentric Castleman Disease (KSHV-MCD) and the KSHV inflammatory cytokine syndrome. Front. Microbiol. 2012, 3, 73. [Google Scholar] [PubMed]
- Cronin, D.M.; Warnke, R.A. Castleman disease: An update on classification and the spectrum of associated lesions. Adv. Anat. Pathol. 2009, 16, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Pluda, J.M.; Yarchoan, R.; Jaffe, E.S.; Feuerstein, I.M.; Solomon, D.; Steinberg, S.M.; Wyvill, K.M.; Raubitschek, A.; Katz, D.; Broder, S. Development of non-Hodgkin lymphoma in a cohort of patients with severe human immunodeficiency virus (HIV) infection on long-term antiretroviral therapy. Ann. Intern. Med. 1990, 113, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Chadburn, A.; Hyjek, E.M.; Tam, W.; Liu, Y.; Rengifo, T.; Cesarman, E.; Knowles, D.M. Immunophenotypic analysis of the Kaposi sarcoma herpesvirus (KSHV; HHV-8)-infected B cells in HIV+ multicentric Castleman disease (MCD). Histopathology 2008, 53, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Tosato, G.; Fonville, T.W.; Pittaluga, S. Serum viral interleukin-6 in AIDS-related multicentric Castleman disease. Blood 2001, 97, 2526–2527. [Google Scholar] [CrossRef] [PubMed]
- Polizzotto, M.N.; Uldrick, T.S.; Wang, V.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Pittaluga, S.; O’Mahony, D.; Whitby, D.; Tosato, G.; et al. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood 2013, 122, 4189–4198. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Narazaki, M.; Kishimoto, T.; Tosato, G. Receptor engagement by viral interleukin-6 encoded by Kaposi sarcoma-associated herpesvirus. Blood 2001, 98, 3042–3049. [Google Scholar] [CrossRef]
- Du, M.Q.; Diss, T.C.; Liu, H.; Ye, H.; Hamoudi, R.A.; Cabecadas, J.; Dong, H.Y.; Harris, N.L.; Chan, J.K.; Rees, J.W.; et al. KSHV- and EBV-associated germinotropic lymphoproliferative disorder. Blood 2002, 100, 3415–3418. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Sato, Y.; Kurata, T.; Mori, S.; Sata, T. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology 2000, 269, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Sasai, M.; Shima, Y.; Nakagawa, M.; Matsumoto, T.; Shirai, T.; Kishimoto, T.; Yoshizaki, K. Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 2000, 95, 56–61. [Google Scholar] [PubMed]
- Nishimoto, N.; Kanakura, Y.; Aozasa, K.; Johkoh, T.; Nakamura, M.; Nakano, S.; Nakano, N.; Ikeda, Y.; Sasaki, T.; Nishioka, K.; et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005, 106, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Suthaus, J.; Stuhlmann-Laeisz, C.; Tompkins, V.S.; Rosean, T.R.; Klapper, W.; Tosato, G.; Janz, S.; Scheller, J.; Rose-John, S. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric Castleman disease in mice. Blood 2012, 119, 5173–5181. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, A.G.; Aaron, L.; Mateus, C.; Gyan, E.; Gorin, I.; Viard, J.P.; Calvez, V.; Dupin, N. Rituximab therapy for HIV-associated Castleman disease. Blood 2003, 102, 2786–2788. [Google Scholar] [CrossRef] [PubMed]
- Knowles, D.M.; Inghirami, G.; Ubriaco, A.; Dalla-Favera, R. Molecular genetic analysis of three AIDS-associated neoplasms of uncertain lineage demonstrates their B-cell derivation and the possible pathogenetic role of the Epstein-Barr virus. Blood 1989, 73, 792–799. [Google Scholar] [PubMed]
- Perez, C.L.; Rudoy, S. Anti-CD20 monoclonal antibody treatment of human herpesvirus 8-associated, body cavity-based lymphoma with an unusual phenotype in a human immunodeficiency virus-negative patient. Clin. Diagn. Lab. Immunol. 2001, 8, 993–996. [Google Scholar] [PubMed]
- Siddiqi, T.; Joyce, R.M. A case of HIV-negative primary effusion lymphoma treated with bortezomib, pegylated liposomal doxorubicin, and rituximab. Clin. Lymphoma Myeloma 2008, 8, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Kliche, S.; Kremmer, E.; Hammerschmidt, W.; Koszinowski, U.; Haas, J. Persistent infection of Epstein-Barr virus-positive B lymphocytes by human herpesvirus 8. J. Virol. 1998, 72, 8143–8149. [Google Scholar] [PubMed]
- Guasparri, I.; Keller, S.A.; Cesarman, E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 2004, 199, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, A.; Anderson, J.; Papanastasiou, A.; Takeuchi, Y.; Boshoff, C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 2005, 105, 2510–2518. [Google Scholar] [CrossRef] [PubMed]
- Judde, J.G.; Lacoste, V.; Briere, J.; Kassa-Kelembho, E.; Clyti, E.; Couppie, P.; Buchrieser, C.; Tulliez, M.; Morvan, J.; Gessain, A. Monoclonality or oligoclonality of human herpesvirus 8 terminal repeat sequences in Kaposi’s sarcoma and other diseases. J. Natl. Cancer Inst. 2000, 92, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the gamma-herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Boshoff, C.; Weiss, R.A.; Chang, Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996, 274, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Tosato, G. Role of vascular endothelial growth factor/vascular permeability factor in the pathogenesis of Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphomas. Blood 1999, 94, 4247–4254. [Google Scholar] [PubMed]
- Aoki, Y.; Jaffe, E.S.; Chang, Y.; Jones, K.; Teruya-Feldstein, J.; Moore, P.S.; Tosato, G. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood 1999, 93, 4034–4043. [Google Scholar]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J.; Ruvolo, V.R.; Burns, W.H.; Sandford, G.; Wan, X.; Ciufo, D.; Hendrickson, S.B.; Guo, H.G.; Hayward, G.S.; Reitz, M.S. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 1997, 3, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Hibi, M.; Nakagawa, N.; Nakagawa, T.; Yasukawa, K.; Yamanishi, K.; Taga, T.; Kishimoto, T. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science 1993, 260, 1808–1810. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Wang, H.; Nicholas, J. Human herpesvirus 8 interleukin-6 (vIL-6) signals through gp130 but has structural and receptor-binding properties distinct from those of human IL-6. J. Virol. 1999, 73, 8268–8278. [Google Scholar] [PubMed]
- Meads, M.B.; Medveczky, P.G. Kaposi’s sarcoma-associated herpesvirus-encoded viral interleukin-6 is secreted and modified differently than human interleukin-6: Evidence for a unique autocrine signaling mechanism. J. Biol. Chem. 2004, 279, 51793–51803. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Sandford, G.; Nicholas, J. Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J. Virol. 2009, 83, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. KSHV infection and the pathogenesis of Kaposi’s sarcoma. Annu. Rev. Pathol. 2006, 1, 273–296. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Jasmin, A.; Eby, M.T.; Hood, L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 1999, 18, 5738–5746. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Espigol-Frigole, G.; Gasperini, P.; Uldrick, T.S.; Yarchoan, R.; Tosato, G. A20/TNFAIP3 inhibits NF-kappaB activation induced by the Kaposi’s sarcoma-associated herpesvirus vFLIP oncoprotein. Oncogene 2013, 32, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Ma, A.; Harhaj, E.W. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010, 327, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Sin, S.H.; Roy, D.; Wang, L.; Staudt, M.R.; Fakhari, F.D.; Patel, D.D.; Henry, D.; Harrington, W.J., Jr.; Damania, B.A.; Dittmer, D.P. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 2007, 109, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, P.; Tosato, G. Targeting the mammalian target of Rapamycin to inhibit VEGF and cytokines for the treatment of primary effusion lymphoma. Leukemia 2009, 23, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Nichols, L.A.; Adang, L.A.; Kedes, D.H. Rapamycin blocks production of KSHV/HHV8: Insights into the anti-tumor activity of an immunosuppressant drug. PLoS One 2011, 6, e14535. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Sin, S.H.; Lucas, A.; Venkataramanan, R.; Wang, L.; Eason, A.; Chavakula, V.; Hilton, I.B.; Tamburro, K.M.; Damania, B.; et al. mTOR inhibitors block Kaposi sarcoma growth by inhibiting essential autocrine growth factors and tumor angiogenesis. Cancer Res. 2013, 73, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Gao, S.J.; Dominguez, G.; Cesarman, E.; Lungu, O.; Knowles, D.M.; Garber, R.; Pellett, P.E.; McGeoch, D.J.; Chang, Y. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcomae. J. Virol. 1996, 70, 549–558. [Google Scholar]
- Stine, J.T.; Wood, C.; Hill, M.; Epp, A.; Raport, C.J.; Schweickart, V.L.; Endo, Y.; Sasaki, T.; Simmons, G.; Boshoff, C.; et al. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood 2000, 95, 1151–1157. [Google Scholar] [PubMed]
- Dairaghi, D.J.; Fan, R.A.; McMaster, B.E.; Hanley, M.R.; Schall, T.J. HHV8-encoded vMIP-I selectively engages chemokine receptor CCR8. Agonist and antagonist profiles of viral chemokines. J. Biol. Chem. 1999, 274, 21569–21574. [Google Scholar]
- Kledal, T.N.; Rosenkilde, M.M.; Coulin, F.; Simmons, G.; Johnsen, A.H.; Alouani, S.; Power, C.A.; Luttichau, H.R.; Gerstoft, J.; Clapham, P.R.; et al. A broad-spectrum chemokine antagonist encoded by Kaposi’s sarcoma-associated herpesvirus. Science 1997, 277, 1656–1659. [Google Scholar] [CrossRef]
- Boshoff, C.; Endo, Y.; Collins, P.D.; Takeuchi, Y.; Reeves, J.D.; Schweickart, V.L.; Siani, M.A.; Sasaki, T.; Williams, T.J.; Gray, P.W.; et al. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science 1997, 278, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Sozzani, S.; Luini, W.; Bianchi, G.; Allavena, P.; Wells, T.N.; Napolitano, M.; Bernardini, G.; Vecchi, A.; D’Ambrosio, D.; Mazzeo, D.; et al. The viral chemokine macrophage inflammatory protein-II is a selective Th2 chemoattractant. Blood 1998, 92, 4036–4039. [Google Scholar] [PubMed]
- Campbell, J.J.; Haraldsen, G.; Pan, J.; Rottman, J.; Qin, S.; Ponath, P.; Andrew, D.P.; Warnke, R.; Ruffing, N.; Kassam, N.; et al. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature 1999, 400, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Katano, H.; Tadagaki, K.; Sato, Y.; Ohsaki, E.; Mori, Y.; Yamanishi, K.; Ueda, K. Novel monoclonal antibodies for identification of multicentric Castleman’s disease; Kaposi’s sarcoma-associated herpesvirus-encoded vMIP-I and vMIP-II. Virology 2012, 425, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Scalley-Kim, M.L.; Hess, B.W.; Kelly, R.L.; Krostag, A.R.; Lustig, K.H.; Marken, J.S.; Ovendale, P.J.; Posey, A.R.; Smolak, P.J.; Taylor, J.D.; et al. A novel highly potent therapeutic antibody neutralizes multiple human chemokines and mimics viral immune modulation. PLoS One 2012, 7, e43332. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Field, N.P.; Gal-Oz, E.; Bonanno, G.A. Continuing bonds and adjustment at 5 years after the death of a spouse. J. Consult. Clin. Psychol. 2003, 71, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Eby, M.T.; Rathore, N.; Sinha, S.K.; Kumar, A.; Chaudhary, P.M. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem. 2002, 277, 13745–13751. [Google Scholar] [CrossRef] [PubMed]
- Field, N.; Low, W.; Daniels, M.; Howell, S.; Daviet, L.; Boshoff, C.; Collins, M. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 2003, 116, 3721–3728. [Google Scholar] [CrossRef]
- Keller, S.A.; Schattner, E.J.; Cesarman, E. Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 2000, 96, 2537–2542. [Google Scholar] [PubMed]
- Hussain, A.R.; Ahmed, S.O.; Ahmed, M.; Khan, O.S.; Al Abdulmohsen, S.; Platanias, L.C.; Al-Kuraya, K.S.; Uddin, S. Cross-talk between NFkB and the PI3-kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis. PLoS One 2012, 7, e39945. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, R.; Matta, H.; Chaudhary, P.M. A purine scaffold HSP90 inhibitor BIIB021 has selective activity against KSHV-associated primary effusion lymphoma and blocks vFLIP K13-induced NF-kappaB. Clin. Cancer Res. 2013, 19, 5016–5026. [Google Scholar] [CrossRef] [PubMed]
- Nayar, U.; Lu, P.; Goldstein, R.L.; Vider, J.; Ballon, G.; Rodina, A.; Taldone, T.; Erdjument-Bromage, H.; Chomet, M.; Blasberg, R.; et al. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood 2013, 122, 2837–2847. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Prodromou, C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 2006, 75, 271–294. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Peyrat, J.F.; Brion, J.D.; Alami, M. Recent advances in Hsp90 inhibitors as antitumor agents. Anti-Cancer Agents Med. Chem. 2008, 8, 761–782. [Google Scholar]
- Chen, W.; Sin, S.H.; Wen, K.W.; Damania, B.; Dittmer, D.P. Hsp90 inhibitors are efficacious against Kaposi Sarcoma by enhancing the degradation of the essential viral gene LANA, of the viral co-receptor EphA2 as well as other client proteins. PLoS Pathog. 2012, 8, e1003048. [Google Scholar] [CrossRef] [PubMed]
- Biamonte, M.A.; Van de Water, R.; Arndt, J.W.; Scannevin, R.H.; Perret, D.; Lee, W.C. Heat shock protein 90: Inhibitors in clinical trials. J. Med. Chem. 2010, 53, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Goloudina, A.R.; Demidov, O.N.; Garrido, C. Inhibition of HSP70: A challenging anti-cancer strategy. Cancer Lett. 2012, 325, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Devin, A.; Miller, A.; Lin, Y.; Rodriguez, Y.; Neckers, L.; Liu, Z.G. Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J. Biol. Chem. 2000, 275, 10519–10526. [Google Scholar] [CrossRef] [PubMed]
- Thangjam, G.S.; Dimitropoulou, C.; Joshi, A.D.; Barabutis, N.; Shaw, M.C.; Kovalenkov, Y.; Wallace, C.M.; Fulton, D.J.; Patel, V.; Catravas, J.D. Novel mechanism of attenuation of LPS-induced NF-kappaB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.J.; Boshoff, C.; Jayachandra, S.; Weiss, R.A.; Chang, Y.; Moore, P.S. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 1997, 15, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Rivas, C.; Thlick, A.E.; Parravicini, C.; Moore, P.S.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 2001, 75, 429–438. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S.; Talbot, S.J.; Boshoff, C.H.; Zarkowska, T.; Godden, K.; Paterson, H.; Weiss, R.A.; Mittnacht, S. Cyclin encoded by KS herpesvirus. Nature 1996, 382, 410. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, E.W.; Hodgson, J.G.; Gray, J.W.; Kogan, S.; Jones, N.; Evan, G.I. The role of p53 in suppression of KSHV cyclin-induced lymphomagenesis. Cancer Res. 2004, 64, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, M.; Watanabe, T.; Yang, A.; Starost, M.F.; Kobayashi, H.; Atkins, A.M.; Borris, D.L.; Hanan, E.A.; Schimel, D.; Bryant, M.A.; et al. Lymphatic dysfunction in transgenic mice expressing KSHV k-cyclin under the control of the VEGFR-3 promoter. Blood 2005, 105, 2356–2363. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T. Small-molecule antagonists of p53-MDM2 binding: Research tools and potential therapeutics. Cell Cycle 2004, 3, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Kurki, S.; Enback, J.; Iotzova, G.; Haas, J.; Laakkonen, P.; Laiho, M.; Ojala, P.M. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J. Clin. Investig. 2007, 117, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Ojala, P.M. p53 reactivation kills KSHV lymphomas efficiently in vitro and in vivo: New hope for treating aggressive viral lymphomas. Cell Cycle 2007, 6, 2205–2209. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sakakibara, S.; Tosato, G. Contribution of Viral Mimics of Cellular Genes to KSHV Infection and Disease. Viruses 2014, 6, 3472-3486. https://doi.org/10.3390/v6093472
Sakakibara S, Tosato G. Contribution of Viral Mimics of Cellular Genes to KSHV Infection and Disease. Viruses. 2014; 6(9):3472-3486. https://doi.org/10.3390/v6093472
Chicago/Turabian StyleSakakibara, Shuhei, and Giovanna Tosato. 2014. "Contribution of Viral Mimics of Cellular Genes to KSHV Infection and Disease" Viruses 6, no. 9: 3472-3486. https://doi.org/10.3390/v6093472