2.1. Discovery, Geographic Distribution and Natural Host range
Classical ISFs have a ubiquitous geographic distribution; these viruses have been isolated from mosquitoes in every continent with the exception of Antarctica (
Table 1). Additionally, cISF-like sequences have been detected in sandflies and midges in several Mediterranean countries as discussed later in this section. The first cISF to be discovered was cell fusing agent virus (CFAV) after its isolation from
Aedes aegypti cell cultures over 40 years ago [
17]. This discovery received little attention as illustrated by the fact that 17 years passed before another article on the virus was published [
18]. CFAV has since been isolated from or detected in field-collected mosquitoes in Indonesia [
19], Mexico [
20], Puerto Rico [
21] and Thailand [
22,
23]. CFAV has also been isolated from laboratory colonies originally established from mosquitoes collected in the United States [
24]. Additionally, CFAV-like sequences have been identified in field-collected mosquitoes in Argentina (Genbank Accession No. DQ335466, DQ335467 and DQ431718) but the sequences are too short (87–110 nt) for reliable analysis.
Table 1.
Geographic distribution and natural host range of classical insect-specific flaviviruses.
Table 1.
Geographic distribution and natural host range of classical insect-specific flaviviruses.
| a Virus | Isolate Available | Geographic Distribution | Natural Host Range | References |
|---|
| Aedes flavivirus (AEFV) | Yes | Japan (2003), Italy (2008), USA (2011), b Thailand (2012) | Ae. albopictus, Ae. flavopictus, Cx. pipiens | [12,19,24,25,26,27] |
| Aedes galloisi flavivirus (AGFV) | Yes | Japan (2003) | Ae. galloisi | [28] |
| Calbertado virus (CLBOV) | Yes | Canada (2003), USA (2006) | Cx. tarsalis, Cx. pipiens | [29,30] |
| Cell fusing agent virus (CFAV) | Yes | Laboratory (1975), Puerto Rico (2002), Indonesia (2004), Mexico (2007), Thailand (2008), bUnited States (2012) | Ae. albopictus, Ae. aegypti, Culex spp. | [17,19,20,21,22,24] |
| Culex flavivirus (CxFV) | Yes | Japan (2003), Indonesia (2004), China (2006), Guatemala (2006), USA (2006), Mexico (2007), Trinidad (2008), Uganda (2008), Argentina (2009) | Cx. interrogator, Cx. maxi, Cx. nigripalpus, Cx. pipiens, Cx. quinquefasciatus, Cx. restuans, Cx. tarsalis, Cx. tritaeniorhynchus, Cx. usquatus | [29,31,32,33,34,35,36,37,38] |
| c Culex theileri flavivirus (CTFV) | Yes | Spain (2006), Portugal (2009–2010), Greece (2010), Thailand (date not specified) | Cx. fuscocephala, Cx. pipiens, Cx. theileri | [25,39,40,41] (Genbank Accession No. AY457040) |
| d Hanko virus (HANKV) | Yes | Finland (2005), Spain (2006), Italy (ca. 2007), Portugal (ca. 2007) | Ae. caspius, Ae. detritus, Ae. vexans, Cx. pipiens, Cx. perexiguus, Cx. theileri | [25,39,42,43] |
| Kamiti River virus (KRV) | Yes | Kenya (1999) | Ae. macintoshi | [44,45] |
| Nakiwogo virus (NAKV) | Yes | Uganda (2008) | Mansonia africana nigerrima | [38] |
| e Nienokoue virus(NIEV) | f Yes | Cote d’Ivoire (2004) | Culex spp. | (Genbank Accession No. NC_024299) |
| Palm Creek virus (PCV) | Yes | Australia (2010) | Coquillettidia xanthogaster | [46] |
| Quang Binh virus (QBV) | Yes | Vietnam (2002), China (2009) | An. sinensis, Cx. tritaeniorhynchus | [47,48,49] |
The next cISF to be discovered was Kamiti River virus (KRV) after its isolation from mosquitoes in Kenya in 1999 (
Table 1) [
44,
45]. A KRV-like sequence has also been detected in mosquitoes in Argentina (Genbank Accession No. DQ335465) but, due to the limited amount of sequence data obtained in the study (a 124-nt region of the NS5 gene was sequenced), additional information is needed to determine whether KRV occurs in this country. The third member of the cISF group to be reported was Culex flavivirus (CxFV). The virus was first isolated from
Culex spp. mosquitoes in Japan and Indonesia in 2003–2004 [
31] and later isolated from
Culex spp. mosquitoes in Argentina [
32], Brazil [
50], China [
33,
51], Guatemala [
34], Mexico [
35,
36,
52], Taiwan [
53], Trinidad [
37], Uganda [
38] and the United States [
29,
37,
54,
55,
56].
In the last ten years, several other cISFs have been described and these include: Aedes flavivirus (AEFV) in Japan [
19], Italy [
12,
25,
57], Thailand [
24] and the United States [
26], Aedes galloisi flavivirus (AGFV) in Japan [
28], Calbertado virus (CLBOV) in Canada [
30,
58] and the USA [
29], Nakiwogo virus (NAKV) in Uganda [
38], Nienokoue virus (NIEV) in Cote d’Ivoire (Genbank Accession No. NC_024299), Palm Creek virus (PCV) in Australia [
46] and Quang Binh virus (QBV) in Vietnam [
47] and China [
48] (
Table 1). Several other novel cISFs have also been identified. However, after performing nucleotide sequence alignments, it is apparent that some of these viruses have been assigned multiple names due to their simultaneous discoveries by independent research groups as discussed below.
Three apparently novel cISFs were reported in the literature within in space of a few months: Hanko virus (HANKV) after its isolation from
Ae. caspius in Finland in 2005 [
42], Ochlerotatus flavivirus (OcFV) after its isolation from various
Aedes and
Culex spp. mosquitoes in Spain, Italy and Portugal from 2007 to 2010 [
25] and Spanish Ochlerotatus flavivirus (SOcFV) after its isolation from
Ae. caspius in Spain in 2006 [
39] (
Table 1). Huhtamo and colleagues sequenced the entire ORF of their virus [
42]; the two other research groups sequenced a 238 to 917 nt region of the NS5 gene [
25,
39]. Pairwise nucleotide sequence alignments of the 163-nt region shared by representative HANKV, OcFV and SOcFV sequences (Genbank Accession Nos., JQ268258, GQ476991 and JF707790 respectively) revealed that these viruses are 91% to 96% identical. It has been proposed that flaviviruses with >84% nucleotide sequence identity should be classified within the same species [
59]. Although these alignments were performed using short sequences in the relatively highly conserved NS5 region, according to the above criterion, HANKV, OcFV and SOcFV are likely to be the same virus species. For the remainder of this review, the virus will be referred to as HANKV because Huhtamo and colleagues [
42] performed the most comprehensive sequence analysis. The following year, an article describing an apparently novel cISF designated Ochlerotatus caspius flavivirus from Portugal (OCFV
PT) was published [
43]. The authors sequenced almost all of the OCFV
PT genome and reported that it has 89% nucleotide identity to the corresponding region of HANKV. In accordance to the criterion for flaviviral species demarcation [
59], this virus is HANKV and not an unrecognized cISF species.
Another cISF has been assigned multiple names: Culex theileri flavivirus (CTFV or CxthFV), Spanish Culex flavivirus (SCxFV) and Wang Thong virus (WTV). Culex theileri flavivirus (CTFV) was isolated from
Cx. theileri in Portugal in 2009–2010 [
40] (
Table 1). The same virus was independently discovered by another research group after its isolation from
Cx. theileri in Portugal and Spain in 2007–2010, and coincidently given the same name but different acronym (e.g., CxthFV) [
25]. SCxFV is the name assigned to several isolates obtained from
Cx. theileri and
Cx. pipiens in Spain in 2006 [
39]. WTV was detected in
Cx. fuscocephala in Thailand on an unspecified date (Genbank Accession No. AY457040). Parreira and colleagues sequenced almost the entire genome of their virus [
40]; the other groups sequenced a 159 to 917 nt region of the NS5 gene [
25,
39]. Pairwise sequence alignments of the 140-nt region shared by representative CTFV, CxthFV, SCxFV and WTV sequences (Genbank Accession Nos. HE574574, EU716420, JF707811 and AY457040, respectively) revealed that these viruses are 91% to 100% identical. Therefore, according to the criterion for flaviviral species demarcation [
59], CTFV, CxthFV, SCxFV and WTV are the same virus. For the remainder of this review, the virus will be referred to as Culex theileri flavivirus since this name was chosen by two of the four research groups that made the discovery [
25,
40]. The acronym selected by Parreira and colleagues (e.g., CTFV) will be used because these researchers performed the most comprehensive sequence analysis [
40].
Quang Binh virus or a novel QBV-like virus (designated Yunnan Culex flavivirus; YNCxFV) was isolated from 10 pools of
Cx. tritaeniorhynchus and one pool of
Anopheles sinensis collected in the Yunnan Province of China in 2009 [
48] (
Table 1). The genome of one isolate was completely sequenced and the ORF was reported to have 83.0% nucleotide identity to the corresponding region of the prototypical QBV isolate. Because this figure is close to the >84% value for flavivirus species demarcation [
59], the authors opted for a conservative approach and considered their isolate to be a strain of QBV [
48]. The authors also pointed out that their isolate was obtained from the same mosquito spp. and geographic region as QBV (Yunnan Province borders Vietnam) and that additional testing (
i.e., neutralization assays) was required before the isolate could be considered the prototypical member of a novel species. For the purpose of this review, the entire genomic sequences (as opposed to the entire ORFs) of the prototypical QBV and YNCxFV isolates (Genbank Accession Nos. FJ644291 and KC464457, respectively) were aligned to shed more light on their genetic relatedness. The two sequences have 83.7% nucleotide identity which is even closer to the threshold value establish by Kuno
et al [
59]. This analysis also revealed that the two genomes are of the exact same length (10,865 nt). Thus, YNCxFV could very well be a divergent isolate of QBV rather than a novel cISF species. A comparison of the lengths of the non-coding regions revealed that the 5' UTR of YNCxFV is one nucleotide shorter than the corresponding region of QBV (111 nt
vs. 112 nt) while its 3' UTR is one nucleotide longer (674 nt
vs. 673 nt). Some cISFs display strain-specific differences in the lengths of their non-coding region lengths; for example, the 5' and 3' UTRs of CxFV Toyama 1431 strain (Genbank Accession No. AB701775) are both one nucleotide shorter than the corresponding regions of CxFV Tokyo strain (Genbank Accession No. AB262759). Because it is unclear whether YNCxFV is an unusual isolate of QBV or a distinct virus species, the conservative approach opted by Zuo and colleagues [
48] will be used for the remainder of this review and their virus will be referred to as QBV.
A potentially novel cISF, designated Aedes vexans flavivirus (AeveFV), was isolated from
Ae. vexans in Italy and the Czech Republic in 2008–2009 [
25]. A short (131 to 263 nt) region of the NS5 gene was sequenced and shown to have no more than 80% nucleotide identity to the corresponding region of its closest relative, consistent with the discovery of a new virus. However, the sequences are short and more comprehensive sequencing experiments are needed to determine whether AeveFV is an unrecognized virus. Likewise, a potentially novel cISF, designated Czech Aedes vexans flavivirus (Czech AeveFV), was isolated from
Ae. vexans in the Czech Republic in 2009 [
25] but the corresponding sequences are too short (209 to 217 nt) for reliable analysis. Another potentially novel cISF, designated Aedes cinereus flavivirus (AeciFV), was detected in
Ae. cinereus in the U.K. in 2010 [
25]. Comprehensive sequence alignments were not performed (the virus was compared to only five other cISFs) and the sequence has not been deposited into the Genbank database. Additional information is needed to determine whether AeciFV is a novel cISF. Although AeveFV, Czech AeveFV and AeciFV could very well represent novel cISFs, they are not listed in
Table 1; the table is restricted to viruses for which more than 300 nt of sequence data are available.
Two 917-nt sequences corresponding to a cISF designated Culex pipiens flavivirus were detected in mosquitoes in Portugal in 2009–2010 (Genbank Accession No. HE997068-HE997069). The sequences are 97% identical to the corresponding region of CTFV and therefore, the species name of Culex pipiens flavivirus should be discontinued. Multiple 165-nt sequences corresponding to a virus denoted as Culicinae flavivirus were also identified in mosquitoes in Portugal (Genbank Accession No. EU716415–EU716419 and EU716421–EU716424). This species name should also be discontinued because these sequences are 90%–99% identical to the corresponding region of HANKV. Other species names that appear in the NCBI taxonomy and Genbank databases that should be discontinued for similar reasons are Mediterranean Culex flavivirus, Mediterranean Ochlerotatus flavivirus and mosquito flavivirus (for example, Genbank Accession No. JF707854, JF707806 and KF882513).
Although cISFs have been isolated exclusively from mosquitoes, cISF-like sequences have been detected by molecular methods in other dipterans indicating that cISFs may not have a mosquito-restricted host range. Novel cISF-like sequences of 157 nt were detected in male
Phlebotomus perniciosus in Algeria in 2006–2007 [
60]. PCR products were not detected when the reverse-transcription step was excluded suggesting that a novel virus, rather than CSA, was identified. Virus isolation experiments were not performed because the sandflies had been preserved in guanidinium thiocyanate. Classical ISF-like sequences were also detected in sandflies in Spain but once again virus isolation experiments were not attempted [
61]. In another study, a 6567-nt cISF-like sequence was identified by RNA deep sequencing in chironomids (non-biting midges) in France, although again isolation of virus particles was not attempted [
62].
2.2. In Vitro and in Vivo Replication Potential in Vertebrates and Arthropod Cells
Classical ISFs have not been isolated from any vertebrates in nature and cannot replicate in any vertebrate cell lines that have been tested; thus, these viruses are assumed to possess a vertebrate-incompetent replication phenotype. The most comprehensive
in vitro host range studies were performed with PCV which was shown to lack the ability to replicate in hamster (BHK-21), human (SW-13), monkey (Vero) and porcine (PS-EK) cells [
46], and CxFV which cannot replicate in avian (DF-1), hamster (BHK-21) or monkey (Vero) cells [
29,
31]. Most other cISFs have been demonstrated to lack the ability to infect hamster (BHK-21) and/or monkey (Vero) cells [
17,
19,
28,
29,
39,
44,
47]. Attempts to infect suckling mice with AeFV and CxFV by intracerebral inoculation were unsuccessful [
26,
37].
Every described cISF possesses the ability to replicate in
Ae. albopictus (C6/36) cells [
17,
19,
28,
30,
31,
38,
40,
42,
45,
46,
47]. Some cISFs induce cytopathic effect (CPE) and form plaques in C6/36 cells whereas others do not. Another determinant of whether CPE occurs is the passage history of the virus. PCV does not induce CPE in C6/36 cells after the first or second passage but often morphological changes (
i.e., syncytia and vacuolation in cells) are observed by the fourth passage [
46]. Moderate CPE was periodically observed in C6/36 cells inoculated with CxFV that had been passed at least twice whereas CPE was usually absent in cells infected with the original inoculum or virus passed once [
31]. CxFV isolates from Japan do not plaque in C6/36 cells [
31] unlike isolates from Guatemala [
63]. CxFV and CFAV both reach maximum titers of approximately 10
7 plaque forming units (pfu)/mL in C6/36 cells while KRV produces a maximum titer of 10
8 pfu/mL in this cell line [
44,
63]. The replicative potentials of select cISFs have also been assessed in other mosquito cell lines including CFAV which replicates in
Ae. albopictus (AA23) and
Ae. aegypti (A20) cells [
21] and KRV which replicates in
Ae. pseudoscutellaris (AP-61) and
Ae. aegypti cells [
44].
In vivo experiments have been performed to characterize the replicative potential and tissue tropisms of CxFV in
Culex spp. mosquitoes [
63,
64]. CxFV establishes a systemic infection in
Cx. pipiens, as indicated by the detection of viral RNA in all tissues examined (salivary glands, ovaries, testes, head, fat bodies and midguts) [
64]. The presence of CxFV RNA in the salivary glands is interesting because, due to the vertebrate-incompetent replication phenotype of this virus, establishment of a salivary gland infection does not appear necessary for its persistence in nature. CxFV was not detected in the saliva of
Cx. quinquefasciatus infected with CxFV alone but was detected in the saliva of mosquitoes co-infected with CxFV and WNV [
63].
2.3. Transmission
Vertical transmission is defined as the process by which an infected female directly transmits a pathogen to her progeny. The detection of cISFs in mosquitoes of all life stages, including adults of both sexes, indicates that vertical transmission is a major mechanism by which these viruses persist in mosquitoes in nature [
21,
26,
29,
45,
52,
65]. The initial isolations of KRV were made from
Ae. mcintoshi larvae and pupae [
45], CxFV RNA has been detected in
Cx. pipiens egg rafts, larvae, adult males and adult females [
29,
65] and AeFV was isolated from a pool of male
Ae. albopictus reared to adults from field-collected larvae [
26]. Additionally, the first isolate of CFAV was obtained from the C6/36 cell line which was derived from
Ae. albopictus larvae [
17,
66].
One mechanism of vertical transmission is transovarial transmission (TOT), defined as the process by which progeny of infected females are directly infected in the egg stage within the ovary before release and subsequent insemination. Experiments performed with field-infected
Cx. pipiens revealed that TOT is an efficient mechanism for CxFV persistence [
64]. Filial infection (FI) and TOT rates of 97.4% and 100%, respectively were reported. These values are considerably greater than the <1% FI and vertical infection rates typically reported in mosquitoes infected with dual-host flaviviruses [
67,
68,
69]. Viral dissemination to the ovaries is necessary for TOT to occur. Accordingly, CxFV RNA was detected in the ovaries of F
1 produced from field-infected
Cx. pipiens [
64]. Interestingly, TOT did not occur when uninfected laboratory-colonized
Cx. pipiens were infected with CxFV by needle inoculation [
64]. One explanation for the different TOT rates between the experimentally and naturally infected
Cx. pipiens could be that mosquitoes with lifelong infections (
i.e., vertically infected mosquitoes) are more susceptible to TOT than mosquitoes infected as adults. The mosquitoes did not possess an ovarian infection barrier because CxFV RNA was detected in their ovaries.
Vertical transmission of KRV has been demonstrated in laboratory-colonized
Ae. aegypti [
70]. The FI rate in the F
1 produced by the infected mosquitoes was 3.9% while the TOT rate was not reported. One likely explanation for the dramatically lower FI rate in this study as compared to the FI rate of 97.4% reported for CxFV is that there is no direct evidence to indicate the
Ae. aegypti is a natural host of KRV. The virus has only been isolated from
Ae. macintoshi in the field [
45] and vertical transmission is presumably more efficient in the natural mosquito host.
The contribution of venereal transmission in cISF persistence was investigated by allowing CxFV-infected male
Cx. pipiens to mate with uninfected females [
65]. Reciprocal mating experiments were also performed. Virus was transmitted to 2.4%–5.3% of the mosquitoes indicating that venereal transmission has a minor role in CxFV persistence. Horizontal transmission among larvae and non-sexual contact transmission among adults were considered unlikely modes of CxFV maintenance [
65]. Efficient
per os transmission of KRV has been reported for
Ae. aegypti [
70]. In these studies, 62.4% of mosquitoes that fed on infectious blood were positive for KRV by virus isolation in cell culture. Virus was also isolated from 90.2% of second instar larvae exposed to KRV-infected C6/36 cells. Efficient
per os infection has also been reported for
Ae. aegypti exposed to Eilat virus (EILV), an insect-specific alphavirus, via infectious blood meal [
71]. Infection and dissemination rates of 63%–78% and 8%–26% were observed.
Ae. albopictus,
An. gambiae and
Cx. quinquefasciatus were also susceptible to EILV infection, albeit at a lower rate. Studies need to be performed to assess whether efficient
per os infection occurs in mosquitoes exposed to cISFs via natural food sources (
i.e., nectar).
Some cISFs exhibit seasonal activity [
29,
37,
52,
54]. These findings could be considered unexpected if vertical transmission was the sole mechanism for their persistence in nature. CxFV was detected in mosquitoes in Texas, U.S.A from November to March but not April to August, even though mosquitoes were abundant at these times [
37]. CLBOV was not detected year-round in
Cx. pipiens and
Cx. tarsalis in Colorado, U.S.A [
29]. These findings could indicate that another mode of transmission has a major role in cISF persistence. Alternatively, these findings could be a consequence of sampling biases, small sample sizes or limitations in viral detection methods.
2.4. Competitive Interaction between cISFs and Dual-Host Flaviviruses
Superinfection exclusion (or homologous interference) is the process by which host cells infected with one virus do not support productive replication of the same or similar virus [
72]. This phenomenon has been observed during infections by a broad range of viruses and can occur in both vertebrate and invertebrate hosts [
73,
74,
75,
76]. Data regarding the abilities of cISFs to induce superinfection exclusion of dual-host flaviviruses in mosquito cells has been variable. Prior infection with PCV significantly reduced WNV and MVEV replication in C6/36 cells [
46]. In contrast, prior exposure to CxFV had no effect on WNV replication in C6/36 cells [
63]. In another study, WNV titers were significantly lower in CxFV-infected C6/36 cells compared to uninfected C6/36 cells at earlier, but not later, time points [
65]. The
in vitro growth kinetics and yields of JEV and DENV did not differ significantly in
Cx. tritaeniorhynchus cells persistently infected with CxFV when compared to cells without pre-existing CxFV infections although JEV superinfection induced severe CPE [
77]. Taken together, the above data indicate that cISFs can suppress the
in vitro replication of dual-host flaviviruses in mosquito cells under some circumstances.
Vector competence experiments were performed with two colonies of
Cx. pipiens, one persistently infected with CxFV and the other not, in order to evaluate the effect of CxFV on WNV transmission in this mosquito spp. [
65]. At 7 days p.i., a significantly lower percentage of CxFV-infected mosquitoes (72%) had disseminated WNV infections compared to single-virus infected mosquitoes (94%). Infection and transmission rates did not differ significantly. At 14 days p.i., WNV infection, dissemination and transmission rates did not differ significantly between the two groups. These data indicate that CxFV can suppress the
in vivo replication of WNV early during infection. However, it should be noted that the mosquito colonies used for these experiments are from different geographic locations (Colorado and Iowa) and therefore, their differential susceptibilities to WNV infection could to due to factors other than co-infection with CxFV. In another study, sequential infection experiments demonstrated that prior infection with CxFV had no significant effect on WNV infection, dissemination or transmission in
Cx. quinquefasciatus [
63]. Coinfection experiments demonstrated that the ability of WNV to be transmitted by
Cx. quinquefasciatus after simultaneous inoculation with WNV and CxFV was strain-specific. A significantly higher percentage of co-inoculated Honduras
Cx. quinquefasciatus transmitted WNV compared to mosquitoes inoculated with WNV alone. In contrast, the percentage of co-inoculated Sebring
Cx. quinquefasciatus that transmitted WNV did not differ significantly from the single-virus infected control group. These experiments indicate that cISFs can enhance the transmissibility of dual-host flaviviruses under some circumstances. In this regard, a positive ecological association between CxFV and WNV was reported in field-collected
Culex spp. mosquitoes in Chicago, U.S.A. in 2006 [
56]. WNV-positive mosquito pools were four times more likely to be positive for CxFV compared to spatiotemporally matched WNV-negative pools.
2.5. Genome Sequencing and Phylogeny
Complete genome sequences are available for five cISFs: AeFV, CFAV, CxFV, KRV and QBV (
Table 2). The prototypical isolates of these viruses possess 5’ UTRs of 91 to 113 nt, consistent with the lengths of the 5’ UTRs of most other flaviviruses [
2]. The 3’ UTRs of CFAV, CxFV and QBV are also of the expected size. However, the 3' UTRs of AeFV and KRV are unusually long. The 3' UTR of AeFV consists of 945 nt while the 3' UTR of KRV consists of 1205 nt which is approximately twice the length of a typical flavivirus 3'UTR [
2]. It has been proposed that the unusually long KRV 3'UTR resulted from an almost complete duplication of a precursor sequence [
78]. According to the Genbank database, the complete genome of NIEV has also been sequenced (Genbank Accession No. NC_024299). However, the 3' UTR of this virus is remarkably short (167 nt) and therefore, we consider it likely that the sequence is truncated at the 3' end. Of the remaining cISFs, complete polyprotein ORF sequences are available for CTFV, HANKV, NAKV and PCV. Limited sequence data are available for AGFV and CLBOV; 556 and 946 nt of their NS5 genes have been sequenced, respectively.
Table 2.
Summary of sequence data available for classical insect-specific flaviviruses.
Table 2.
Summary of sequence data available for classical insect-specific flaviviruses.
| Virus | Sequence Data Available | Length of Genome (nt) | Length of 5’ UTR (nt) | Length of 3’ UTR (nt) | a Genbank Accession No. |
|---|
| Aedes flavivirus | Genome | 11,064 | 96 | 945 | NC_012932 |
| Aedes galloisi flavivirus | Partial NS5 | b - | - | - | AB639347 |
| Calbertado virus | Partial NS5 | - | - | - | EU569288 |
| Cell fusing agent virus | Genome | 10,695 | 113 | 556 | NC_001564 |
| Culex flavivirus | Genome | 10,834 | 91 | 657 | NC_008604 |
| Culex theileri flavivirus | ORF | - | - | - | HE574574 |
| Hanko virus | ORF | - | - | - | JQ268258 |
| Kamiti River virus | Genome | 11,375 | 96 | 1205 | NC_005064 |
| Nakiwogo virus | ORF | - | - | - | GQ165809 |
| Nienokoue virus | ORF | - | - | - | NC_024299 |
| Palm Creek virus | ORF | - | - | - | KC505248 |
| Quang Binh virus | Genome | 10,865 | 112 | 673 | NC_012671 |
The codon and dinucleotide usage preferences of cISFs are consistent with their apparent vertebrate-incompetent replication phenotype [
79,
80]. Vertebrates and invertebrates preferentially have certain codon and dinucleotide usage biases, and studies performed with RNA viruses have shown that their preferences often mimic those of their hosts [
81,
82,
83,
84]. Vertebrates display a strong under-representation of UpA and CpG, and over-representation of UpG and CpA. Mosquitoes also display a strong under-representation of UpA but have no bias for CpG depletion or for UpG and CpA excess [
85]. A comparison of the dinucleotide usage preferences of representative viruses from the cISF, NKV and dual-host groups (CxFV, MODV and WNV) revealed that MODV and to a lesser extent WNV demonstrate a CpG decrease while CxFV has no bias against this dinucleotide [
79]. All three viruses demonstrate an underutilization of UpA. Similar observations were reported when the dinucleotide usage preferences of CFAV, CxFV and KRV were compared to that of multiple NKV and dual-host flaviviruses [
80].
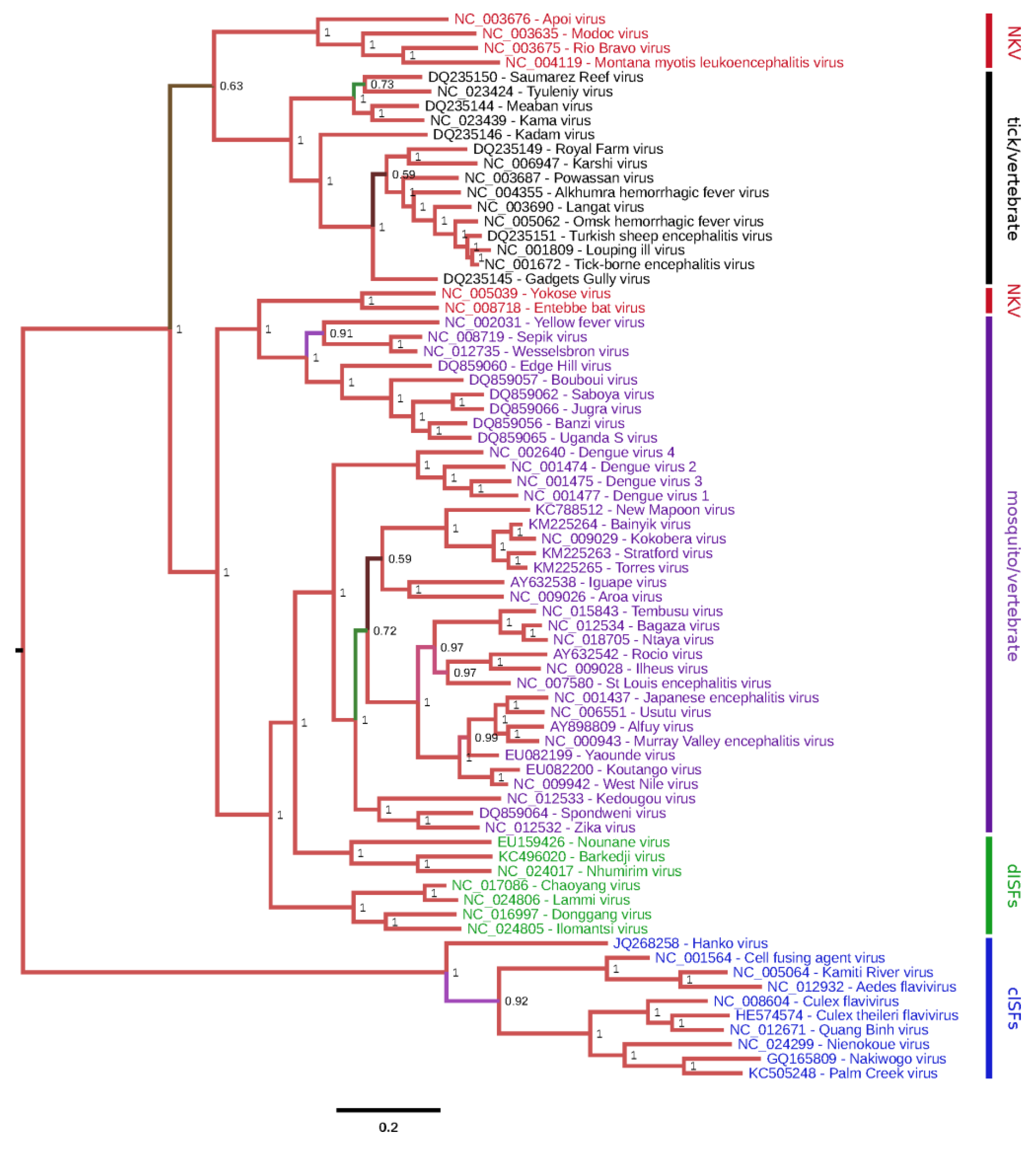
Classical ISFs are phylogenetically distinct from all other known flaviviruses (
Figure 1). These viruses currently separate into two main clades (
Figure 2). Clade 1 is composed of cISFs usually associated with
Aedes spp. mosquitoes (AEFV, AGFV, CFAV and KRV). Subclade 2 contains
Culex-associated viruses (CLBOV, CTFV CxFV, NIEV and QBV) in addition to NAKV and PCV, which were isolated from
Mansonia and
Coquillettidia spp. mosquitoes, respectively. HANKV is highly divergent from both clades 1 and 2 and may be regarded as forming a third clade. Although this virus has been detected in
Culex spp. mosquitoes, it is more frequently associated with
Aedes spp. mosquitoes [
25,
39,
42].
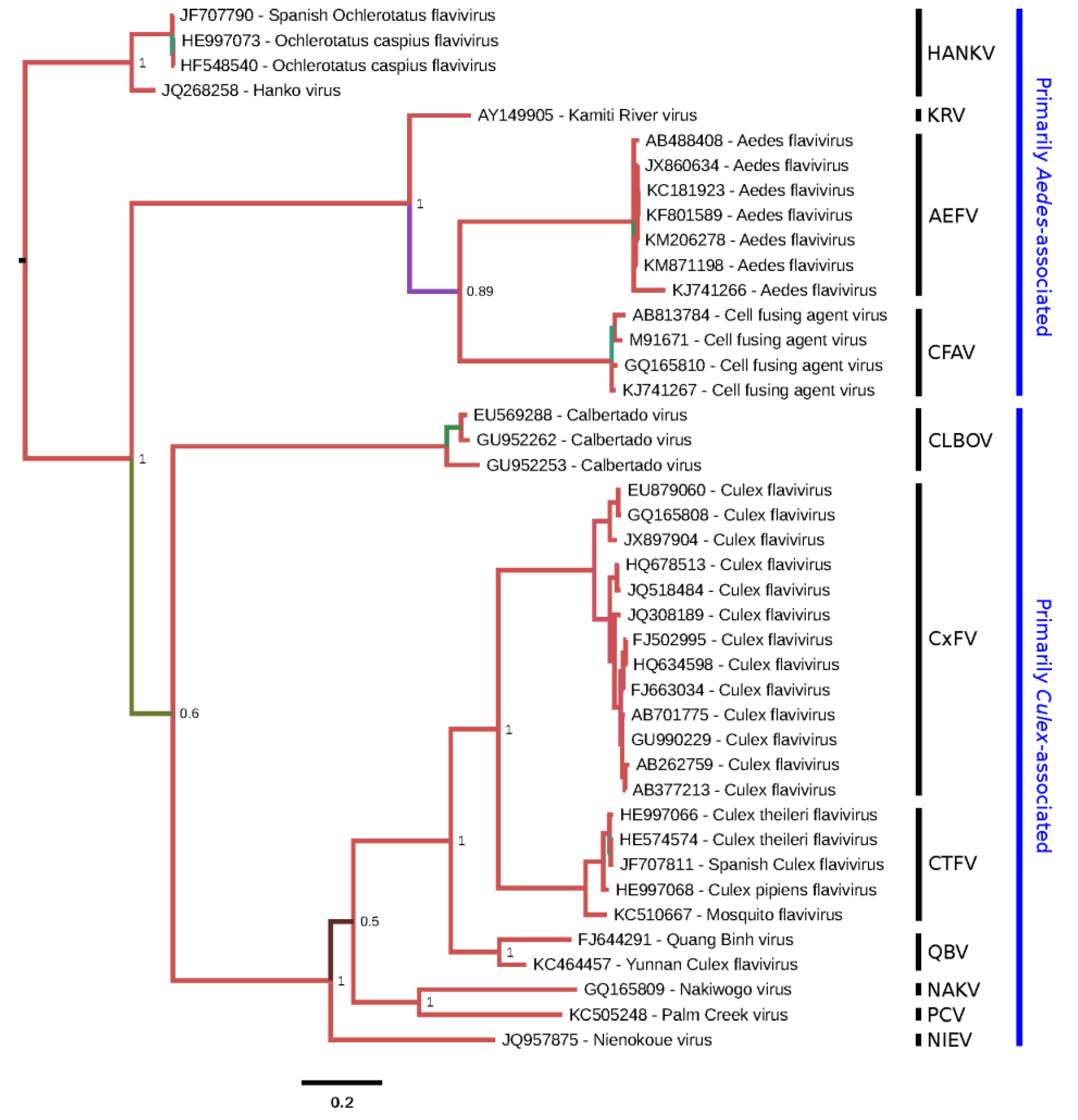
Figure 2.
Phylogenetic tree for selected cISF partial NS5 sequences. A 795-nt region of NS5 corresponding to nt 8916-9710 of M91671.1 (CFAV) was used in order to include CLBOV, for which only partial NS5 sequences are available. The corresponding amino acid sequences were aligned with MUSCLE [
14] and this amino acid alignment was used to guide a nucleotide sequence alignment. A maximum likelihood phylogenetic tree was estimated using the Bayesian Markov chain Monte Carlo method implemented in MrBayes version 3.2.3 [
16] using the general time reversible (GTR) substitution model with gamma-distributed rate variation across sites and a proportion of invariable sites. Chains were run for 10 million generations, with the first 25% discarded as burn-in. The figure was produced using FigTree (
http://tree.bio.ed.ac.uk/software/figtree/). Based on the full-genus tree (
Figure 1), HANKV was selected as an outgroup to root the tree. Nodes are labelled with posterior probability values and poorly supported branches are also highlighted with alternative colors. Tips are labelled with isolate names as provided in original publications or, if unpublished, in sequence records. Species (as defined in this review) are grouped (vertical black bars) and annotated at right.
Figure 2.
Phylogenetic tree for selected cISF partial NS5 sequences. A 795-nt region of NS5 corresponding to nt 8916-9710 of M91671.1 (CFAV) was used in order to include CLBOV, for which only partial NS5 sequences are available. The corresponding amino acid sequences were aligned with MUSCLE [
14] and this amino acid alignment was used to guide a nucleotide sequence alignment. A maximum likelihood phylogenetic tree was estimated using the Bayesian Markov chain Monte Carlo method implemented in MrBayes version 3.2.3 [
16] using the general time reversible (GTR) substitution model with gamma-distributed rate variation across sites and a proportion of invariable sites. Chains were run for 10 million generations, with the first 25% discarded as burn-in. The figure was produced using FigTree (
http://tree.bio.ed.ac.uk/software/figtree/). Based on the full-genus tree (
Figure 1), HANKV was selected as an outgroup to root the tree. Nodes are labelled with posterior probability values and poorly supported branches are also highlighted with alternative colors. Tips are labelled with isolate names as provided in original publications or, if unpublished, in sequence records. Species (as defined in this review) are grouped (vertical black bars) and annotated at right.
![Viruses 07 01927 g002]()
2.6. Ribosomal Frameshifting
Programmed -1 ribosomal frameshifting (-1 PRF) is the process by which specific mRNA sequences induce a proportion of ribosomes to shift -1 nt and continue translating in the new reading frame [
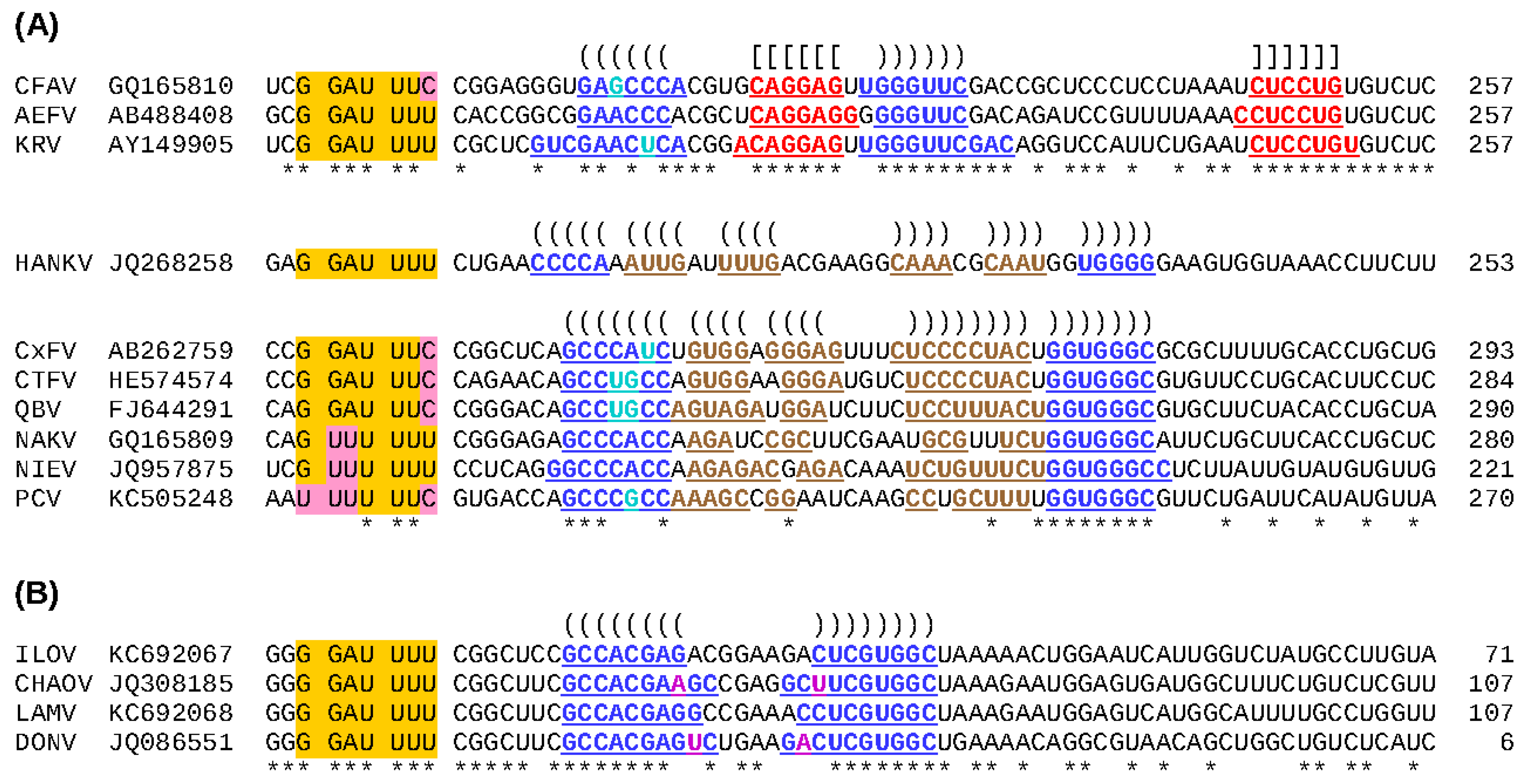
86]. PRF is utilized by many RNA viruses to control gene expression and to increase the number of protein products that can be expressed from a limited number of mRNA transcripts. The eukaryotic -1 frameshift site usually consists of a ‘slippery’ heptanucleotide fitting the motif X XXY YYZ (where XXX normally represents any three identical nucleotides although certain exceptions such as UCC, GGA, GUU and GGU also occur; YYY represents AAA or UUU; Z represents A, C or U; and spaces separate zero-frame codons), followed by a 5 to 9 nt ‘spacer’ region and then a stable RNA secondary structure such as a pseudoknot or stem-loop.
Figure 3.
Predicted -1 frameshift sites in ISFs. (
A) Apparently all cISFs contain a -1 PRF site just downstream of the predicted junction between the regions encoding NS1 and NS2A. Frameshifting results in translation of a long overlapping ORF, termed
fifo. The ‘slippery’ heptanucleotide sequence at which the -1 nt shift occurs is highlighted in orange, with nucleotide variations highlighted in pink. Ribosomes that shift -1 nt read the last nucleotide of the heptanucleotide twice. Predicted frameshift stimulatory elements (an RNA pseudoknot structure in the CFAV clade and an RNA stem-loop structure in the HANKV and CxFV clades) are annotated: nucleotides predicted to be involved in base-pairing interactions are colored and underlined, and predicted base-pairings are indicated with “()”s and “[]”s (see also
Figure 4). Conserved positions are indicated with “*”s. The length (in codons) of the
fifo ORF in each sequence is indicated at right; (
B) There is strong comparative genomic evidence that members of the dISF clade encompassing ILOV, CHAOV, LAMV and DONV contain a functionally utilized -1 PRF site towards the 3' end of the region encoding NS2B.
Figure 3.
Predicted -1 frameshift sites in ISFs. (
A) Apparently all cISFs contain a -1 PRF site just downstream of the predicted junction between the regions encoding NS1 and NS2A. Frameshifting results in translation of a long overlapping ORF, termed
fifo. The ‘slippery’ heptanucleotide sequence at which the -1 nt shift occurs is highlighted in orange, with nucleotide variations highlighted in pink. Ribosomes that shift -1 nt read the last nucleotide of the heptanucleotide twice. Predicted frameshift stimulatory elements (an RNA pseudoknot structure in the CFAV clade and an RNA stem-loop structure in the HANKV and CxFV clades) are annotated: nucleotides predicted to be involved in base-pairing interactions are colored and underlined, and predicted base-pairings are indicated with “()”s and “[]”s (see also
Figure 4). Conserved positions are indicated with “*”s. The length (in codons) of the
fifo ORF in each sequence is indicated at right; (
B) There is strong comparative genomic evidence that members of the dISF clade encompassing ILOV, CHAOV, LAMV and DONV contain a functionally utilized -1 PRF site towards the 3' end of the region encoding NS2B.
![Viruses 07 01927 g003]()
Previous studies have provided evidence that all known cISFs utilize -1 PRF to express a novel overlapping gene (designated
fifo) in the NS2A-NS2B regions of their genomes [
87]. However, CTFV, HANKV, NIEV and PCV had not been discovered when this conclusion was reached. To investigate whether -1 PRF is a universal feature of cISFs, the nucleotide sequences of all cISFs for which NS2A-NS2B data are available (as of 22 January 2015) were analyzed. All sequences were shown to possess a heptanucleotide sequence that conforms to the requirements of a ‘slippery’ -1 PRF motif (
Figure 3A) and a downstream
fifo ORF ranging from 221 (NIEV) to 293 (CxFV) codons. The exception to this is the laboratory-adapted isolate of CFAV; the
fifo ORF of this isolate is disrupted by three premature termination codons suggesting that the gene is dispensable for
in vitro replication. In addition to the slippery heptanucleotide frameshift site sequence, all cISF sequences were found to contain a potential stem-loop (CxFV and HANKV clades) or pseudoknot (CFAV clade) structure at the appropriate spacing downstream of the slippery heptanucleotide (
Figure 3A and
Figure 4A). It is interesting to note that this overlapping gene is unique to viruses in the cISF group; it is not encoded by the genomes of any other flaviviruses. Nevertheless, -1 PRF is utilized by various other flaviviruses. Apparently all viruses in the Japanese encephalitis (JE) serogroup, except for SLEV, utilize efficient -1 PRF to produce a larger NS1-related protein (NS1’) and to reduce synthesis of the 3'-encoded non-structural proteins relative to the proteins encoded upstream of the frameshift site [
88,
89,
90,
91]. -1 PRF has also been predicted to occur in many dISFs [
87] (see
Section 3f) and in Wesselsbron and Sepik viruses [
92].
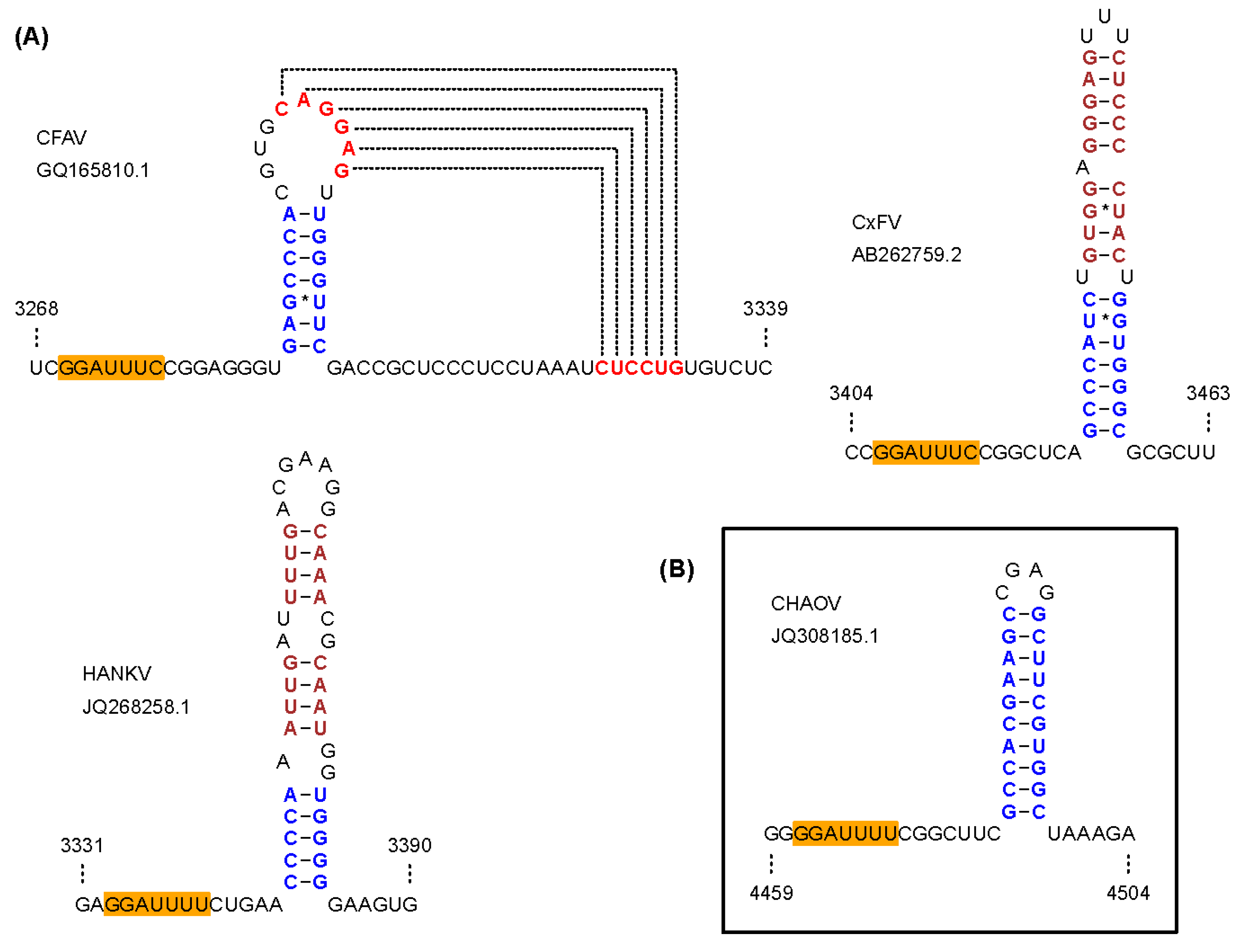
Figure 4.
Predicted frameshift-stimulatory RNA structures in ISFs. (A) Frameshifting in cISFs is predicted to be stimulated by an RNA pseudoknot structure in the CFAV clade, and an RNA stem-loop structure in the CxFV and HANKV clades; (B) Frameshifting in the CHAOV clade of dISFs is predicted to be stimulated by an RNA stem-loop structure.
Figure 4.
Predicted frameshift-stimulatory RNA structures in ISFs. (A) Frameshifting in cISFs is predicted to be stimulated by an RNA pseudoknot structure in the CFAV clade, and an RNA stem-loop structure in the CxFV and HANKV clades; (B) Frameshifting in the CHAOV clade of dISFs is predicted to be stimulated by an RNA stem-loop structure.
a Table 3.
Predicted cleavage sites in the polyproteins of cISFs.
a Table 3.
Predicted cleavage sites in the polyproteins of cISFs.
| Junction | AEFV | CFAV | CxFV | CTFV | HANKV | KRV | NAKV | NIEV | PCV | QBV | Dual-Host Flaviviruses |
|---|
| Virion C/Anch | b LEAQR↓SHSPV | c LESRR↓TTGNP | d LEAKR↓SAKNA | LEVRR↓SANNP | LEKER↓SHPRK | e LEKQR↓SGPNL | LEKRR↓GVWSP | LEQRR↓GAQRG | LEKKR↓DGRAA | LENRR↓SANPL | After dibasic residues |
| C/prM | b GLALS↓ETLRY | j VLCGC↓VVIDM | n MMVLG↓AVVID | VLCGC↓VIIDM | IVVTG↓LSIEL | e GLCYG↓EMLRY | VGIFS↓LNVVD | MVTFA↓AVVDV | FGVMG↓VVVID | TLCGT↓MVIDM | Signalase-like cleavage |
| pr/M | b PRKRR↓SSPQR | KREKR↓SREPP | d KRERR↓VASTN | KRVKR↓APETP | ERETR↓QKVDD | e VRRRR↓APQPQ | NRKQR↓SVKDE | RPVRR↓DVTPA | TRAKR↓VAPDG | KRVKR↓ATEQP | Furin |
| prM/E | b NVVRA↓TSIEP | j TTVKG↓EFVEP | d TTVKG↓EFVEP | TTVKG↓EFVEP | NVVKG↓EFVEP | e NVVKA↓SSIEP | TTVRG↓EFMEP | TTVSG↓EYLEP | TTVRG↓EYMEP | STVKG↓EFVEP | Signalase-like cleavage |
| E/NS1 | f RRVAG↓DIGCG | c YYVRA↓DLGCG | d VYTKA↓DVGCG | YFARA↓DVGCG | VYVKA↓DVGCG | e RSVSA↓DVGCG | YTVRA↓DFGCG | YYVRA↓DVGCG | YFVRA↓DFGCG | YYTRA↓DVGCG | Signalase-like cleavage |
| NS1/NS2A | b GKADA↓TADFH | c GKANA↓QSDFR | ° PPVEG↓SYPDF | PGTGA↓FPDFQ | YRVPS↓TNAED | e GKAHA↓CSDFR | PPSGA↓EKLQQ | GGAEA↓TQSFF | PMGET↓AKIQN | PGAEA↓LLQDF | Signalase-like cleavage |
| NS2A/NS2B | g KSSYR↓TSGRS | k RNGYR↓DSGAN | p RSGLR↓ASRRS | KSGLR↓ASKSS | RSGYR↓ALCSS | s KNGYR↓DYGAS | ASGLR↓KPRPH | KSGLR↓SITSW | GDGLR↓APRPH | KSGLR↓ASKRS | After dibasic residues |
| NS2B/NS3 | b NEHCR↓SDDLL | c TASNR↓SDDLL | q VSVFR↓SNEVN | STAYR↓AGVND | TNAFR↓SDELI | e SEQNR↓SDDLL | EFAQR↓SSSEL | STAQR↓SDLLL | AMSQR↓ANSEL | TSNRR↓SGVND | After dibasic residues |
| NS3/NS4A | h YINTR↓SSASL | l YMNCR↓GGPTL | r YLKQR↓SNFNF | FLKQR↓SGANF | YMGTR↓SFLSV | t YLNCR↓SSQTF | FLKQR↓SVLPF | FLKQR↓SLFID | FLKQR↓SLYFD | FLKQR↓SVLNF | After dibasic residues |
| NS4A/2K | AAGNR↓SYLDS | SIGNR↓SYMDS | NNVHR↓AYTTD | NNVHR↓AYTGD | SAGQR↓SYVDI | AIGNR↓SYMDS | GGSQR↓GILDS | ANSQR↓GFAEN | GGSQR↓GVLDS | TNVHR↓AYTGD | After dibasic residues |
| 2K/NS4B | b CSVLA↓WEMRL | c CGVLA↓WEMRM | d MGVVA↓WEMDL | MGVVA↓WELNL | IGVIC↓WELRL | e CGVLA↓WEMRL | IGIAA↓WELQL | SAVVA↓WELNL | IGVTA↓WELEL | MGIVA↓WELEL | Signalase-like cleavage |
| NS4B/NS5 | i FSKFR↓ALEKS | m FNQFR↓ALEKS | dRMALR↓SLVKT | RGGLR↓SLVKT | NITTR↓SLEKS | u FNQFR↓ALEKS | RLSVR↓SLVKS | LDMRR↓SLMKT | RLGVR↓SLVKS | RLATR↓SLVKT | After dibasic residues |
Immunoflorescence assays performed using polyclonal antibodies raised against two predicted CxFV FIFO antigens detected a protein product in CxFV-infected, but not mock-infected, C6/36 cells [
87]. However, bands of the expected size were not detected when lysates from CxFV-infected cells were analyzed by Western blot using these same antibodies. Thus, it is not known whether
fifo is expressed as a frameshift fusion simply with the N-terminal few amino acids of NS2A (
i.e., NS2A
N-FIFO) or, as in the case of the JE serogroup NS1' protein, as a fusion also with NS1 (
i.e., NS1-NS2A
N-FIFO). It is also possible that the fusion products are internally cleaved. Additional work is needed to investigate the expression and functional relevance of the
fifo product in cISFs.
2.7. Predicted Polyprotein Cleavage Sites
The predicted proteolytic cleavage sites of all cISFs for which complete polyprotein ORF data are available are shown in
Table 3. For the most part, these sites conform to the rules established for dual-host flaviviruses although there are some exceptions. Studies performed with dual-host flaviviruses have revealed that a host signal peptidase mediates cleavage between C/prM, prM/E, E/NS1 and 2K/NS4B and that these junctions typically conform to predicted signalase cleavage sites [
93]. Similar sites were identified at the predicted C/prM, prM/E, E/NS1 and 2K/NS4B junctions of most cISFs. The NS1/NS2A cleavage site of dual-host flaviviruses is signalase-like with respect to the '-1, -3' rule, but an upstream hydrophobic domain is absent. Previous work has demonstrated that, at least in DENV, cleavage requires translation of substantial parts of NS2A and it has been proposed that the hydrophobic domains in NS2A lead to a conformation that presents the NS1/NS2A cleavage site to an endoplasmic reticulum-resident host protease that may be signalase [
94]. In the dual-host flaviviruses, NS1/NS2A cleavage usually occurs after a Val-X-Ala site. Although the predicted cISFs cleavage sites generally lack one or other of these residues, they are still compatible with the signalase '-1, -3' rule [
95]. In dual-host flaviviruses, the cellular protease furin cleaves prM to generate the mature form of the protein [
93,
96]. Furin normally cleaves after the motif Arg-X-Lys/Arg-Arg but cleavage can also occur after Arg-X-X-Arg [
97]. As for dual-host flaviviruses, the predicted pr/M junction of every cISF is preceded by RXKR or RXRR, except for HANKV and NAKV which contain only the minimal furin cleavage site RXXR.
The virion C/anchor, NS2A/NS2B, NS2B/NS3, NS3/NS4A, NS4A/2K and NS4B/NS5 junctions of dual-host flaviviruses are cleaved by the viral NS2B/NS3 serine protease, which normally cleaves after two basic amino acid residues (KR, RR, RK) or sometimes after QR at the P
2 and P
1 positions, followed by a small amino acid (G, A or S) at the P'
1 position [
93,
98,
99]. The corresponding cISF cleavage sites are not always obvious and frequently appear to deviate from these motifs. It should be noted that only two cISF cleavage sites have been experimentally determined (viz. CFAV anchorC/prM and prM/E) both of which are signalase rather than viral protease cleavage sites [
18]. Alignment between cISF and dual-host or dISF flavivirus sequences at the NS2B/NS3, NS3/NS4A, NS4A/2K and NS4B/NS5 junctions suggests that cleavage in cISFs occurs between R at the P
1 position and G, A or S at the P'
1 position, but there seems to be substantial flexibility at the P
2 position (
Table 3). The exact cleavage site at the virion C/anchor junction was difficult to predict due to a cluster of basic amino acids; most cISFs contain a potential Arg-Gly/Ala/Ser (P
1-P'
1) cleavage site at the C-terminal end of the cluster of basic residues, while a few may use alternative motifs in this region. Prediction of the NS2A/NS2B cleavage site in cISFs was particularly problematic: while some species (e.g., CxFV) contain several Arg-Gly/Ala/Ser (P
1-P'
1) motifs in the critical region between two predicted transmembrane regions (aligning to the corresponding NS2A/NS2B junction in dual-host and dISF flaviviruses), other species contained no such motifs in this region. A conserved Arg residue was annotated as a potential cleavage site in
Table 3, notwithstanding that in CFAV, KRV, AEFV and NAKV it is followed by Asp, Asp, Thr and Lys, respectively, while in several species (including AEFV) there are closely spaced alternative cleavage sites. It should be noted that, due to the additional constraints imposed on this sequence region in the cISFs as a result of the overlapping
fifo ORF, it is possible that NS2A/NS2B cleavage in the cISFs may have evolved to take place at a somewhat different location than we have inferred from comparison to other flavivirus sequences.
In some instances, the predicted cleavage sites listed in
Table 3 are different from those reported by others. For example, the NS2A/NS2B cleavage sites that we proposed for AEFV, CFAV and KRV are located 4 to 25 residues upstream of the sites previously proposed for these viruses [
18,
19,
31,
44]. Our predicted NS4B/NS5 cleavage sites are located one residue upstream of the sites originally proposed for AEFV, CFAV and KRV [
18,
19,
44]. Our analysis was performed using additional cISF sequences, thus facilitating the identification of conserved sites. Nevertheless, our data are subjective and experimental data (e.g., amino acid sequencing) are needed to conclusively identify the cleavage sites in the polyproteins of cISFs.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



