
Mechanisms of Cancer Cell Killing by the Adenovirus E4orf4 Protein

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. The Unique Mode of E4orf4-Induced Cell Death
2.1. Cancer Specificity of E4orf4-Induced Cell Death
2.2. Caspase-Independent Cell Death Signaling that Feeds into Known Cell Death Pathways
2.3. Evolutionary Conservation of E4orf4 Cell Death Signaling

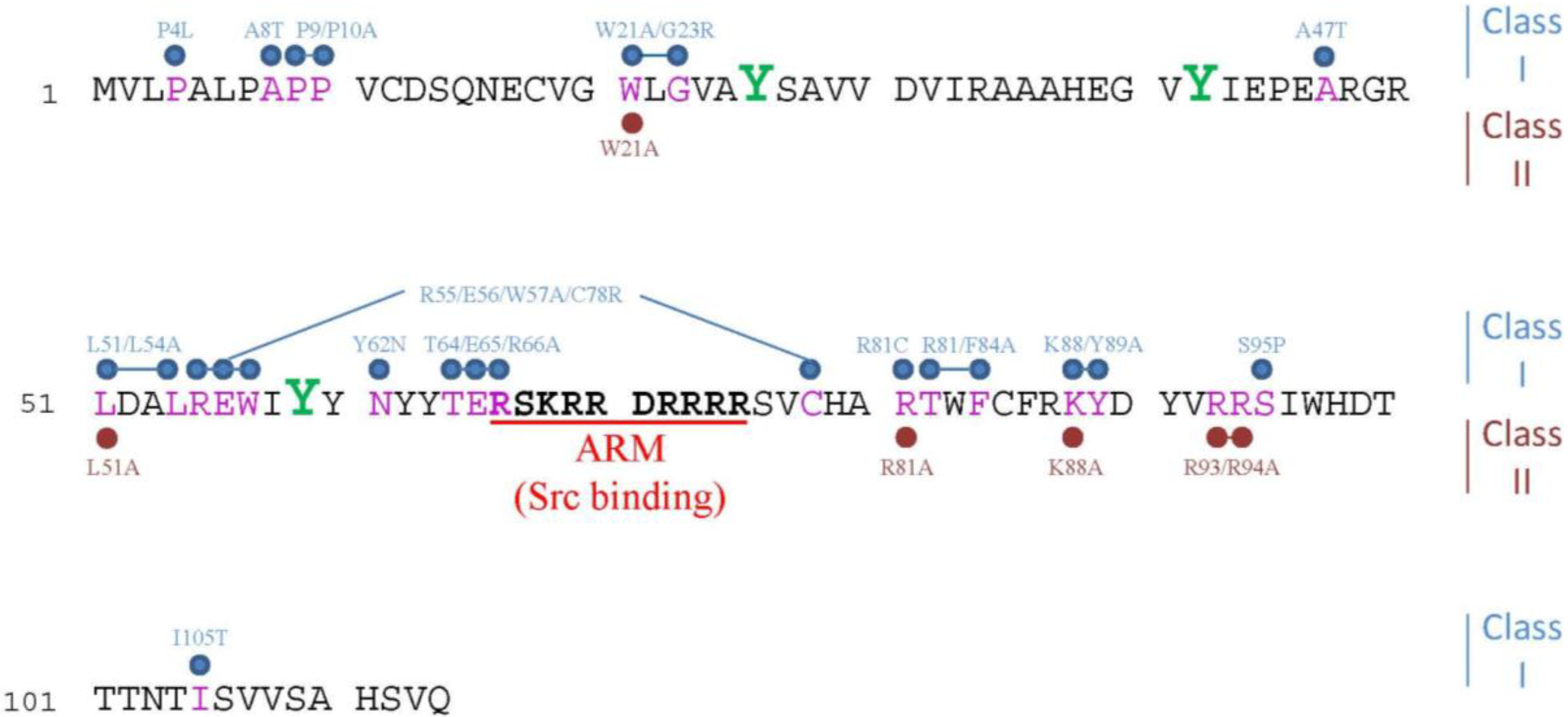
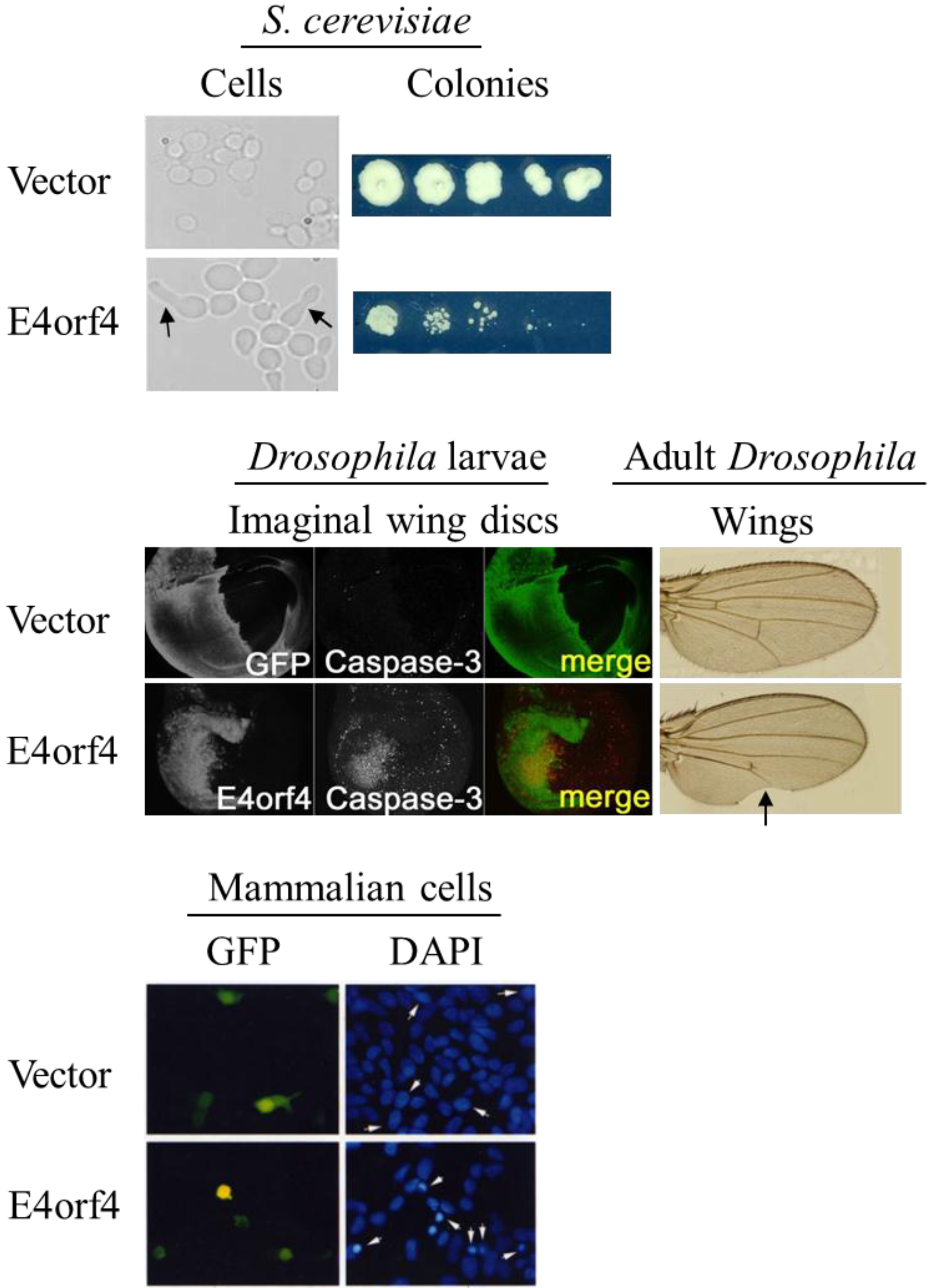
2.4. Morphological Hallmarks of E4orf4-Induced Cell Death and Assays for Measuring E4orf4-Induced Cell Killing
2.5. Perturbations of the Cell Cycle Precede E4orf4-Induced Cell Death
2.6. E4orf4 Induces Nuclear and Cytoplasmic Cell Death Programs
3. Mechanisms Underlying the Contribution of E4orf4-Associating Proteins to E4orf4-Induced Cell Death
3.1. PP2A
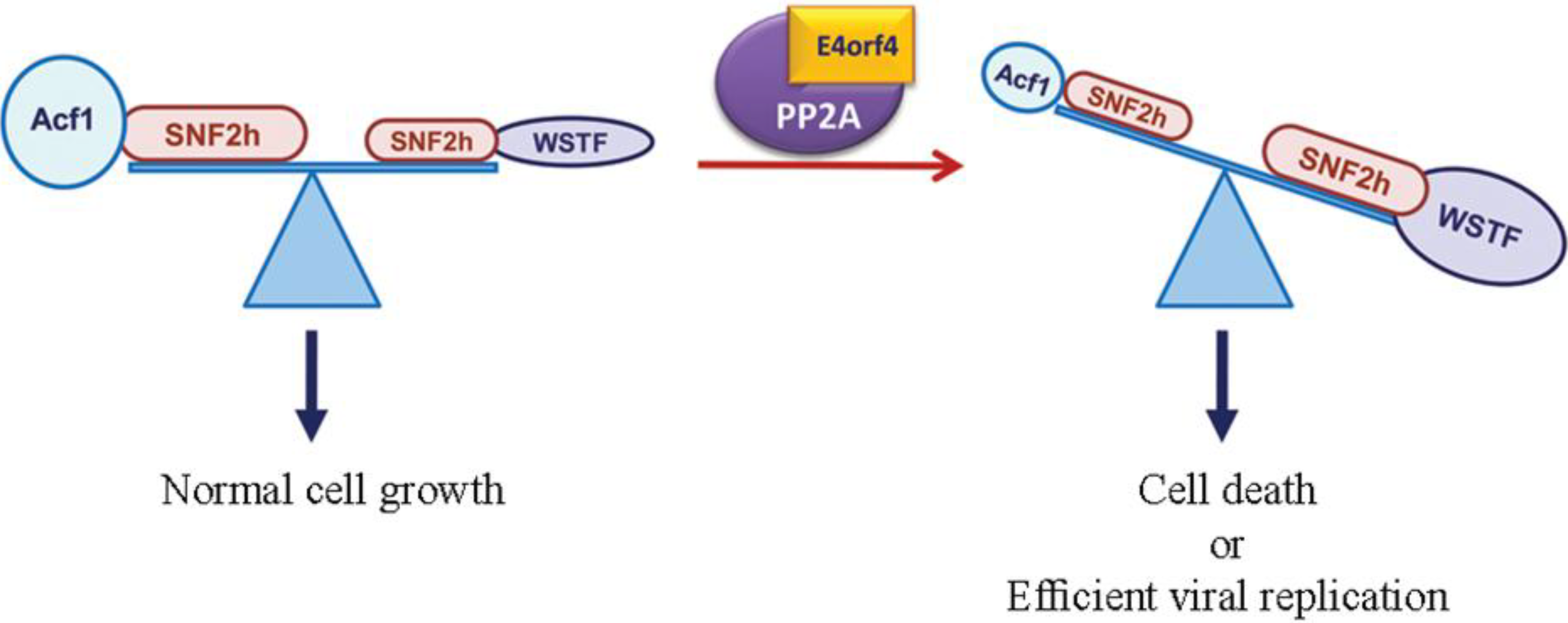
3.2. The ATP-Dependent Chromatin Remodeling Factor, ACF

3.3. The APC/C Ubiquitin Ligase Complex
3.4. The Golgi UDPase, Ynd1
3.5. Src Family Kinases (SFKs)
4. E4orf4-Induced Cell Death in an Animal Model
5. Perspectives

Acknowledgments
Conflicts of Interest
References
- Ben-Israel, H.; Sharf, R.; Rechavi, G.; Kleinberger, T. Adenovirus E4orf4 protein downregulates MYC expression through interaction with the PP2A-B55 subunit. J. Virol. 2008, 82, 9381–9388. [Google Scholar] [CrossRef] [PubMed]
- Bondesson, M.; Ohman, K.; Mannervik, M.; Fan, S.; Akusjarvi, G. Adenovirus E4 open reading 4 protein autoregulates E4 transcription by inhibiting E1A transactivation of the E4 promoter. J. Virol. 1996, 70, 3844–3851. [Google Scholar] [PubMed]
- Mannervik, M.; Fan, S.; Strom, A.C.; Helin, K.; Akusjarvi, G. Adenovirus E4 open reading frame 4-induced dephosphorylation inhibits E1A activation of the E2 promoter and E2F-1-mediated transactivation independently of the retinoblastoma tumor suppressor protein. Virology 1999, 256, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Muller, U.; Kleinberger, T.; Shenk, T. Adenovirus E4orf4 protein reduces phosphorylation of c-fos and E1A proteins while simultaneously reducing the level of AP-1. J. Virol. 1992, 66, 5867–5878. [Google Scholar] [PubMed]
- Estmer Nilsson, C.; Petersen-Mahrt, S.; Durot, C.; Shtrichman, R.; Krainer, A.R.; Kleinberger, T.; Akusjarvi, G. The adenovirus E4-orf4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J. 2001, 20, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Kanopka, A.; Muhlemann, O.; Petersen-Mahrt, S.; Estmer, C.; Ohrmalm, C.; Akusjarvi, G. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature 1998, 393, 185–187. [Google Scholar] [CrossRef] [PubMed]
- O'Shea, C.; Klupsch, K.; Choi, S.; Bagus, B.; Soria, C.; Shen, J.; McCormick, F.; Stokoe, D. Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J. 2005, 24, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Bridge, E.; Medghalchi, S.; Ubol, S.; Leesong, M.; Ketner, G. Adenovirus early region 4 and viral DNA synthesis. Virology 1993, 193, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Medghalchi, S.; Padmanabhan, R.; Ketner, G. Early region 4 modulates adenovirus DNA replication by two genetically separable mechanisms. Virology 1997, 236, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.M.; Hearing, P. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J. Virol. 1989, 63, 2605–2615. [Google Scholar] [PubMed]
- Marcellus, R.C.; Chan, H.; Paquette, D.; Thirlwell, S.; Boivin, D.; Branton, P.E. Induction of p53-independent apoptosis by the adenovirus E4orf4 protein requires binding to the Balpha subunit of protein phosphatase 2A. J. Virol. 2000, 74, 7869–7877. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, B.; Sharf, R.; Avital-Shacham, M.; Pechkovsky, A.; Kleinberger, T. Structure- and modeling-based identification of the adenovirus E4orf4 binding site in the protein phosphatase 2A B55alpha subunit. J. Biol. Chem. 2013, 288, 13718–13727. [Google Scholar] [CrossRef] [PubMed]
- Miron, M.J.; Gallouzi, I.E.; Lavoie, J.N.; Branton, P.E. Nuclear localization of the adenovirus E4orf4 protein is mediated through an arginine-rich motif and correlates with cell death. Oncogene 2004, 23, 7458–7468. [Google Scholar] [CrossRef] [PubMed]
- Champagne, C.; Landry, M.C.; Gingras, M.C.; Lavoie, J.N. Activation of adenovirus type 2 early region 4 ORF4 cytoplasmic death function by direct binding to Src kinase domain. J. Biol. Chem. 2004, 279, 25905–25915. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, T.; Shenk, T. Adenovirus E4orf4 protein binds to protein phosphatase 2A, and the complex down regulates E1A-enhanced junB transcription. J. Virol. 1993, 67, 7556–7560. [Google Scholar] [PubMed]
- Haesen, D.; Sents, W.; Lemaire, K.; Hoorne, Y.; Janssens, V. The basic biology of PP2A in hematologic cells and malignancies. Front. Oncol. 2014, 4, e347. [Google Scholar] [CrossRef]
- Koren, R.; Rainis, L.; Kleinberger, T. The scaffolding A/Tpd3 subunit and high phosphatase activity are dispensable for Cdc55 function in the Saccharomyces cerevisiae spindle checkpoint and in cytokinesis. J. Biol. Chem. 2004, 279, 48598–48606. [Google Scholar] [CrossRef] [PubMed]
- Roopchand, D.E.; Lee, J.M.; Shahinian, S.; Paquette, D.; Bussey, H.; Branton, P.E. Toxicity of human adenovirus E4orf4 protein in Saccharomyces cerevisiae results from interactions with the Cdc55 regulatory B subunit of PP2A. Oncogene 2001, 20, 5279–5290. [Google Scholar] [CrossRef] [PubMed]
- Shtrichman, R.; Sharf, R.; Barr, H.; Dobner, T.; Kleinberger, T. Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc. Natl. Acad. Sci. USA 1999, 96, 10080–10085. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Brignole, C.; Marcellus, R.; Thirlwell, S.; Binda, O.; McQuoid, M.J.; Ashby, D.; Chan, H.; Zhang, Z.; Miron, M.J.; et al. The adenovirus E4orf4 protein induces G2/M arrest and cell death by blocking PP2A activity regulated by the B55 subunit. J. Virol. 2009, 83, 8340–8352. [Google Scholar] [CrossRef] [PubMed]
- Shtrichman, R.; Sharf, R.; Kleinberger, T. Adenovirus E4orf4 protein interacts with both Bα and B' subunits of protein phosphatase 2A, but E4orf4-induced apoptosis is mediated only by the interaction with Bα. Oncogene 2000, 19, 3757–3765. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, A.; Lahav, M.; Bitman, E.; Salzberg, A.; Kleinberger, T. E4orf4 induces PP2A- and Src-dependent cell death in Drosophila melanogaster and at the same time inhibits classic apoptosis pathways. Proc. Natl. Acad. Sci. USA 2013, 110, E1724–E1733. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.N.; Nguyen, M.; Marcellus, R.C.; Branton, P.E.; Shore, G.C. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J. Cell Biol. 1998, 140, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Marcellus, R.C.; Lavoie, J.N.; Boivin, D.; Shore, G.C.; Ketner, G.; Branton, P.E. The early region 4 orf4 protein of human adenovirus type 5 induces p53-independent cell death by apoptosis. J. Virol. 1998, 72, 7144–7153. [Google Scholar] [PubMed]
- Shtrichman, R.; Kleinberger, T. Adenovirus type 5 E4 open reading frame 4 protein induces apoptosis in transformed cells. J. Virol. 1998, 72, 2975–2982. [Google Scholar] [PubMed]
- Miron, M.J.; Blanchette, P.; Groitl, P.; Dallaire, F.; Teodoro, J.G.; Li, S.; Dobner, T.; Branton, P.E. Localization and importance of the adenovirus E4orf4 protein during lytic infection. J. Virol. 2009, 83, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Branton, P.E.; Roopchand, D.E. The role of adenovirus E4orf4 protein in viral replication and cell killing. Oncogene 2001, 20, 7855–7865. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, H.O.; Rodriguez, J.; Govek, E.E.; Guo, W.; Kobayashi, R.; Hannon, G.; Lazebnik, Y.A. Oncogene-dependent apoptosis is mediated by caspase-9. Proc. Natl. Acad. Sci. USA 1998, 95, 13664–13669. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, A.; Salzberg, A.; Kleinberger, T. The adenovirus E4orf4 protein induces a unique mode of cell death while inhibiting classical apoptosis. Cell Cycle 2013, 12, 2343–2344. [Google Scholar] [CrossRef] [PubMed]
- Landry, M.C.; Champagne, C.; Boulanger, M.C.; Jette, A.; Fuchs, M.; Dziengelewski, C.; Lavoie, J.N. A functional interplay between the small GTPase Rab11a and mitochondria-shaping proteins regulates mitochondrial positioning and polarization of the actin cytoskeleton downstream of Src family kinases. J. Biol. Chem. 2014, 289, 2230–2249. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; Sanz-Gonzalez, S.M.; Moll, U.M.; Andres, V. Classic and novel roles of p53: Prospects for anticancer therapy. Trends Mol. Med. 2007, 13, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Mitrus, I.; Missol-Kolka, E.; Plucienniczak, A.; Szala, S. Tumour therapy with genes encoding apoptin and E4orf4. Anticancer Res. 2005, 25, 1087–1090. [Google Scholar] [PubMed]
- Wang, D.M.; Zhou, Y.; Xie, H.J.; Ma, X.L.; Wang, X.; Chen, H.; Huang, B.R. Cytotoxicity of a recombinant fusion protein of adenovirus early region 4 open reading frame 4 (E4orf4) and human epidermal growth factor on p53-deficient tumor cells. Anticancer Drugs 2006, 17, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, H.; Ma, X.L.; Xie, H.J.; Wang, C.L.; Zhang, S.H.; Wang, X.; Huang, B.R. Fusion protein of adenovirus E4orf4 and human epidermal growth factor inhibits tumor cell growth. Int. J. Cancer 2009, 125, 1186–1192. [Google Scholar] [CrossRef] [PubMed]
- Galioot, A.; Godet, A.N.; Maire, V.; Falanga, P.B.; Cayla, X.; Baron, B.; England, P.; Garcia, A. Transducing properties of a pre-structured alpha-helical DPT-peptide containing a short canine adenovirus type 2 E4orf4 PP2A1-binding sequence. Biochim. et Biophys. Acta 2013, 1830, 3578–3583. [Google Scholar] [CrossRef]
- Wolkersdorfer, G.W.; Morris, J.C.; Ehninger, G.; Ramsey, W.J. Trans-complementing adenoviral vectors for oncolytic therapy of malignant melanoma. J. Gene Med. 2004, 6, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Marcellus, R.C.; Teodoro, J.G.; Wu, T.; Brough, D.E.; Ketner, G.; Shore, G.; Branton, P.E. Adenovirus type 5 early region 4 is responsible for E1A-induced p53-independent apoptosis. J. Virol. 1996, 70, 6207–6215. [Google Scholar] [PubMed]
- Livne, A.; Shtrichman, R.; Kleinberger, T. Caspase activation by adenovirus E4orf4 protein is cell line-specific and is mediated by the death receptor pathway. J. Virol. 2001, 75, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Miron, M.J.; Champagne, C.; Gingras, M.C.; Branton, P.E.; Lavoie, J.N. Distinct cell death pathways triggered by the adenovirus early region 4 orf 4 protein. J. Cell Biol. 2002, 158, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Szymborski, A.; Miron, M.J.; Marcellus, R.; Binda, O.; Lavoie, J.N.; Branton, P.E. The adenovirus E4orf4 protein induces growth arrest and mitotic catastrophe in H1299 human lung carcinoma cells. Oncogene 2009, 28, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Janssens, V.; Goris, J. Protein phosphatase 2a: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Segawa, Y.; Suga, H.; Iwabe, N.; Oneyama, C.; Akagi, T.; Miyata, T.; Okada, M. Functional development of Src tyrosine kinases during evolution from a unicellular ancestor to multicellular animals. Proc. Natl. Acad. Sci. USA 2006, 103, 12021–12026. [Google Scholar] [CrossRef] [PubMed]
- Kornitzer, D.; Sharf, R.; Kleinberger, T. Adenovirus E4orf4 protein induces PP2A-dependent growth arrest in S. Cerevisiae and interacts with the anaphase promoting complex/cyclosome. J. Cell Biol. 2001, 154, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Ben-Israel, H.; Kleinberger, T. Adenovirus and cell cycle control. Front. Biosci. 2002, 7, d1369–d1395. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Rudner, A.D.; Murray, A.W. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 2000, 149, 1377–1390. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Protopopov, Y.; Listovsky, T.; Brandeis, M.; Hershko, A. Phosphorylation of the cyclosome is required for its stimulation by Fizzy/cdc20. Biochem. Biophys. Res. Commun. 1999, 260, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Mui, M.Z.; Roopchand, D.E.; Gentry, M.S.; Hallberg, R.L.; Vogel, J.; Branton, P.E. Adenovirus protein E4orf4 induces premature APCCdc20 activation in Saccharomyces cerevisiae by a protein phosphatase 2A-dependent mechanism. J. Virol. 2010, 84, 4798–4809. [Google Scholar] [CrossRef] [PubMed]
- Moshe, Y.; Bar-On, O.; Ganoth, D.; Hershko, A. Regulation of the action of early mitotic inhibitor 1 on the anaphase-promoting complex/cyclosome by cyclin-dependent kinases. J. Biol. Chem. 2011, 286, 16647–16657. [Google Scholar] [CrossRef] [PubMed]
- Guardavaccaro, D.; Kudo, Y.; Boulaire, J.; Barchi, M.; Busino, L.; Donzelli, M.; Margottin-Goguet, F.; Jackson, P.K.; Yamasaki, L.; Pagano, M. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev. Cell 2003, 4, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Margottin-Goguet, F.; Hsu, J.Y.; Loktev, A.; Hsieh, H.M.; Reimann, J.D.; Jackson, P.K. Prophase destruction of EMI1 by the SCF(betaTrcp/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev. Cell 2003, 4, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Cabon, L.; Sriskandarajah, N.; Mui, M.Z.; Teodoro, J.G.; Blanchette, P.; Branton, P.E. Adenovirus E4orf4 protein-induced death of p53-/- H1299 human cancer cells follows a G1 arrest of both tetraploid and diploid cells due to a failure to initiate DNA synthesis. J. Virol. 2013, 87, 13168–13178. [Google Scholar] [CrossRef] [PubMed]
- Sriskandarajah, N.; Blanchette, P.; Kucharski, T.J.; Teodoro, J.; Branton, P.E. Analysis by live imaging of effects of the adenovirus E4orf4 protein on passage through mitosis of H1299 tumor cells. J. Virol. 2015, 89, 4685–4689. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.; Poot, R.A.; Kukimoto, I.; Garcia-Jimenez, C.; Dellaire, G.; Varga-Weisz, P.D. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat. Genet. 2002, 32, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Brestovitsky, A.; Sharf, R.; Mittelman, K.; Kleinberger, T. The adenovirus E4orf4 protein targets PP2A to the ACF chromatin-remodeling factor and induces cell death through regulation of SNF2h-containing complexes. Nucleic Acids Res. 2011, 39, 6414–6427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yang, R.; Wang, S.; Dong, Z. Paclitaxel: New uses for an old drug. Drug Des. Dev. Ther. 2014, 8, 279–284. [Google Scholar]
- Sun, B.; Cai, Y.; Li, Y.; Li, J.; Liu, K.; Yang, Y. The nonstructural protein NP1 of human bocavirus 1 induces cell cycle arrest and apoptosis in Hela cells. Virology 2013, 440, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Begemann, M.; Kashimawo, S.A.; Lunn, R.M.; Delohery, T.; Choi, Y.J.; Kim, S.; Heitjan, D.F.; Santella, R.M.; Schiff, P.B.; Bruce, J.N.; et al. Growth inhibition induced by Ro 31–8220 and calphostin C in human glioblastoma cell lines is associated with apoptosis and inhibition of CDC2 kinase. Anticancer Res. 1998, 18, 3139–3152. [Google Scholar] [PubMed]
- Zhou, B.-B.; Li, H.; Yuan, J.; Kirschner, M.W. Caspase-dependent activation of Cyclin-dependent kinases during Fas-induced apoptosis in Jurkat cells. Proc. Natl. Acad. Sci. USA 1998, 95, 6785–6790. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Kroemer, G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002, 9, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Mitotic arrest and cell fate: Why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 2007, 6, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.T.; Karlseder, J. DNA damage associated with mitosis and cytokinesis failure. Oncogene 2013, 32, 4593–4601. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.N.; Champagne, C.; Gingras, M.-C.; Robert, A. Adenovirus E4 open reading frame 4-induced apoptosis involves dysregulation of Src family kinases. J. Cell Biol. 2000, 150, 1037–1055. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, T. Induction of cancer-specific cell death by the adenovirus E4orf4 protein. Adv. Exp. Med. Biol. 2014, 818, 61–97. [Google Scholar] [PubMed]
- Gingras, M.C.; Champagne, C.; Roy, M.; Lavoie, J.N. Cytoplasmic death signal triggered by Src-mediated phosphorylation of the adenovirus E4orf4 protein. Mol. Cell. Biol. 2002, 22, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, H.; Hsieh, T.C.; Pallas, D.C. Cdc55p-mediated E4orf4 growth-inhibition in S. cerevisiae is mediated only in part via the catalytic subunit of PP2A. J. Virol. 2008, 82, 3612–3623. [Google Scholar] [CrossRef] [PubMed]
- Mui, M.Z.; Kucharski, M.; Miron, M.J.; Hur, W.S.; Berghuis, A.M.; Blanchette, P.; Branton, P.E. Identification of the adenovirus E4orf4 protein binding site on the B55alpha and Cdc55 regulatory subunits of PP2A: Implications for PP2A function, tumor cell killing and viral replication. PLOS Pathog. 2013, 9, e1003742. [Google Scholar] [CrossRef] [PubMed]
- Swingle, M.; Ni, L.; Honkanen, R.E. Small-molecule inhibitors of ser/thr protein phosphatases: Specificity, use and common forms of abuse. Methods Mol. Biol. 2007, 365, 23–38. [Google Scholar] [PubMed]
- Katayose, Y.; Li, M.; Al-Murrani, S.W.; Shenolikar, S.; Damuni, Z. Protein phosphatase 2A inhibitors, I(1)(PP2A) and I(2)(PP2A), associate with and modify the substrate specificity of protein phosphatase 1. J. Biol. Chem. 2000, 275, 9209–9214. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Brautigan, D.L. The alpha4 regulatory subunit exerts opposing allosteric effects on protein phosphatases PP6 and PP2A. J. Biol. Chem. 2006, 281, 30503–30511. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, Y.; Zhang, P.; Jeffrey, P.D.; Shi, Y. Structure of a protein phosphatase 2A holoenzyme: Insights into B55-mediated Tau dephosphorylation. Mol. Cell 2008, 31, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Burke, D.J. Cdc55p, the B-type regulatory subunit of protein phosphatase 2A, has multiple functions in mitosis and is required for the kinetochore/spindle checkpoint in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997, 17, 620–626. [Google Scholar] [PubMed]
- Maoz, T.; Koren, R.; Ben-Ari, I.; Kleinberger, T. YND1 interacts with CDC55 and is a novel mediator of E4orf4-induced toxicity. J. Biol. Chem. 2005, 280, 41270–41277. [Google Scholar] [CrossRef] [PubMed]
- Vernieri, C.; Chiroli, E.; Francia, V.; Gross, F.; Ciliberto, A. Adaptation to the spindle checkpoint is regulated by the interplay between Cdc28/Clbs and PP2ACdc55. J. Cell Biol. 2013, 202, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Smolders, L.; Teodoro, J.G. Targeting the anaphase promoting complex: Common pathways for viral infection and cancer therapy. Expert Opin. Ther. Targets 2011, 15, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Afifi, R.; Sharf, R.; Shtrichman, R.; Kleinberger, T. Selection of apoptosis-deficient adenovirus E4orf4 mutants in S. cerevisiae. J. Virol. 2001, 75, 4444–4447. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.C.; Sevigny, J.; Zimmermann, H. The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinerg. Signal. 2006, 2, 409–430. [Google Scholar] [CrossRef]
- Zhong, X.; Guidotti, G. A yeast Golgi E-type ATPase with an unusual membrane topology. J. Biol. Chem. 1999, 274, 32704–32711. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H. Ectonucleotidases: Some recent developments and a note on nomenclature. Drug Dev. Res. 2001, 52, 44–56. [Google Scholar] [CrossRef]
- Avital-Shacham, M.; Sharf, R.; Kleinberger, T. NTPDASE4 gene products cooperate with the adenovirus E4orf4 protein through PP2A-dependent and -independent mechanisms and contribute to induction of cell death. J. Virol. 2014, 88, 6318–6328. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, K.; Ziv, K.; Maoz, T.; Kleinberger, T. The cytosolic tail of the Golgi apyrase Ynd1 mediates E4orf4-induced toxicity in Saccharomyces cerevisiae. PLOS ONE 2010, 5, e15539. [Google Scholar] [CrossRef] [PubMed]
- Landry, M.C.; Sicotte, A.; Champagne, C.; Lavoie, J.N. Regulation of cell death by recycling endosomes and golgi membrane dynamics via a pathway involving Src-family kinases, Cdc42 and Rab11a. Mol. Biol. Cell 2009, 20, 4091–4106. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Smadja-Lamere, N.; Landry, M.C.; Champagne, C.; Petrie, R.; Lamarche-Vane, N.; Hosoya, H.; Lavoie, J.N. Adenovirus E4orf4 hijacks rho GTPase-dependent actin dynamics to kill cells: A role for endosome-associated actin assembly. Mol. Biol. Cell 2006, 17, 3329–3344. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.N.; Landry, M.C.; Faure, R.L.; Champagne, C. Src-family kinase signaling, actin-mediated membrane trafficking and organellar dynamics in the control of cell fate: Lessons to be learned from the adenovirus E4orf4 death factor. Cell Signal 2010, 22, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Smadja-Lamere, N.; Boulanger, M.C.; Champagne, C.; Branton, P.E.; Lavoie, J.N. Jnk-mediated phosphorylation of paxillin in adhesion assembly and tension-induced cell death by the adenovirus death factor E4orf4. J. Biol. Chem. 2008, 283, 34352–34364. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Chen, F. Jnk-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res. 2012, 72, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Lin, A. Activation of the JNK signaling pathway: Breaking the brake on apoptosis. Bioessays 2003, 25, 17–24. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleinberger, T. Mechanisms of Cancer Cell Killing by the Adenovirus E4orf4 Protein. Viruses 2015, 7, 2334-2357. https://doi.org/10.3390/v7052334
Kleinberger T. Mechanisms of Cancer Cell Killing by the Adenovirus E4orf4 Protein. Viruses. 2015; 7(5):2334-2357. https://doi.org/10.3390/v7052334
Chicago/Turabian StyleKleinberger, Tamar. 2015. "Mechanisms of Cancer Cell Killing by the Adenovirus E4orf4 Protein" Viruses 7, no. 5: 2334-2357. https://doi.org/10.3390/v7052334
APA StyleKleinberger, T. (2015). Mechanisms of Cancer Cell Killing by the Adenovirus E4orf4 Protein. Viruses, 7(5), 2334-2357. https://doi.org/10.3390/v7052334